
Abstract
Tyrosine kinases play significant roles in tumor progression and therapy resistance. Inhibitors of tyrosine kinases are on the forefront of targeted therapy. For prostate cancer, tyrosine kinases play an additional role in the development of castration-resistant disease state, the most troubling aspect of prostate cancinogenesis which presently defies any effective treatment. Among the 30 or so tyrosine kinases expressed in a typical prostate cancer cell, nearly one third of them have been implicated in prostate carcinogenesis. Interestingly, most of them channel signals through a trio of non-receptor tyrosine kinases, Src/Etk/FAK, referred here as Src tyrosine kinase complex. This complex has been shown to play a significant role in the aberrant activation of androgen receptor (AR) mediated by growth factors (e.g., epidermal growth factor (EGF)), cytokines (interleukin (IL)-6), chemokines (IL-8), and neurokines (gastrin-releasing peptide). These factors are induced and released from the prostate cancer to the stromal cells upon androgen withdrawal. The Src kinase complex has the ability to phosphorylate androgen receptor, resulting in the nuclear translocation and stabilization of un-liganded androgen receptor. Indeed, tyrosine kinase inhibitors targeting Src can inhibit androgen-independent growth of prostate cancer cells in vitro and in preclinical xenograft model. While effective in inducing growth arrest and inhibiting metastasis of castration-resistant tumors, Src inhibitors rarely induce a significant level of apoptosis. This is also reflected by the general ineffectiveness of tyrosine kinase inhibitors as monotherapy in clinical trials. One of the underlying causes of apoptosis resistance is “autophagy,” which is induced by tyrosine kinase inhibitors and by androgen withdrawal. Autophagy is a self-digesting process to regenerate energy by removal of long-lived proteins and retired organelles to provide a survival mechanism to cells encountering stresses. Excessive autophagy, sometimes, could lead to type II programmed cell death. We demonstrated that autophagy blockade sensitizes prostate cancer cells toward Src tyrosine kinase inhibitor. Thus, a combination therapy based on Src tyrosine kinase inhibitor and autophagy modulator deserves further attention as a potential treatment for relapsed prostate cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Tyrosine Kinases and Prostate Cancer
There is very strong evidence that tyrosine kinases (TKs) are involved in the growth and metastasis of prostate cancer (for example [1, 2]). TKs also play key roles in tumor sensitivity to radiation and chemical-induced apoptosis. There are a total of 90 human TKs, 58 receptor TKs and 32 non-receptor TKs. Based on our extensive profilings of CaP cells, we found that on average, about 30 to 40 tyrosine kinases are expressed in prostate cancer cells [3]. For example in LNCap, we identified approximately 22 receptor kinases (ErbB1, ErbB2, ErbB3, EphA1, EphB1, EphB2, EphB3, EphB4, Sky, Mer, Ddr1, Ddr2, PDGFRB, FGFR2, FGFR3, FGFR4, IGFR, MET, RON, RET, TrkA, VGFR) and 14 non-receptor tyrosine kinases (SRC, FYN, YES, LYN, BRK, FER, CSK, ETK, ABL, ARG, JAK1, TYK2, FAK, PYK2). This is likely an underestimate due to the sensitivity of the RT-PCR conditions used.
Consistent with our profiling results, erbB/EGF-R, erbB2/HER2, erbB3/HER3, FGF-R, insulin-like growth factor Insulin-like Growth Factor (IGF)-R Met, Ron, Axl, Mer/Nyk, Kit, RET, and NGF-R have been implicated in prostate carcinogenesis [4–11]. For instance, EGF and IGF were shown to activate AR in the absence of androgen and induce androgen-independent growth of LNCaP cells via Src pathway [12, 13]. ErbB2/HER2 was reported to be overexpressed in androgen independent xenografts and confers androgen independence [14–17]. It’s elevated expression in clinically localized prostate cancer identifies metastatic potential [8]. Likewise, erbB3 is found to be required for the growth [18] and malignant progession of prostate cancer [19]. ErbB3 was shown to signal through Src and Etk/BMX non-receptor tyrosine kinases [18]. We first recognized the diverse pathways and varied effects of erbB2/erbB3 signaling [20, 21] and found that they also play a role in channeling signals transmitted by cytokines such as IL-6. Earlier work also showed that Mer (also called Nyk) is overexpressed in prostate carcinomas and is able to activate Src and increase the expression of a host of cytokines and chemokines ([22] and unpublished work). Recent studies showed that erbB2/erbB3 as well as Mer signaled through Ack1 non-receptor tyrosine kinase to phosphorylate and activate AR [23]. Likewise, Ret overexpression is associated with high-grade PIN and prostate cancer [24], and was an activator of Src [25].
Kit, usually found in hematopoietic stem cells, was shown to be aberrantly expressed as a truncated, activated receptor, which channels its effects by activating Src [26, 27]. IGF-1 receptor and Met receptor, as well as their ligands have been implicated in the development of advanced stages of CaP [27], although the expression of c-met is suppressed by AR [28, 29] and that of IGF-1R is augmented by androgen treatment. The ligand for Met, HGF, is involved in drug resistance [30] and metastasis [31] of CaP, with the former process involving Src signaling. The transcriptional activation of IGF-1R by androgen is via Src kinase [32]. These results suggest that different receptor TKs are implicated in different sets of CaPs and perhaps at various stages of the development. There is unlikely a common receptor TK target for all CaPs [33]. One interesting common thread of many of these receptor TKs is their ability to activate Src and its companion TKs, which will be the focus of our discussion below. While under normal conditions, c-Src activity is required for channeling multiple signals by these receptors and regulated by their ligands, elevated and constitutive Src activity, presumably due to the deregulation of the receptor activities, is a hallmark of end-stage CRPCs [34, 35].
