
Abstract
To identify microRNAs (miRNAs) associated with estrogen receptor (ESR1) status, we profiled luminal A, ESR1+ breast cancer cell lines versus triple negative (TN), which lack ERα, progesterone receptor and Her2/neu. Although two thirds of the differentially expressed miRNAs are higher in ESR1+ breast cancer cells, some miRNAs, such as miR-222/221 and miR-29a, are dramatically higher in ESR1− cells (∼100- and 16-fold higher, respectively). MiR-222/221 (which target ESR1 itself) and miR-29a are predicted to target the 3′ UTR of Dicer1. Addition of these miRNAs to ESR1+ cells reduces Dicer protein, whereas antagonizing miR-222 in ESR1− cells increases Dicer protein. We demonstrate via luciferase reporter assays that these miRNAs directly target the Dicer1 3′ UTR. In contrast, miR-200c, which promotes an epithelial phenotype, is 58-fold higher in the more well-differentiated ERα+ cells, and restoration of miR-200c to ERα− cells causes increased Dicer protein, resulting in increased levels of other mature miRNAs typically low in ESR1− cells. Together, our findings explain why Dicer is low in ERα negative breast cancers, since such cells express high miR-221/222 and miR-29a levels (which repress Dicer) and low miR-200c (which positively affect Dicer levels). Furthermore, we find that miR-7, which is more abundant in ERα+ cells and is estrogen regulated, targets growth factor receptors and signaling intermediates such as EGFR, IGF1R, and IRS-2. In summary, miRNAs differentially expressed in ERα+ versus ERα− breast cancers actively control some of the most distinguishing characteristics of the luminal A and TN subtypes, such as ERα itself, Dicer, and growth factor receptor levels.
Similar content being viewed by others


Introduction
Since specific microRNAs (miRNAs) are capable of regulating hundreds of mRNAs simultaneously, it was not unexpected to find that miRNA profiling can distinguish breast cancer subtypes [5]. MiRNAs function by binding to the 3′ untranslated region (UTR) of their targets and either prevent translation or cause mRNA degradation. The human RNase III-type nuclease Dicer performs the final step of biogenesis of miRNAs in which the pre-miRNA stem loop is cleaved to produce a mature miRNA. The mature miRNA is then incorporated into the RNA induced silencing complex, consisting of Dicer, Tar RNA-binding protein (TRBP), argonaute proteins, and several other proteins, which guide the mature miRNA to specific target mRNAs. Conditional deletion of Dicer enhances transformation and tumorigenesis, and Dicer functions as a haploinsufficient tumor suppressor [36, 37]. Three separate studies of estrogen receptor alpha (ESR1) positive (ESR1+) versus negative (ESR1−) breast cancers found that the majority of differentially expressed miRNAs are less abundant in ESR1− tumors [5, 26, 45]. It is likely that reduced Dicer expression is related to the global down-regulation of the miRNAome observed in cancer, and it is thought that the reduced number and abundance of miRNAs in human cancers reflects an altered differentiation state [7, 43]. Expression of Dicer is lower in breast cancer cell lines and clinical samples that have undergone epithelial to mesenchymal transition (EMT) [23], and Dicer is differentially expressed between ESR1 positive versus negative breast tumors [11]. Similarly, lower Dicer levels are associated with loss of ESR1 in ovarian cancers [18].
Dicer levels are regulated by let-7 via binding sites in the Dicer1 3′ UTR and coding region [19, 60]. MiR-103/107 was recently reported to repress Dicer1 through three sites in the Dicer 3′ UTR [44]. We observed that the 3′ UTR of Dicer1 also contains well-conserved binding sites for miR-221/222, which directly target ESR1 [16, 65] and for miR-29a. We find these to be the most differentially expressed miRNAs higher in ERα- negative versus ERα+ breast cancer cells. We hypothesized that miR-221/222 directly represses not only ESR1, but also Dicer itself, and that miR-29a also directly targets Dicer, possibly explaining why Dicer is lower in ERα negative breast cancers.
In contrast to miR-221/222 and miR-29a, the majority of differentially expressed miRNAs are higher in ERα+ cells, and of these, miR-200c is the most differentially expressed. We previously observed that restoration of miR-200c to dedifferentiated endometrial cancer cells increased Dicer1 mRNA levels [14]. We now demonstrate that restoration of miR-200c to triple negative (TN) breast cancer cells (that lack ERα, progesterone receptors, and Her2neu expression) causes an increase in Dicer protein resulting in an increase in the mature form of some of the miRNAs that are typically lower ERα− cells.
Materials and Methods
Cell Culture and Hormone Treatments
MCF7 and T47D breast cancer cells, which belong to the luminal A subtype, were grown in DMEM, l-glutamine, penicillin/streptomycin, and fetal bovine serum (FBS). MDA-MB-231 breast cancer cells (triple negative subtype) were grown in MEM containing FBS, HEPES, NEAA, l-glutamine, penicillin/streptomycin, and insulin. BT549 breast cancer cells (triple negative subtype) were grown in RPMI containing FBS and insulin. Hec50 cells were grown in DMEM containing FBS and penicillin/streptomycin. Cells were maintained at 37°C and 5% CO2. The identity of all cell lines was confirmed using the Identifiler DNA profiling kit (ABI) in the University of Colorado Cancer Center Sequencing Core Facility.