Among non-receptor TKs, Src kinase complex, Src, Etk and FAK, as well as their respective family members figure prominently in CaPs and will be described separately in the next section. Little is known about the functions of other families of non-receptor tyrosine kinases in prostate carcinogenesis. Ack was found to be able to directly phosphorylate AR and the Ack/AR complex is recruited to the ARE sites in the nucleus [36]. C-abl is generally not expressed or expressed at very low levels in various cell lines, based on our profile results, and virtually nothing is known about its role in CaP. Brk, a Src-related kinase, was shown to have different subnuclear localization in CaP cells vs normal counterpart, although its role in prostate carcinogenesis is not clear [37]. For a more detailed description on non-receptor TK, please see our recent review [1].
The Src/FAK/Etk Tyrosine Kinase Complex
Src is found to form a complex (or complexes) with another two TKs FAK and Etk. Src is known to interact with FAK through SH2–phosphor-Y397 interaction [38, 39] and we showed that Etk interacts with FAK through PH–FERM domain interaction [40]. Recent structural data revealed that FERM domain serves as an autoinhibitory domain of FAK. Interaction of FERM with other proteins (in this case Etk) would “open” up the FAK kinase domain and exposes Y397 for binding to Src, resulting in the activation of both Src and FAK. We showed Src phosphorylates and activates Etk [41]. These three kinases, thus, cross-activate one another, and signal received by one would enhance the others. That said, these kinases have their own signal pathways and cellular functions. Their individual connections to CaP are briefly summarized below.
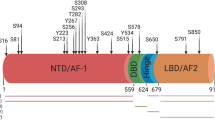
Although the first oncogene identified, Src’s role in human malignancies has not been fully appreciated. This is likely due to the lack of frequent genetic mutations of this gene in human cancers, although rare mutation has been reported in colon cancer [42]. Src is ubiquitously expressed in all prostate cancer samples analyzed ([43], and unpublished data). Its activity is upregulated in hormone refractory CaP [35]. In addition to Src, two other Src family kinases, Fyn and Lyn, are also implicated in CaP progression [44, 45]. Inhibitors for Src family kinases, Dasatinib [35] and Saracatinib [46, 47], both arrest CaP cell growth and migration in vitro with the effect on metastasis more pronounced in vivo. Strong evidence suggests that Src is involved in invasion and metastasis especially bone metastasis [45, 48–50]. Src kinase channel signals through the conventional Ras/Raf/ERK and PI3K/Akt, but also activates Stat3, c-myc, and beta-catenin [51–53] and Id-1 [54]. The activation of beta-catenin and Id-1 by Src are relevant to invasion and metastasis [50, 54] so is its phosphorylation of FAK [55, 56]. Prostate cells have a propensity to undergo neuroendocrine differentiation in which Src pathway may be involved, as recent reports showed that RPTPalpha and PTP1B, phosphatases which activate Src, induce neuroendocrine differentiation [57, 58]. Probably, most relevant to the development of CRPC (castration-resistant prostate cancer) is Src’s ability to activate androgen receptor and translocate AR into the nucleus [59–61] upon activation by neuropeptides [60, 62, 63] by chemokine IL-8 [64] and by EGF [44] (Fig. 1). Recently, it was shown that Src directly phosphorylates, stabilizes, and translocates AR, which is the major underlying mechanism whereby Src activates AR’s transcriptional activity and how growth factors and cytokines activate AR under androgen-depleted conditions [23, 63, 65, 66]. While Src is able to activate AR and serves as a coactivator, Src is reciprocally activated by AR and serves as a downstream effector of AR signaling [67, 68]. The latter is usually referred to as the non-genomic action of androgen receptor. The above discussion illustrates the role of Src and family members in prostate carcinogenesis; they are signal integrators of many growth factors and appropriate targets for therapy.

Src kinase complex is an integrator of signals from growth factor (EGF), cytokine (IL-6) to chemokines (GRP and IL-8). Src, Etk, and FAK cross-activate one another. Src and Etk kinases activate AR by direct phosphorylation leading to androgen-independent growth; Src and Etk signals suppress autophagy and apoptosis providing survival factors and together with FAK, they promote invasion and metastasis. (GRP) gastrin-releasing peptide; (G) G protein, (AR*) aberrantly activated receptor in the absence of natural ligand
FAK was originally identified as an Src substrate, with subsequent recognition of its role in focal adhesions upon integrin engagement. Src’s association with and phosphorylation of FAK increases FAK’s activity which then phosphorylates a host of proteins involved in cell adhesion and migration [38]. They include paxillin and p130cas [69]. FAK is involved in both the formation and dissolution of the focal adhesion plaques. FAK inhibitor inhibits the metastasis of PC3 in vivo, but does not affect its growth and survival [70]. Src interactions with FAK depend on FAK’s autophosphorylation of Y397, which is also the binding site for PI3K [71–74]. FAK is overexpressed, activated, and associated with paxillin in prostatic tissues from CaP patients with metastases [75, 76]. FAK is activated by neuropeptides and cell attachment, presumably via the RhoA pathway [77]. In the TRAMP mouse CaP model, FAK was shown to be required for androgen-independent formation of neuroendocrine carcinoma [78].
Etk belongs to the Btk family of TKs, typified by an N-terminal PH domain, followed by an SH3, SH2, and kinase domain. It was first reported as Bmx (X-linked bone marrow kinase) [79]. We independently cloned this kinase from the CWR22 CaP xenograft [80]. Etk is expressed in epithelial and endothelial cells, and hence its name. Our lab has carried out extensive structural and functional studies of this kinase in CaP [61, 81, 82]. The multidomain structure of Etk allows it to interact with a variety of signal molecules, including FAK, Src, RhoA, paxillin, STAT3, PAK1, PTPD1, VEGFR2, and p53. These molecules are known to be involved in migration, survival, and angiogenesis [83]. Through its PH domain, which binds PIP3, Etk is activated by PI3K [82], but in turn, it is a strong activator of PI3K pathway [84]. Etk’s autokinase activity is further augmented by Src phosphorylation at Y566 (the Src 418 equivalent) [41]. We and others showed that Etk activation can be triggered by a variety of ligands, including IL-6 [82], bombesin [61], integrin [40], EGF [85], neuregulin (ErbB2/erbB3 activator) [18], and vascular endothelial growth factor (VEGF) [86]. Upon activation by VEGF, Etk in turn augments the transcription of VEGF, providing a strong positive feedback loop during angiogenesis. Regarding cellular processes in CaP, Etk is involved in neuroendocrine differentiation, DNA-damage responses, hypoxia response, and the development of hormone refractory tumors. In most cases, it provides strong survival functions and protects CaP from thapsigargin and photodynamic therapy-induced apoptosis [87] and from p53-induced apoptosis through direct interaction with p53 [88], but under certain conditions (e.g., cleaved by Caspase), it can accelerate apoptosis [89], thus serving as a gatekeeper for cell survival. Etk was shown to be overexpressed in CaP and transgenic mice with prostate-specific expression of Etk developed PIN (prostatic intraepithelial neoplasia) [81, 88]. In a recent report, Etk is shown to be overexpressed in hormone-resistant tumors and correlates well with the phosphorylation of AR at Y534 site, the phosphoacceptor of Src and Etk kinases (see below) [90]. Its potential role in the development of CRPC is shown by its ability to mediate the activation of androgen receptor by IL-6 [91] and its association, phosphorylation, and stabilization of androgen receptor in a manner similar to Src [90]. Etk is found to phosphorylate AR at Y534 and Y551/552, although not as potent as Src. The detailed mechanism of AR activation by Etk is not all identical to Src; for instance, Etk’s association with AR requires phosphorylated tyrosine 551/552 of AR as anchor sites for the SH2 domain of Etk. By contrast, the association between Src and AR rely on the SH3/proline rich domains of Src and AR, respectively. In addition, the expression of Etk, but not Src, is in response of androgen deprivation, thus serving as a natural protector of prostate epithelial cells. That said, Etk is able to activate FAK and Src, and as such, the entire complex is involved in CRPC development. Both Etk and Src are important targets for therapeutic intervention.
Src Tyrosine Kinase Inhibitors and Prostate Cancer Therapy
There have been relatively few studies regarding the effects of Src family kinases inhibitors on CaP cell lines and xenografts. Dasatinib, a clinically approved thiazole and pyrimidine-based inhibitor, inhibits Src and Lyn TK at low nanomolar and effectively suppresses cell adhesion, invasion, and migration of DU145 [43]. This inhibition is accompanied by the decreased phosphorylation of FAK and p130cas, and decreased expression of MMP-9. Similarly, a pyrrolopyrimidine-based Src inhibitor inhibits the growth, adhesion, motility, and invasion of PC3 [92]. Another pyrazolo–pyrimidine-based Src inhibitor was shown to inhibit EGF-induced migration of PC3 [93]. A peptide inhibitor for Lyn inhibits the in vitro growth of DU145 and PC3 at low micromolar range. It reduces the tumor volume of in vivo DU145 explant in nude mice [44]. Thus, the Src family inhibitors appear to primarily affect migration and invasion of CaP cells and, to a lesser extent, growth. Our lab has been studying the efficacy of Saracatinib (AstraZeneca, formerly AZD0530) [94] and PP2 (Pfizer). The former is a drug under clinical development, whereas the latter is primarily a research tool. We found that the dual use of PP2 and Saracatinib will help us pinpoint the intended target better. In androgen-sensitive LNCaP cells, neuropeptides and EGF are able to activate Src, which in turn, translocates AR into the nucleus and activates AR target genes, resulting in androgen-independent growth of the cells [60, 62, 66]. Both PP2 and Saracatinib treatments reverse this course, resulting in the growth inhibition of these cells. Likewsie, Dasatinib was also found to inhibit phosphorylation-activated AR and androgen-independent growth of CaP under various treatment conditions [23]. Saracatinib inhibits Src family kinases with an IC50 at a high nanomolar range [46], but also targets c-abl with less potency. As a result, we initially chose DU145 for studies of signal attenuation, since this cell line expresses undetectable levels of c-abl and c-arg. To understand the general role of Src kinase in prostate cancer cell biology, the expression and activation were profiled in CWR22Rv1, DU145, LAPC-4, LNCaP, and PC3, as well as immortalized prostate epithelial cells such as RWPE-1. Saracatinib (and similarly PP2) was found to inhibit the growth of all cell lines with various IC50s. The IC50 for growth inhibition is around 6 μM for LNCaP, CWR22Rv1, and DU145, but more than 16 μM for RWPE-1. These results suggest that Src activation has oncogenic effects in both androgen receptor-dependent and independent CaPs, broadening the application of Src inhibitor as a treatment option for this disease. Both proliferation and migration of CaP cells are inhibited by the treatment, with little evidence of apoptosis, based on the appearance of sub-G1 fraction and caspase activation assays. These inhibitions were accompanied by reduced activation of the ERK1/2 pathway and reduced expression of β-catenin, c-myc, and cyclin D1, consistent with their being the downstream targets of Src and the arrest of growth at the G0/G1 phase. Likewise, the activation of cell motility factor such as FAK, p130CAS, and paxillin activation were inhibited. Using an in vivo androgen-independent xenograft model we developed, Saracatinib is found to be highly effective in inhibiting metastasis and to a much lesser extent, tumor growth [62]. It also exhibits efficacy in inhibiting bone metastasis of prostate cancer [49]. Despite these benefits, Src inhibitors do not seem to induce extensive killings of tumor cells, which compromise its efficacy as an anti-cancer agent. This, however, is not unique, as most of the tyrosine kinase inhibitors failed as a monotherapeutic agent. The key question then is how to enhance the killing effects of the Src inhibitor specifically and the tyrosine kinase inhibitors in general.