MCF7 cells were grown in phenol red-free media containing charcoal stripped serum for 24 h prior to hormone treatments. The cells were treated with ethanol, 10 nM estradiol, or a combination of 10 nM estradiol and 1 μM ICI 182,780 (ICI, Tocris Bioscience) for 24 h before harvesting total RNA using Trizol (Invitrogen), which retains both small RNA species such as miRNAs and larger RNAs such as mRNAs and rRNAs.
Immunoblotting
Whole cell lysates made with RIPA buffer were separated on SDS PAGE gels and transferred to PVDF membranes, blocked, and probed overnight at 4°C. Primary antibodies used were: ERα (clone AER611,NeoMarkers), E-cadherin (clone NCH-38, DAKO), ZEB1 (rabbit polyclonal, Dr. Doug Darling, University of Louisville); N-cadherin (clone 13A9, Upstate), Vimentin (clone V9, Sigma), Dicer (rabbit polyclonal, Sigma), α-tubulin (clone B-5-1-2, Sigma), EGFR (rabbit polyclonal, Cell Signaling Technology), IGF1Rβ (rabbit polyclonal (C-20), Santa Cruz Biotechnology), IRS-1 [56], IRS-2 (rabbit polyclonal (H-205), Santa Cruz Biotechnology), ERK1/2 (MAPK), phospho-specific and total (rabbit polyclonal, Cell Signaling Technology). After incubation with HRP-conjugated secondary antibodies, results were detected using Western Lightning Chemiluminescence Reagent Plus (Perkin Elmer).
MiRNA Microarray Profiling
Total RNA was prepared using Trizol (Invitrogen). Labeling, hybridization to miRNA microarray slides, and feature extraction was performed by ThermoFisher using the Agilent miRNA microarray platform containing all miRNAs in the Sanger version 10 database. Each miRNA probe is spotted in seven locations to allow for statistical analysis to be performed. Relative intensity data for the multiple probes for each miRNA was subjected to statistical filtering. Probes with p values ≤0.05 in at least two of the eight slides were retained for further analysis. For the luminal versus triple negative screen, the filtered array data was analyzed and clustering was performed using GeneSpring GX 10 (Agilent Technologies). Data was filtered using a twofold change cutoff and a p value of 0.05 (ANOVA, Benjamini Hochberg FDR multiple testing correction). For the graphical representation of the data, averages were taken for T47D and MCF7 to generate the ERα+ values and averages for MDA-MB-231 and BT549 were used to generate the ERα− values.
Real Time RT-PCR
TaqMan MiRNA Reverse Transcription kit was used to generate cDNA from total RNA using a miR-7, miR-29a, miR-221, miR-22, miR-193, miR-148a, or U6 specific primers (Applied Biosystems). For normalization, real time RT-PCR was performed on the cDNA using 18S rRNA primers and probe (Applied Biosystems). For miR-34a, miR-19b, miR-20a, and miR-106a, poly A tailing and reverse transcription was performed using the NCode miRNA qRT-PCR kit (Invitrogen). SYBR green real time RT-PCR was performed using the Universal Forward Primer (Invitrogen) and a miRNA specific primer. For normalization, levels of β-actin were quantified using gene-specific primers. The relative miRNA levels were calculated using the comparative Ct method (ΔΔCt). Briefly, the Ct (cycle threshold) values for the rRNA, U6, or actin were subtracted from Ct values of the miRNA to achieve the ΔCt value. The 2−ΔCt was calculated and then divided by a control sample to achieve the relative miRNA levels (ΔΔCt). Reported values are the means and standard errors of three biological replicates.
Immunohistochemistry
Sections were cut at 4 μm and heat immobilized. After deparaffinization and antigen retrieval, endogenous peroxidase was blocked. Sections were incubated with primary antibody for 1 h. Primary antibody used was Estrogen Receptor alpha (clone 1D5, Dako). Vectastain Elite ABC kit (Vector Labs) was used for serum blocking and antibody detection, followed by incubation with 3, 3′-diaminobenzidine (Dako) for protein visualization.
In Situ Hybridization
Sections of paraffin-embedded specimens were deparaffinized in xylene, rehydrated with ethanol, and subjected to proteinase K digestion (10 μg/ml, 5 min) and 0.2% glycine treatment. Samples were refixed in 4% paraformaldehyde and treated with acetylation solution, rinsing with PBS between treatments. Slides were prehybridized at 53°C for 1 h in hybridization solution (50% formamide, 5× SSC, 0.5 mg/ml yeast tRNA, heparin). Double-DIG LNA-modified DNA probe complementary to mature miR-222 or scramble control (Exiqon) at 40 nM in hybridization solution was incubated overnight at 50°C and washed in SSC at increasing stringency (5 to 0.2× SCC) at 50°C, then with PBST at room temperature. Slides were incubated for 1 h with blocking solution (TBST, 1% BSA, 0.1% FBS) and then 1 h with 1:2,000 dilution of anti-digoxigenin antibody (Roche). After washing with TBST, slides were incubated with AP solution for 15 min and then BM purple-substrate until color development was evident (24 to 72 h). The reaction was stopped when positive controls developed purple color while negative controls remained colorless.