Src Tyrosine Kinase Inhibitor and Autophagy
To search for the potential cause of apoptosis-resistance in Src inhibitor-treated cells, we looked for prosurvival signals still active or even induced after treatment. The most consistent observation is the induction by Src inhibitors of a high level of autophagy or macroautophagy, which is generally viewed as a survival mechanism for cells in response to stress (nutritional, genotoxic, hypoxia, etc.) [47]; however, when deregulated, excessive autophagy leads to the postulated type II programmed cell death. Modulating autophagy has recently emerged as a possible means to benefit cancer therapy [95]. Autophagy or macroautophagy is a catabolic process involving the degradation of a cell’s own components through the lysosomal machinery. It begins with the formation of a double-membrane vesicle, or autophagosome, around a targeted region of the cell, separating the contents from the rest of the cytoplasm [96]. The resultant vesicle then fuses with a lysosome where the contents are degraded. A series of genes (ATG, autophagy genes), highly conserved from yeast to mammals, have been implicated in autophagic processes. For instance, autophagosome is formed by Atg5 and Atg12 conjugation, which then recruits microtubule-associated protein 1 light chain 3 (LC3), a mammalian homologue of Atg8. LC3 is cleaved by autophagin (Atg4) to produce the active cytosolic form LC3-I (18 kDa), which is then further activated by Atg7, transferred to Atg3, and posttranslationally modified into the membrane-bounded active form, LC3-II. The conversion of LC3 isoforms has been used as a biochemical index in autophagosome formation, and the number of autophagosomes (presented as cytoplasmic puncta) provides a semiquantitative measure of autophagy induction. A standard way of measuring autophagosome is to incorporate a GFP-LC3 construct into the cells to be studied. Upon autophagosome formation, the number of green dots represented by GFP-LC3-II embedded in autophagosome membrane provides a measure of the beginning step of autophagy. The cellular pathways that control the autophagic process are not well understood. The PTEN/PI3-kinase (Class I)/Akt/mTOR/S6K pathway have been strongly implicated as a negative regulator of autophagy. It is thought that S6K which regulates protein translation restricted the translation of some of the Atgs. Since Src/FAK tyrosine kinase complex associates with the p85 subunit of PI3 kinase and activates PI3 kinase, Src activation is expected to suppress autophagy and its inhibition to cause autophagy. Indeed, Saracatinib or PP2 both inhibit the activation of PI3 kinase downstream effectors, AKT, mTOR, and S6K [47]. Although the AKT-mTOR/S6K pathway is likely to contribute to the suppression of autophagy, it may not be the only one. Src kinase is known to enhance intracellular energy resource in favor of survival and growth. For instance, Src activates AKT and PI3 kinase, which are capable of transporting GLUT4 to the plasma membrane, resulting in glucose influx [97, 98]. Recently, Src activation is also seen to be responsible for thrombin-induced p38MAPK-mediated activation of glucose transporter [99]. Likewise, Src is known to activate HIF-1alpha via the degradation of VHL, an E3 Ub ligase for HIF-1alpha [100], and HIF-1alpha transcriptionally activates GLUT1, also resulting in a rich supply of glucose [40]. Src inhibition may thus result in glucose depletion, a condition known to induce autophagy. In addition, Src may also function to provide an amino acid balance for the anaerobic glycolysis pathway by increasing the higher level of glutamine via c-myc, a target gene of Src [101]. Src inhibition would thus lead to an energy imbalance, another condition known to induce autophagy. Thus, Src inhibition may engender a nutritional starvation condition, leading to autophagy. Finally, as discussed above, Src inhibition severely compromises AR activity, which is another potential cause for autophagy. Li et al. previously showed that androgen depletion which attenuates AR activity results in autopahgy [102]. The exact mechanism as to how Src inhibitor induces autophagy, deserves further attention. Our results that Src inhibitor induces autophagy may not be unique to this system. Recently, imatinib, an inhibitor of Abl/Src has also been found to induce a significant level of autophagy, which may account for its lower efficiency as a monotherapeutic agent [103].
Autophagy Blockade Enhances the Killing Effects of Src Inhibitors
Having demonstrated that Src inhibitors induced profound autophagy in prostate cancer cells, we asked whether autophagy may contribute to the apoptosis-resistance observed for tyrosine kinase inhibitors therapy. To test this thesis, we looked for inhibitors of the autophagy process and assessed its biological effects when used in combination with Saracatinib. The idea behind these experiments is that if autophagy is the barrier to apoptosis, autophagy inhibitor would enhance the Saracatinib-induced killing of prostate cancer cells. We chose both a genetic inhibitor, shRNA against Beclin, a critical ingredient of autophagy and a pharmacological inhibitor, chloroquine (CQ) for this study. Chloroquine is a lysotropic agent which disrupts lysosomal functions by blocking autophagosome fusion and inability to degrade autophagosome and its cargo. Chloroquine is a well-tolerated, clinically approved antimalarial agent, increasing its prospect as an adjunctive therapy. Using shRNA against Beclin (Atg8) and CQ, we showed unambiguously that inhibition of autophagy dramatically enhanced the apoptosis-inducing effect of Src tyrosine kinase inhibitor. In vivo testing using PC3 xenograft confirmed a synergistic effect of Saracatinib and CQ on the inhibition of tumor growth and enhanced apoptosis in the tumor tissues. This finding suggests that autophagy modulator may serve as an effective adjunctive therapeutic agent to complement tyrosine kinase inhibitor [47].