Transfections
Transfections of 50 nM miR-221, miR-222, miR-29a, miR-200c, and miR-7 mimics (Ambion) were performed as described previously [14]. Protein and RNA were harvested 72 h post-transfection.
Luciferase Assays
Fragments of the ESR1 3′ UTR containing the putative binding sites for miR-203, miR-221, and miR-22 (nucleotides 2126–2472, ESR1 A) and a region that does not contain any miR-221 or miR-22 binding sites (nucleotides 3585–4249, ESR1 B) were amplified by PCR from HeLa genomic DNA (New England Biolabs). Fragments of the Dicer1 3′ UTR containing putative binding sites for miR-29a (nucleotides 1096–1752, Dicer 3′ UTR A) or miR-221/222 (nucleotides 2636–3028, Dicer 3′ UTR B) were also amplified. These fragments were cloned into pMIR-REPORT (Ambion). Site-directed mutagenesis was used to introduce a three-nucleotide mutation into the location where the miRNA see sequence binds (Dicer A mut and Dicer B mut). For ESR1, MCF7s were used, and for Dicer, Hec50 (an endometrial cell line) was used. Cells (20,000) per well were plated into a 96 well plate. The cells were mock transfected, transfected with 50 nM negative control mimic, with mimics for miR-221 or miR-222 (for the ESR1 fragments), miR-29a, miR-222, or antagonists for miR-29a or miR-222 (for the Dicer fragments) (Dharmacon). After 24 h, firefly reporter plasmid (0.196 μg) and a Renilla luciferase normalization plasmid pRL-SV40 (0.004 μg) were introduced using Lipofectamine 2000. Cells were harvested 48 h later for analysis using the Dual Luciferase Reporter assay system (Promega).
Generation of Stable Cell Lines
Cell lines stably expressing shRNAs targeting ZEB1 or luciferase were generated using SMARTvectorTM shRNA Lentiviral Particles (Thermo Scientific Dharmacon) as described previously [15]. For stable expression of the miR-222 antagonist, pmiR-222-Zip, or pGreenPuro Scramble Control (System Biosciences Inc.), lentiviral vectors were packaged in 293FT cells and virus was harvested after 48 h. Virus was added to MDA-MB-231 or BT549 cells at 1:10 or 1:1 virus:media and selection was performed using puromycin.
Results
MiRNAs are Differentially Expressed in ERα+ and ERα− Breast Cancer Cell Lines, the Majority Being More Abundant in ERα+ Cells
We performed miRNA microarray profiling of two breast cancer cell lines representing the luminal A subtype (MCF7 and T47D) and two representing the TN subtype (MDA-MB-231 and BT549). Luminal A cells are relatively well differentiated and retain expression of ERα and E-cadherin, while the TN cells, in particular the basal-like or claudin low subset (which the MDA-MB-231 and BT549 cells represent) [24], have lost expression of these luminal markers and express mesenchymal markers such as ZEB1, N-cadherin, and vimentin (Fig. 1a) and have thus undergone EMT. We also observe that Dicer levels are higher in luminal A cell lines (MCF7 and T47D) compared to TN cell lines (MDA-231 and BT549) (Fig. 1b). Previous reports have indicated that Dicer1 mRNA expression is lower in carcinoma cells with a mesenchymal phenotype [11, 23]. To determine if this is true in large scale datasets, we mined four breast cancer microarray datasets for Dicer1 expression separating the data into ESR1+ and ESR1− cohorts. Dicer1 mRNA levels are significantly lower in the ESR1− breast cancers in all four studies (Supplemental Fig. 1). We find that 53 miRNAs are differentially expressed in luminal A versus TN cell lines (Fig. 1c). Consistent with previous reports that the majority of miRNAs are downregulated in aggressive breast cancers [5, 26, 45], two thirds (31) of the 53 differentially expressed miRNAs that we identify are higher in ERα+ cells compared to ERα− (Fig. 1c).

MiRNAs differentially expressed in luminal versus triple negative breast cancer cell lines. a Protein expression of epithelial markers (ERα, E-cadherin) and mesenchymal markers (ZEB1, vimentin, N-cadherin) in luminal A (MCF7 and T47D) and triple negative (MDA-MB-231 and BT549) cell lines. PSTAIR is shown as a loading control. b Immunoblot of Dicer in luminal A (MCF7 and T47D) and triple negative (MDA-MB-231 and BT549) cells, with α-tubulin as a loading control. c MiRNA microarray analysis performed in luminal A versus triple negative cell lines. Biological duplicate samples for each cell line were hybridized to Agilent miRNA microarrays. Heatmap of miRNAs that exhibit a 1.5-fold differential expression between luminal and triple negative cell lines
MiR-221/222 and miR-29a are the Most Differentially Expressed miRNAs More Abundant in ERα− Cells
Of the miRNAs higher in ERα− cells, the most differentially expressed and most abundant were the highly homologous miR-221 and miR-222, as well as miR-29a (Fig. 2a). Real time PCR on independent samples confirmed that these miRNAs are more abundant in ERα− (Fig. 2b). The most differentially expressed miRNA that is higher in ERα+ cells is miR-200c, which has been previously demonstrated to be lost in high grade cancers [6, 14, 58], followed by the other miR-200 family members (Fig. 2c). The differential expression of miR-200c in ERα+ and ERα− was also confirmed by RT-PCR (Fig. 2d).