Conclusion
Since the identification of the first tyrosine kinase profile in prostate cancer in 1996, numerous studies have implicated aberrant expression and activation of tyrosine kinases in the progression of prostate cancer, especially during the transition to a castration-resistant state. A common signal integrator of many of these tyrosine kinases is the Src tyrosine kinase complex, including Src, FAK, and Etk/BMX tyrosine kinases, which control growth, migration, survival, and tumor resistance. They are also involved in the phosphorylation and aberrant activation of androgen receptor in a ligand-independent manner. Targeting Src tyrosine kinase complex, thus represents a potential treatment option for castration-resistant prostate cancer, in conjunction with hormone therapy. As amply evident from recent exciting discoveries, tyrosine kinases are promising targets for cancer therapeutics and are on the frontline of targeted therapy; however, as monotherapy, tyrosine kinase inhibitors have not been found to be particularly effective. One possible reason is their propensity of inducing autophagy, which confers resistance to apoptosis. Several clinically approved drugs which block autophagy were shown to enhance cell killing, when used in combination with tyrosine kinase inhibitors. We look forward to the future development of more specific tyrosine kinase inhibitors as well as autophagy modulators and their applications to castration-resistant prostate cancers.
References
Chang YM, Kung HJ, Evans CP (2007) Nonreceptor tyrosine kinases in prostate cancer. Neoplasia 9:90–100
Kung H, Tepper C, de Vere WR (2001) Tyrosine kinases and cellular signaling in prostate cancer. In: Chung LWK, Isaacs W, Simons J (eds) Prostate cancer: biology, genetics and the new therapeutics. Humana Press, Inc., New Jersey, pp 241–266
Robinson D, He F, Pretlow T, Kung HJ (1996) A tyrosine kinase profile of prostate carcinoma. Proc Natl Acad Sci USA 93:5958–5962
Damon SE, Plymate SR, Carroll JM et al (2001) Transcriptional regulation of insulin-like growth factor-I receptor gene expression in prostate cancer cells. Endocrinology 142:21–27
Davies G, Jiang WG, Mason MD (2001) HGF/SF modifies the interaction between its receptor c-Met, and the E-cadherin/catenin complex in prostate cancer cells. Int J Mol Med 7:385–388
Kwabi-Addo B, Ropiquet F, Giri D, Ittmann M (2001) Alternative splicing of fibroblast growth factor receptors in human prostate cancer. Prostate 46:163–172
Gimm O, Armanios M, Dziema H, Neumann HP, Eng C (2000) Somatic and occult germ-line mutations in SDHD, a mitochondrial complex II gene, in nonfamilial pheochromocytoma. Cancer Res 60:6822–6825
Ricciardelli C, Jackson MW, Choong CS et al (2008) Elevated levels of HER-2/neu and androgen receptor in clinically localized prostate cancer identifies metastatic potential. Prostate 68:830–838
Shiozawa Y, Pedersen EA, Patel LR et al (2010) GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia 12:116–127
Thobe MN, Gurusamy D, Pathrose P, Waltz SE (2010) The Ron receptor tyrosine kinase positively regulates angiogenic chemokine production in prostate cancer cells. Oncogene 29:214–226
Wiesner C, Nabha SM, Dos Santos EB et al (2008) C-kit and its ligand stem cell factor: potential contribution to prostate cancer bone metastasis. Neoplasia 10:996–1003
Culig Z, Hobisch A, Cronauer MV et al (1994) Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res 54:5474–5478
Maecker HL, Yun Z, Maecker HT, Giaccia AJ (2002) Epigenetic changes in tumor Fas levels determine immune escape and response to therapy. Cancer Cell 2:139–148
Craft N, Shostak Y, Carey M, Sawyers CL (1999) A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat Med 5:280–285
Gregory CW, Whang YE, McCall W et al (2005) Heregulin-induced activation of HER2 and HER3 increases androgen receptor transactivation and CWR-R1 human recurrent prostate cancer cell growth. Clin Cancer Res 11:1704–1712
Wen Y, Hu MC, Makino K et al (2000) HER-2/neu promotes androgen-independent survival and growth of prostate cancer cells through the Akt pathway. Cancer Res 60:6841–6845
Yeh S, Lin HK, Kang HY, Thin TH, Lin MF, Chang C (1999) From HER2/Neu signal cascade to androgen receptor and its coactivators: a novel pathway by induction of androgen target genes through MAP kinase in prostate cancer cells. Proc Natl Acad Sci USA 96:5458–5463
Jiang X, Borgesi RA, McKnight NC, Kaur R, Carpenter CL, Balk SP (2007) Activation of nonreceptor tyrosine kinase Bmx/Etk mediated by phosphoinositide 3-kinase, epidermal growth factor receptor, and ErbB3 in prostate cancer cells. J Biol Chem 282:32689–32698
Soler M, Mancini F, Meca-Cortes O et al (2009) HER3 is required for the maintenance of neuregulin-dependent and -independent attributes of malignant progression in prostate cancer cells. Int J Cancer 125:2565–2575
Grasso AW, Wen D, Miller CM, Rhim JS, Pretlow TG, Kung HJ (1997) ErbB kinases and NDF signaling in human prostate cancer cells. Oncogene 15:2705–2716
Qiu Y, Ravi L, Kung HJ (1998) Requirement of ErbB2 for signalling by interleukin-6 in prostate carcinoma cells. Nature 393:83–85