MiR-222 and miR-29a are higher in ERα- cell lines compared to ERα+. a Graphical representation of miRNAs more highly expressed in ERα+ breast cancer cell lines that are 1.5 different with P < 0.05. The blue bars are the average values for the ERα+ cells (T47D and MCF7), while the red bars are the average values for the ERα− cells (MDA-MB-231 and BT549). b Real time PCR validation of miR-222 (left) and miR-29a (right) expression levels, relative to the BT549 values. Error bars represent standard error of the mean. c Graphical representation of miRNAs more highly expressed in ERα+ cell lines and (d) real time PCR validation of miR-200c expression levels, relative to BT549 values. Error bars represent standard error of the mean
MiR-222 and miR-22 Act Additively to Decrease ESR1
Study of the miRNAs predicted to target the ESR1 3′ UTR using Miranda, PicTar, and Targetscan target prediction programs indicates that the miR-221/222 and miR-22 target sites are in close proximity (Fig. 3a). These miRNAs have been previously reported to target ESR1 [48, 52, 65], and we find that they are both higher in ESR1− cell lines (indicated in red). In contrast, we find that many of the other miRNAs predicted to bind the ESR1 3′ UTR are paradoxically more abundant in ESR1+ (indicated in blue) (Fig. 3a). MiRNAs can cooperate to downregulate a target when their binding sites are closely located as the miR-221/222 and miR-22 sites are [10, 57]. While the addition of each miRNA alone to ESR1+ MCF7 cells causes a marked decrease in ERα protein, an additive effect was observed when both miRNAs are combined (Fig. 3b). To demonstrate direct targeting of the miRNAs to the 3′ UTR of the ESR1 transcript, we utilized luciferase reporter assays in which two regions of the ESR1 3′ UTR (termed ESR1 A and B) were cloned into the region 3′ of the luciferase gene on a reporter vector. The region denoted ESR1 A contains the miR-221/222 and miR-22 target sites, while ESR1 B is predicted not to be targeted by miR-221/222, miR-22, or any of the miRNA that are higher in ERα− cells and serves as a negative control. Empty luciferase reporter vector containing no target sequences downstream of luciferase also serves as a negative control. These vectors were transfected into MCF7 cells (which lack miR-221/222) in combination with either a scrambled negative control, the miR-22 or miR-221 mimics alone or both miR-22 and miR-221 in combination (Fig. 3c). We observe an 18.8% decrease in luciferase activity in the cells transfected with ESR1 A and the miR-22 mimic compared to the scrambled negative control. With the miR-221 mimic, there is a 13.7% decrease in luciferase activity versus the negative control. When both mimics are combined, we observe a 32.7% decrease in luciferase activity, demonstrating an additive effect when the two miRNAs are combined. We performed in situ hybridization for miR-222 (the homolog of miR-221) on luminal A versus TN (confirmed ERα, progesterone receptor and Her2/neu negative) breast cancer clinical samples obtained from the University of Colorado Breast Cancer Tissue Bank (protocol 04-0066). Figure 3c shows in situ hybridization for miR-222 and IHC for ERα on five representative TN and five luminal A breast cancers. We find miR-222 expression only in TN tumors, whereas in luminal A tumors, miR-222 staining is absent (Fig. 3c). Examples of the levels of staining in cells positive (MDA-MB-231) and negative (MCF7) for miR-222 are shown in Supplemental Fig. 2.

MiR-22 and miR-221 act additively to decrease ERα levels. a Map of 3′ UTR of ESR1 showing putative miRNA binding sites. Target sites for miRNAs that have higher expression in ESR1− cells are circled in red, while target sites for miRNAs more highly expressed in ESR1+ cells are in blue. b Western blot of MCF7 cells treated with a mock transfection, a scrambled negative control, miR-22 mimic, a miR-221 mimic or a combination of miR-221 and miR-22 mimics. Protein was harvested 72 h after transfection, transferred, and probed for ERα and α-tubulin as a loading control. The experiment was repeated three times; shown is a representative blot. c The region of the ESR1 3′ UTR containing the miR-22 and 221 binding sites (ESR1 A) and a separate region of the ESR1 3′ UTR not containing miR-22 or miR-221 binding sites (ESR1 B) were each cloned downstream of luciferase in a reporter vector. These constructs or the empty reporter vector were transfected into cells treated with a scrambled negative control, miR-22 mimic, miR-221 mimic or both, and a luciferase assay performed. Error bars represent standard error of the mean for five replicates. Single asterisk indicates a statistically significant difference, P < 0.05, compared to ESR1 A and two asterisks indicate a statistically significant difference, P < 0.01, compared to EV and ESR1 B (two-way ANOVA, Bonferroni post test). d In situ hybridization for miR-222 and immunohistochemistry for ESR1 in luminal and triple negative clinical samples (ESR1 staining is brown and miR-222 staining is purple). MiR-222 in situ staining with a scrambled negative control is shown at the bottom (×400 magnification)
MiR-221, -222, and miR-29a Target Dicer1
In order to test our hypothesis that a direct link exists between miRNAs overexpressed in ESR1− cells and low Dicer levels, we transfected mimics for miR-221/222 and miR-29a into ESR1+ T47D cells and found that they each decrease Dicer protein to almost undetectable levels (Fig. 4a). Real time PCR for each of these miRNAs in the transfected cells is shown in Supplemental Fig. 3. Furthermore, inhibition of miR-222 by stable expression of the antagonist miR-222-ZIP results in increased Dicer protein in both MDA-MB-231 and BT549 cells (Fig. 4b). The Dicer1 3′ UTR contains well-conserved predicted target sites for miR-221/222 and miR-29a in close proximity to previously characterized let-7 sites (Fig. 4c). We cloned two regions of the Dicer1 3′ UTR containing the putative miR-29a or miR-221/222 binding sites (Dicer A and B) as well as those same fragments containing mutated miRNA target sites (Dicer A and B mut) downstream of luciferase (Fig. 4c). There is a decrease in luciferase activity only in the cells treated with miR-29a or miR-222 with the appropriate Dicer construct (Fig. 4d). This effect is abrogated when the target site is mutated, showing that the binding site is functional. Furthermore, antagonists of miR-29a and miR-222 are able to prevent binding, showing that the effect is specific to these miRNAs.

MiR-29a and miR-221 or 222 reduce Dicer protein expression by directly targeting Dicer. a Immunoblot for Dicer in T47D cells mock transfected, transfected with a scrambled negative control, miR-29a, miR-221, or miR-222 mimic. b Immunoblot for Dicer in MDA-MB-231 and BT529 cells stably expressing miR-222 antagonist (222-Zip) or scrambled negative control (SCR-Zip). c Map of 3′ UTR of Dicer1 showing putative miRNA binding sites. Target sites for miRNAs that have higher expression in triple negative cells are in red, target sites for miRNAs more highly expressed in luminal cells are in blue. d Luciferase assay on fragments of the Dicer1 3′ UTR containing the miR-29a binding site (left) or the miR-221/222 binding site (right), fragments containing mutated binding sites or an empty vector. Hec50 cells were mock transfected, transfected with a scrambled negative control, miR-29a or miR-222 mimics, antagonists of miR-29a or miR-222, or a combination of both. The mutations introduced into the putative miRNA binding sites are pictured below the graphs. Asterisk indicates P < 0.05, Student's t test
Dicer is Positively Regulated by miR-200c
We previously observed that miR-200c increases Dicer1 message [14]. We also observed that due to reciprocal repression between miR-200c and ZEB1 [6], reducing ZEB1 expression with shRNA causes an increase in endogenous miR-200c [15]. We find that increasing endogenous miR-200c in MDA-MB-231 cells by using shZEB (which we have shown previously to relieve repression of endogenous miR-200c [15] (Supplemental Fig. 4) or adding exogenous miR-200c mimic increases Dicer protein in MDA-MB-231 and BT549 cells (Fig. 5a, b). Since miRNAs usually function in a repressive manner, the mechanism by which miR-200c increases Dicer protein is likely through an indirect mechanism. We hypothesized that since many mature miRNAs are low in ERα− cells (perhaps due to inefficient maturation as a result of low Dicer), increasing Dicer might increase levels of mature miRNAs typically low in the TN cells. To test this hypothesis we measured levels of the mature forms of miRNAs originally observed to be low in TN cells in cells transfected with miR-200c mimic (in which endogenous Dicer levels had increased). We find that in MDA-MB-231 cells, miR-193b, miR-34a, and miR-148a are increased with miR-200c mimic compared to the negative control (Fig. 5a). Several other miRNAs (miR-15b, miR-103, miR-301a, and miR-106b), which we also find to be more abundant in ERα+ cells, also demonstrated increased levels in the miR-200c treated cells (data not shown). In BT549 cells, we observe an increase in miR-34a, miR-148a, and miR-301a when transfected with the miR-200c mimic (Fig. 5b). However, addition of miR-200c does not repress miR-221/222 levels (data not shown).

Restoration of miR-200c to ESR1− breast cancer cells increases Dicer protein. Immunoblot for Dicer in MDA-MB-231 (a) and BT549 (b) cells mock transfected, transfected with a scrambled negative control or a miR-200c mimic for 72 h. Bottom, real time PCR for miR-193b, miR-34a, and miR-148a in MDA-MB-231 cells and mR-34a, miR-148a, and miR-301a in BT549 cells transfected with a scrambled negative control or a miR-200c mimic. An asterisk indicates P < 0.05, Student's t test
MiR-7 is an Estrogen-Regulated miRNA that Targets Growth Factor Receptors Overexpressed in TN Breast Cancers
To identify miRNAs not only associated with ERα positivity, but actually regulated by estradiol-bound ERα, we performed miRNA microarray profiling of MCF7 cells treated for 24 h with 10 nM estradiol or ethanol vehicle control (Fig. 6a). At 24 h, the expression of six miRNAs significantly decreased while eight significantly increased with estrogen treatment. MiR-7 and miR-324-5p are both higher in ESR1+ cells and positively regulated by estrogen. We confirmed in independent samples by real time RT-PCR that several members of the miR-17-92 cluster or the paralog miR-106a-363 cluster are estrogen regulated (Fig. 6b). MiR-20a and one of the copies of miR-19b appear in the miR-17-92 cluster, while miR-106a and the other copy of miR-19b are in the miR-106a-363 cluster. We confirm that miR-7 is expressed more highly in ESR1+ cell lines and estrogen increases miR-7 levels in an ESR1-dependent manner (Fig. 6c). Bioinformatic analysis predicts epidermal growth factor receptor (EGFR), insulin-like growth factor 1 receptor (IGF1R), and insulin receptor substrates 1 and 2 (IRS-1, IRS-2) as putative miR-7 targets. IGF1R contains three predicted miR-7 binding sites and IRS-2 contains two putative binding sites. Addition of a miR-7 mimic to ESR1− cells dramatically decreased EGFR and IGF1Rβ at the protein level with no effect on the insulin receptor (Fig. 6d). There is also a profound decrease in IRS-2 protein following the addition of the miR-7 mimic, but no effect on IRS-1. Finally, we observe a decrease in the amount of phosphorylated ERK1/2 (MAPK) with no effect on total MAPK.