Wu YM, Robinson DR, Kung HJ (2004) Signal pathways in up-regulation of chemokines by tyrosine kinase MER/NYK in prostate cancer cells. Cancer Res 64:7311–7320
Liu Y, Karaca M, Zhang Z, Gioeli D, Earp HS, Whang YE (2010) Dasatinib inhibits site-specific tyrosine phosphorylation of androgen receptor by Ack1 and Src kinases. Oncogene 29:3208–3216
Dawson DM, Lawrence EG, MacLennan GT et al (1998) Altered expression of RET proto-oncogene product in prostatic intraepithelial neoplasia and prostate cancer. J Natl Cancer Inst 90:519–523
Encinas M, Crowder RJ, Milbrandt J, Johnson EM Jr (2004) Tyrosine 981, a novel ret autophosphorylation site, binds c-Src to mediate neuronal survival. J Biol Chem 279:18262–18269
Paronetto MP, Farini D, Sammarco I et al (2004) Expression of a truncated form of the c-Kit tyrosine kinase receptor and activation of Src kinase in human prostatic cancer. Am J Pathol 164:1243–1251
Marelli MM, Moretti RM, Procacci P, Motta M, Limonta P (2006) Insulin-like growth factor-I promotes migration in human androgen-independent prostate cancer cells via the alphavbeta3 integrin and PI3-K/Akt signaling. Int J Oncol 28:723–730
Hagmar L, Stromberg U, Bonassi S et al (2004) Impact of types of lymphocyte chromosomal aberrations on human cancer risk: results from Nordic to Italian cohorts. Cancer Res 64:2258–2263
Verras M, Lee J, Xue H, Li TH, Wang Y, Sun Z (2007) The androgen receptor negatively regulates the expression of c-Met: implications for a novel mechanism of prostate cancer progression. Cancer Res 67:967–975
Fan S, Meng Q, Laterra JJ, Rosen EM (2009) Role of Src signal transduction pathways in scatter factor-mediated cellular protection. J Biol Chem 284:7561–7577
Dai Y, Siemann DW (2010) BMS-777607, a small-molecule met kinase inhibitor, suppresses hepatocyte growth factor-stimulated prostate cancer metastatic phenotype in vitro. Mol Cancer Ther 9:1554–1561
Wu JD, Haugk K, Woodke L, Nelson P, Coleman I, Plymate SR (2006) Interaction of IGF signaling and the androgen receptor in prostate cancer progression. J Cell Biochem 99:392–401
Fizazi K (2007) The role of Src in prostate cancer. Ann Oncol 18(11):1765–1773
Mendiratta P, Mostaghel E, Guinney J et al (2009) Genomic strategy for targeting therapy in castration-resistant prostate cancer. J Clin Oncol 27:2022–2029
Tatarov O, Mitchell TJ, Seywright M, Leung HY, Brunton VG, Edwards J (2009) SRC family kinase activity is up-regulated in hormone-refractory prostate cancer. Clin Cancer Res 15:3540–3549
Mahajan NP, Whang YE, Mohler JL, Earp HS (2005) Activated tyrosine kinase Ack1 promotes prostate tumorigenesis: role of Ack1 in polyubiquitination of tumor suppressor Wwox. Cancer Res 65:10514–10523
Derry JJ, Prins GS, Ray V, Tyner AL (2003) Altered localization and activity of the intracellular tyrosine kinase BRK/Sik in prostate tumor cells. Oncogene 22:4212–4220
Mitra SK, Schlaepfer DD (2006) Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol 18:516–523
Lietha D, Cai X, Ceccarelli DF, Li Y, Schaller MD, Eck MJ (2007) Structural basis for the autoinhibition of focal adhesion kinase. Cell 129:1177–1187
Chen R, Kim O, Li M et al (2001) Regulation of the PH-domain-containing tyrosine kinase Etk by focal adhesion kinase through the FERM domain. Nat Cell Biol 3:439–444
Tsai YT, Su YH, Fang SS et al (2000) Etk, a Btk family tyrosine kinase, mediates cellular transformation by linking Src to STAT3 activation. Nat Cell Biol 20:2043–2054
Irby RB, Mao W, Coppola D et al (1999) Activating SRC mutation in a subset of advanced human colon cancers. Nat Genet 21:187–190
Nam S, Kim D, Cheng JQ et al (2005) Action of the Src family kinase inhibitor, dasatinib (BMS-354825), on human prostate cancer cells. Cancer Res 65:9185–9189
Goldenberg-Furmanov M, Stein I, Pikarsky E et al (2004) Lyn is a target gene for prostate cancer: sequence-based inhibition induces regression of human tumor xenografts. Cancer Res 64:1058–1066
Saito YD, Jensen AR, Salgia R, Posadas EM (2010) Fyn: a novel molecular target in cancer. Cancer 116:1629–1637
Chang YM, Bai L, Liu S, Yang JC, Kung HJ, Evans CP (2008) Src family kinase oncogenic potential and pathways in prostate cancer as revealed by AZD0530. Oncogene 27:6365–6375
Wu Z, Chang P, Yang J et al (2010) Autophagy blockade sensitizes prostate cancer cell towards Src family kinase inhibitors. Genes Cancer 1:40–49
Lee YC, Huang CF, Murshed M et al (2010) Src family kinase/abl inhibitor dasatinib suppresses proliferation and enhances differentiation of osteoblasts. Oncogene 29:3196–3207
Yang JC, Bai L, Yap S, Gao AC, Kung HJ, Evans CP (2010) Effect of the specific Src family kinase inhibitor saracatinib on osteolytic lesions using the PC-3 bone model. Mol Cancer Ther 9:1629–1637
Park SI, Zhang J, Phillips KA et al (2008) Targeting SRC family kinases inhibits growth and lymph node metastases of prostate cancer in an orthotopic nude mouse model. Cancer Res 68:3323–3333
Bowman T, Broome MA, Sinibaldi D et al (2001) Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc Natl Acad Sci USA 98:7319–7324
Furstoss O, Dorey K, Simon V, Barila D, Superti-Furga G, Roche S (2002) c-Abl is an effector of Src for growth factor-induced c-myc expression and DNA synthesis. EMBO J 21:514–524
Krizbai IA, Bauer H, Bresgen N et al (2005) Effect of oxidative stress on the junctional proteins of cultured cerebral endothelial cells. Cell Mol Neurobiol 25:129–139
Gautschi O, Tepper CG, Purnell PR et al (2008) Regulation of Id1 expression by SRC: implications for targeting of the bone morphogenetic protein pathway in cancer. Cancer Res 68:2250–2258
Hauck CR, Hsia DA, Schlaepfer DD (2002) The focal adhesion kinase—a regulator of cell migration and invasion. IUBMB Life 53:115–119
Planas-Silva MD, Bruggeman RD, Grenko RT, Stanley Smith J (2006) Role of c-Src and focal adhesion kinase in progression and metastasis of estrogen receptor-positive breast cancer. Biochem Biophys Res Commun 341:73–81
Wu C, Zhang L, Bourne PA et al (2006) Protein tyrosine phosphatase PTP1B is involved in neuroendocrine differentiation of prostate cancer. Prostate 66:1125–1135
Zhang XQ, Kondrikov D, Yuan TC, Lin FF, Hansen J, Lin MF (2003) Receptor protein tyrosine phosphatase alpha signaling is involved in androgen depletion-induced neuroendocrine differentiation of androgen-sensitive LNCaP human prostate cancer cells. Oncogene 22:6704–6716
Asim M, Siddiqui IA, Hafeez BB, Baniahmad A, Mukhtar H (2008) Src kinase potentiates androgen receptor transactivation function and invasion of androgen-independent prostate cancer C4-2 cells. Oncogene 27:3596–3604
Desai SJ, Ma AH, Tepper CG, Chen HW, Kung HJ (2006) Inappropriate activation of the androgen receptor by nonsteroids: involvement of the Src kinase pathway and its therapeutic implications. Cancer Res 66:10449–10459
Lee LF, Guan J, Qiu Y, Kung HJ (2001) Neuropeptide-induced androgen independence in prostate cancer cells: roles of nonreceptor tyrosine kinases Etk/Bmx, Src, and focal adhesion kinase. Mol Cell Biol 21:8385–8397
Yang JC, Ok JH, Busby JE, Borowsky AD, Kung HJ, Evans CP (2009) Aberrant activation of androgen receptor in a new neuropeptide-autocrine model of androgen-insensitive prostate cancer. Cancer Res 69:151–160
DaSilva J, Gioeli D, Weber MJ, Parsons SJ (2009) The neuroendocrine-derived peptide parathyroid hormone-related protein promotes prostate cancer cell growth by stabilizing the androgen receptor. Cancer Res 69:7402–7411
Lee LF, Louie MC, Desai SJ et al (2004) Interleukin-8 confers androgen-independent growth and migration of LNCaP: differential effects of tyrosine kinases Src and FAK. Oncogene 23:2197–2205
Kraus S, Gioeli D, Vomastek T, Gordon V, Weber MJ (2006) Receptor for activated C kinase 1 (RACK1) and Src regulate the tyrosine phosphorylation and function of the androgen receptor. Cancer Res 66:11047–11054
Guo Z, Dai B, Jiang T et al (2006) Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell 10:309–319
Migliaccio A, Di Domenico M, Castoria G et al (2005) Steroid receptor regulation of epidermal growth factor signaling through Src in breast and prostate cancer cells: steroid antagonist action. Cancer Res 65:10585–10593
Zhoul J, Hernandez G, Tu SW, Huang CL, Tseng CP, Hsieh JT (2005) The role of DOC-2/DAB2 in modulating androgen receptor-mediated cell growth via the nongenomic c-Src-mediated pathway in normal prostatic epithelium and cancer. Cancer Res 65:9906–9913
Schaller MD (2004) FAK and paxillin: regulators of N-cadherin adhesion and inhibitors of cell migration? J Cell Biol 166:157–159
Slack-Davis JK, Martin KH, Tilghman RW et al (2007) Cellular characterization of a novel focal adhesion kinase inhibitor. J Biol Chem 282:14845–14852
Chen HC, Appeddu PA, Isoda H, Guan JL (1996) Phosphorylation of tyrosine 397 in focal adhesion kinase is required for binding phosphatidylinositol 3-kinase. J Biol Chem 271:26329–26334
Hoosein NM, Logothetis CJ, Chung LW (1993) Differential effects of peptide hormones bombesin, vasoactive intestinal polypeptide and somatostatin analog RC-160 on the invasive capacity of human prostatic carcinoma cells. J Urol 149:1209–1213
Larran J, Salido M, Aparicio J, Lopez A, de Palacio ML, Vilches J (1996) In vitro characterization of bombesin and calcitonin on the proliferation of PC3, DU 145 and LNCaP cancer prostatic cell lines. Int J Dev Biol Suppl:275S–276S
Parsons JT, Martin KH, Slack JK, Taylor JM, Weed SA (2000) Focal adhesion kinase: a regulator of focal adhesion dynamics and cell movement. Oncogene 19:5606–5613
Tremblay L, Hauck W, Aprikian AG, Begin LR, Chapdelaine A, Chevalier S (1996) Focal adhesion kinase (pp 125FAK) expression, activation and association with paxillin and p50CSK in human metastatic prostate carcinoma. Int J Cancer 68:164–171
Tremblay L, Hauck W, Nguyen LT et al (1996) Regulation and activation of focal adhesion kinase and paxillin during the adhesion, proliferation, and differentiation of prostatic epithelial cells in vitro and in vivo. Mol Endocrinol 10:1010–1020
Rodriguez-Fernandez JL, Rozengurt E (1998) Bombesin, vasopressin, lysophosphatidic acid, and sphingosylphosphorylcholine induce focal adhesion kinase activation in intact Swiss 3T3 cells. J Biol Chem 273:19321–19328
Slack-Davis JK, Hershey ED, Theodorescu D, Frierson HF, Parsons JT (2009) Differential requirement for focal adhesion kinase signaling in cancer progression in the transgenic adenocarcinoma of mouse prostate model. Mol Cancer Ther 8:2470–2477