MiR-7, which is associated with ERα positivity and is upregulated by estrogen, targets growth factor receptors and downstream signaling molecules. a MiRNA microarray analysis of miRNAs differentially regulated by 10 nM estradiol (white bars) at 24 h versus the ethanol vehicle controls (black bars) in MCF7 cells. Shown are the miRNAs that have a 1.5-fold difference and P < 0.05. Error bars represent the range of biological duplicates. b Real time PCR for miR-106a, miR-19b, and miR-20a in MCF7 cells treated with the ethanol vehicle control, 10 nM estradiol (E2) or estradiol, and 1 μM ICI (E2 + ICI) for 24 h. Shown are the averages of three replicate samples, and error bars represent standard error of the mean. Asterisk indicates a statistically significant difference between E2 treated and the vehicle control, with P < 0.05, Student's t test. c Real time PCR for miR-7 was performed in two ESR1+ cell lines (MCF7 and T47D) and two ESR1− cell lines (BT549 and MDA-MB-231), top, and in cells treated with the ethanol vehicle control, 10 nM estradiol (E2) or estradiol, and 1 μM ICI (E2 + ICI) for 24 h, bottom. Shown are the averages of three replicate samples, and error bars represent standard error of the mean. d MDA-MB-231 cells were mock transfected, transfected with a scrambled negative control, or a miR-7 mimic for 72 h. Protein was harvested and blots probed for EGFR, IGF1Rβ, InRβ, IRS-1, IRS-2, phospho-MAPK ,and total MAPK (also used as a loading control)
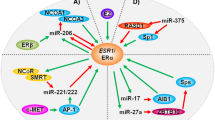
A schematic of the regulation of key distinguishing features of TN versus luminal cancers by miRNAs is shown in Fig. 7. Both Dicer and ERα are expressed at high levels in luminal breast cancers and are markers of a differentiated epithelial phenotype. MiR-221 and miR-222 are high in TN breast cancers and target both Dicer and ERα. MiR-29a is also high in TN breast cancers and targets Dicer. MiR-200c is high in luminal breast cancers and increases Dicer expression. MiR-7 is expressed at high levels in luminal A cells and limits the expression of growth factors receptors such as EGFR and IGF1R that are often overexpressed in TN cancers, and the signaling intermediate IRS-2.

MiRNA regulation of key proteins in luminal and triple negative breast cancers. Dicer and ERα are expressed at high levels in luminal breast cancers. MiR-221 and miR-222 are high in TN breast cancers and target both Dicer and ERα. MiR-29a is also high in TN breast cancers and targets Dicer. MiR-200c is high in luminal breast cancers and increases Dicer expression. EGFR, IGF1R, and IRS-2 are often activated or overexpressed in TN cancers and are all targeted by miR-7
Discussion
MiRNA profiling of ERα+ versus ERα− breast cancer cell lines reveals that the majority of miRNAs are lower in the ERα− cells. This is consistent with previous reports of a global decrease in miRNA expression in cancer [5, 26, 45]. While many miRNAs are located in fragile sites that are often lost in cancer [7], it is also possible that decreased expression of component(s) of the miRNA processing machinery prevent efficient miRNA processing. Indeed, cancer cells can have decreased expression of mature miRNAs, while maintaining expression of precursors [39, 47, 59] and impairment of miRNA processing results in transformation and increased tumorigenesis [36, 37]. MiRNAs are essential for differentiation and maintenance of a differentiated state. Dicer-deficient stem cells are unable to properly differentiate [27, 28], and loss of Dicer causes apoptosis in differentiated neural crest cells [63] and prostate epithelial cells [64]. The loss of differentiation and increased aggressive behavior that accompanies EMT may be in part due to decreased Dicer expression and a resultant decrease in mature miRNA expression.
The data presented herein as well as that of others [11, 23] suggests that high Dicer levels in breast cancer are associated with a well-differentiated epithelial, ERα+ phenotype, while lower Dicer levels are found in the less differentiated ERα- cells. Furthermore, Dicer has been shown to be positively regulated by estradiol [1]. Our findings demonstrate that while the majority of miRNAs are more abundant in ERα+ cells, miR-221/222 and miR-29a are striking exceptions. ESR1 is directly targeted by miR-221/222 and miR-22 [52, 65], and we demonstrate that these miRNAs cooperate to decrease ERα. In clinical samples, we find that expression of miR-222 and ERα is mutually exclusive, consistent with previous reports of miR-222 repressing ESR1 [48, 65] and the reciprocal negative regulatory loop whereby ERα represses miR-222 [16].