Tamagnone L, Lahtinen I, Mustonen T et al (1994) BMX, a novel nonreceptor tyrosine kinase gene of the BTK/ITK/TEC/TXK family located in chromosome Xp22.2. Oncogene 9:3683–3688
Robinson D, He F, Pretlow T, Kung HJ (1996) A tyrosine kinase profile of prostate carcinoma. Proc Natl Acad Sci USA 93:5958–5962
Dai B, Kim O, Xie Y et al (2006) Tyrosine kinase Etk/BMX is up-regulated in human prostate cancer and its overexpression induces prostate intraepithelial neoplasia in mouse. Cancer Res 66:8058–8064
Qiu Y, Robinson D, Pretlow TG, Kung HJ (1998) Etk/Bmx, a tyrosine kinase with a pleckstrin-homology domain, is an effector of phosphatidylinositol 3′-kinase and is involved in interleukin 6-induced neuroendocrine differentiation of prostate cancer cells. Proc Natl Acad Sci USA 95:3644–3649
Qiu Y, Kung HJ (2000) Signaling network of the Btk family kinases. Oncogene 19:5651–5661
Chau CH, Clavijo CA, Deng HT et al (2005) Etk/Bmx mediates expression of stress-induced adaptive genes VEGF, PAI-1, and iNOS via multiple signaling cascades in different cell systems. Am J Physiol Cell Physiol 289:C444–C454
Chen KY, Huang LM, Kung HJ, Ann DK, Shih HM (2004) The role of tyrosine kinase Etk/Bmx in EGF-induced apoptosis of MDA-MB-468 breast cancer cells. Oncogene 23:1854–1862
Chau CH, Chen KY, Deng HT et al (2002) Coordinating Etk/Bmx activation and VEGF upregulation to promote cell survival and proliferation. Oncogene 21:8817–8829
Xue LY, Qiu Y, He J, Kung HJ, Oleinick NL (1999) Etk/Bmx, a PH-domain containing tyrosine kinase, protects prostate cancer cells from apoptosis induced by photodynamic therapy or thapsigargin. Oncogene 18:3391–3398
Jiang T, Guo Z, Dai B et al (2004) Bi-directional regulation between tyrosine kinase Etk/BMX and tumor suppressor p53 in response to DNA damage. J Biol Chem 279:50181–50189
Wu YM, Huang CL, Kung HJ, Huang CY (2001) Proteolytic activation of ETK/Bmx tyrosine kinase by caspases. J Biol Chem 276:17672–17678
Dai B, Chen H, Guo S et al (2010) Compensatory upregulation of tyrosine kinase Etk/BMX in response to androgen deprivation promotes castration-resistant growth of prostate cancer cells. Cancer Res 70(13):5587–5596
Kim O, Jiang T, Xie Y, Guo Z, Chen H, Qiu Y (2004) Synergism of cytoplasmic kinases in IL6-induced ligand-independent activation of androgen receptor in prostate cancer cells. Oncogene 23:1838–1844
Recchia I, Rucci N, Festuccia C et al (2003) Pyrrolopyrimidine c-Src inhibitors reduce growth, adhesion, motility and invasion of prostate cancer cells in vitro. Eur J Cancer 39:1927–1935
Angelucci A, Schenone S, Gravina GL et al (2006) Pyrazolo[3, 4-d]pyrimidines c-Src inhibitors reduce epidermal growth factor-induced migration in prostate cancer cells. Eur J Cancer 42:2838–2845
Hennequin LF, Allen J, Breed J et al (2006) N-(5-chloro-1, 3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5- (tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. J Med Chem 49:6465–6488
Carew JS, Nawrocki ST, Cleveland JL (2007) Modulating autophagy for therapeutic benefit. Autophagy 3(5):464–467
Mortimore GE, Hutson NJ, Surmacz CA (1983) Quantitative correlation between proteolysis and macro- and microautophagy in mouse hepatocytes during starvation and refeeding. Proc Natl Acad Sci USA 80:2179–2183
Klip A (2009) The many ways to regulate glucose transporter 4. Appl Physiol Nutr Metab 34:481–487
Czech MP, Corvera S (1999) Signaling mechanisms that regulate glucose transport. J Biol Chem 274:1865–1868
Kanda Y, Watanabe Y (2005) Thrombin-induced glucose transport via Src-p38 MAPK pathway in vascular smooth muscle cells. Br J Pharmacol 146:60–67
Chou MT, Anthony J, Bjorge J, Fujita DJ (2010) The von Hippel-Lindau tumor suppressor protein is destabilized by src-implications for tumor angiogenesis and progression. Genes Cancer 1:225
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033
Li M, Jiang X, Liu D, Na Y, Gao GF, Xi Z (2008) Autophagy protects LNCaP cells under androgen deprivation conditions. Autophagy 4:54–60
Bellodi C, Lidonnici MR, Hamilton A et al (2009) Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest 119:1109–1123
Acknowledgements
The author is indebted to his students, postdoctors, colleagues, and collaborators who have contributed to the work described in this review. I also wish to thank the organization committee for the invitation to deliver this work in the Sixth International Symposium; Dr. Suresh Mohla for his confidence, encouragement, and help in my research career. The thanks are also extended to Ms. Fran Richardson for her patience and expert help in the preparation of this manuscript. The work was supported by grants from NIH (CA150197, CA114575, DK52659, DK078243) and DOD (PC093350).
Disclosure of Potential Conflict of Interest
No potential conflict of interest was disclosed.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kung, HJ. Targeting Tyrosine Kinases and Autophagy in Prostate Cancer. HORM CANC 2, 38–46 (2011). https://doi.org/10.1007/s12672-010-0053-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-010-0053-3