We show that miR-221/222 and miR-29a directly target Dicer, and these miRNAs are likely responsible for repressing Dicer expression and function in ERα breast cancer. We find that let-7 is higher in ERα breast cancer cells, and it also directly targets and represses Dicer1 [19, 60]. Since there are let-7 sites in close proximity to both the miR-221/222 and miR-29a binding sites, it is possible that these miRNAs work cooperatively.
MiR-200c represses a program of mesenchymal genes to maintain an epithelial state [6, 14, 15, 22, 25, 34], and here, we show that it also positively regulates Dicer, likely through an indirect, yet to be identified mechanism. Importantly, this may represent an additional means by which miR-200c promotes a well-differentiated epithelial phenotype. Our studies indicate that a subset of miRNAs may be low due to insufficient Dicer. While lower levels of mature miRNAs in ERα− cells can be explained by decreased Dicer levels, this begs the question as to how some miRNAs (such as miR-221/222 and miR-29a) are abundant in ERα− breast cancers in the face of low Dicer expression. In lower organisms, such as Drosophila, there are two Dicer proteins; however, in humans, only one Dicer gene exists (Dicer1). Not all miRNAs are equally affected by Dicer depletion [21, 35], suggesting either that miRNA stability is a factor, or perhaps another enzyme exists that can process certain miRNA precursors when Dicer levels are low. For example, miR-451 can be fully processed by Ago2 [9, 13], which is higher in ERα− breast cancers [11] and personal communication (Dorraya El-Ashry and Phillip Miller).
Since both Dicer and ESR1 and their protein products are low or absent in TN breast cancer cells, it makes sense that both are targeted by miRNAs abundant in TN cells. However, both the ESR1 and Dicer 3′ UTRs also have putative target sites for miRNAs that are highly expressed in ERα+ cells. It is likely that other factors are interfering with the miRNA–mRNA interaction. For instance, RNA-binding proteins can bind 3′ UTRs and prevent or recruit miRNA binding [3, 4, 29, 30] or target sites can be mutated or absent due to shortening of the 3′ UTR [20, 46, 50, 55]. Non-coding RNAs or pseudogenes can act as decoys to soak up miRNAs and prevent them from interacting with a target [54, 61]. For instance miR-193b (5.5-fold higher in ESR1+ cells in our study) directly targets ESR1 when it is transfected into MCF7 cells [40]. Perhaps overexpression of this miRNA can overcome whatever is preventing the already abundant endogenous miR-193b from targeting ESR1. Similarly, miR-103/107 was recently reported to directly target Dicer1 [44]. However, in our study and others [42, 45], miR103/107 is higher in ERα+ cells (which have high Dicer) as compared to ERα- cells. While miR-193b and miR-103/107 can target ESR1 and Dicer if overexpressed, these miRNA are already expressed at higher levels in ERα+ cells that express substantial Dicer. Nevertheless, it is possible that these miRNAs naturally fluctuate under certain conditions in order to fine tune or limit ERα or Dicer protein levels.
We sought to determine if any of the miRNAs differentially expressed in ERα+ versus negative breast cancer cells are differentially expressed because they are regulated by estradiol-bound ERα [2, 8, 32]. Several miRNAs located in the miR-17-92 cluster or its paralog clusters are upregulated by estrogen. The miR-17-92 cluster (also known as oncomir-1) has been implicated in several types of cancers [12, 17, 51]. MiR-7 was also both estrogen regulated and more abundant in ERα+ cells. MiR-7 targets EGFR and decreases proliferation [41, 62]. We further demonstrate that miR-7 can also reduce IGF1R and IRS-2 protein expression. EGFR and IGF1R are often overexpressed and constitutively active in TN breast cancers and contribute to an aggressive phenotype [33, 38]. Similarly, IGF1R is often activated in aggressive cancers with poor prognosis, and overexpression of IGF1R in a mouse model results in mammary gland tumors with a basal-like phenotype [31]. Since IRS-2 is a signaling intermediate in the IGF1R pathway [49, 53], miR-7 could be a very effective means by which to abrogate this pathway. Our data suggest that effective re-introduction of miR-7 into TN breast cancer could offer an advantage over inhibitors targeting either EGFR or IGF1R since it would target both pathways simultaneously.
In summary, we demonstrate that the most highly differentially expressed miRNAs more abundant in ERα− breast cancers, namely miR-221/222 and miR-29a, directly repress Dicer1. In contrast, miR-200c, which is more abundant in ERα+ breast cancer cells, increases Dicer protein levels. We conclude that miRNAs differentially expressed in ERα+ versus negative breast cancer cells function to control some of the most distinguishing characteristics of the luminal A as compared to TN breast cancer subtypes such as ERα status, Dicer protein levels, and EGFR and IGF1R growth factor receptor expression.
Abbreviations
- EMT:
-
Epithelial to mesenchymal transition
- ESR1:
-
Estrogen receptor alpha gene
- ERα:
-
Estrogen receptor alpha protein
- miRNA:
-
MicroRNA
- RISC:
-
RNA induced silencing complex
- TRBP:
-
Tar RNA-binding protein
- TN:
-
Triple negative
- UTR:
-
Untranslated region
References
Bhat-Nakshatri P, Wang G, Collins NR, Thomson MJ, Geistlinger TR, Carroll JS, Brown M et al (2009) Estradiol-regulated microRNAs control estradiol response in breast cancer cells. Nucleic Acids Res 37(14):4850–4861
Bhat-Nakshatri P, Wang G, Collins NR, Thomson MJ, Geistlinger TR, Carroll JS, Brown M et al (2009) Estradiol-regulated microRNAs control estradiol response in breast cancer cells. Nucleic Acids Res 37:4850–4861
Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W (2006) Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 125(6):1111–1124
Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W (2006) Stress-induced reversal of MicroRNA repression and mRNA P-body localization in human cells. Cold Spring Harb Symp Quant Biol 71:513–521
Blenkiron C, Goldstein LD, Thorne NP, Spiteri I, Chin SF, Dunning MJ, Barbosa-Morais NL et al (2007) MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome Biol 8(10):R214
Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T (2008) A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep 9(6):582–589
Calin GA, Croce CM (2006) MicroRNA signatures in human cancers. Nat Rev Cancer 6(11):857–866
Castellano L, Giamas G, Jacob J, Coombes RC, Lucchesi W, Thiruchelvam P, Barton G et al (2009) The estrogen receptor-{alpha}-induced microRNA signature regulates itself and its transcriptional response. Proc Natl Acad Sci USA 106(37):15732–15737
Cheloufi S, Dos Santos CO, Chong MM, Hannon GJ (2010) A dicer-independent miRNA biogenesis pathway that requires ago catalysis. Nature 465(7298):584–589
Chen H, Sun JG, Cao XW, Ma XG, Xu JP, Luo FK, Chen ZT (2009) Preliminary validation of ERBB2 expression regulated by miR-548d-3p and miR-559. Biochem Biophys Res Commun 385(4):596–600
Cheng C, Fu X, Alves P, Gerstein M (2009) mRNA expression profiles show differential regulatory effects of microRNAs between estrogen receptor-positive and estrogen receptor-negative breast cancer. Genome Biol 10(9):R90
Chow TF, Mankaruos M, Scorilas A, Youssef Y, Girgis A, Mossad S, Metias S et al (2010) The miR-17-92 cluster is over expressed in and has an oncogenic effect on renal cell carcinoma. J Urol 183(2):743–751
Cifuentes D, Xue H, Taylor DW, Patnode H, Mishima Y, Cheloufi S, Ma E et al (2010) A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science 328(5986):1694–1698
Cochrane DR, Spoelstra NS, Howe EN, Nordeen SK, Richer JK (2009) MicroRNA-200c mitigates invasiveness and restores sensitivity to microtubule-targeting chemotherapeutic agents. Mol Cancer Ther
Cochrane D, Howe E, Spoelstra N, Richer J (2009) Loss of miR-200c: a marker of aggressiveness and chemoresistance in female reproductive cancers. J Oncol 2010:821717. doi:10.1155/2010/821717
Di Leva G, Gasparini P, Piovan C, Ngankeu A, Garofalo M, Taccioli C, Iorio MV et al (2010) MicroRNA Cluster 221–222 and estrogen receptor alpha interactions in breast cancer. J Natl Cancer Inst
Diosdado B, van de Wiel MA, Terhaar Sive Droste JS, Mongera S, Postma C, Meijerink WJ, Carvalho B, Meijer GA (2009) MiR-17-92 cluster is associated with 13q gain and c-myc expression during colorectal adenoma to adenocarcinoma progression. Br J Cancer 101(4):707–714
Faggad A, Budczies J, Tchernitsa O, Darb-Esfahani S, Sehouli J, Muller BM, Wirtz R et al (2010) Prognostic significance of Dicer expression in ovarian cancer-link to global microRNA changes and oestrogen receptor expression. J Pathol 220(3):382–391. doi:10.1002/path.2658
Forman JJ, Legesse-Miller A, Coller HA (2008) A search for conserved sequences in coding regions reveals that the let-7 microRNA targets Dicer within its coding sequence. Proc Natl Acad Sci USA 105(39):14879–14884
Gao Y, He Y, Ding J, Wu K, Hu B, Liu Y, Wu Y et al (2009) An insertion/deletion polymorphism at miRNA-122-binding site in the interleukin-1alpha 3′ untranslated region confers risk for hepatocellular carcinoma. Carcinogenesis 30(12):2064–2069
Giraldez AJ, Cinalli RM, Glasner ME, Enright AJ, Thomson JM, Baskerville S, Hammond SM, Bartel DP, Schier AF (2005) MicroRNAs regulate brain morphogenesis in zebrafish. Science 308(5723):833–838
Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ (2008) The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 10:593–601
Grelier G, Voirin N, Ay AS, Cox DG, Chabaud S, Treilleux I, Leon-Goddard S et al (2009) Prognostic value of Dicer expression in human breast cancers and association with the mesenchymal phenotype. Br J Cancer 101(4):673–683
Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, Gilcrease MZ, Krishnamurthy S, Lee JS, Fridlyand J et al (2009) Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res 69(10):4116–4124
Hurteau GJ, Carlson JA, Spivack SD, Brock GJ (2007) Overexpression of the microRNA hsa-miR-200c leads to reduced expression of transcription factor 8 and increased expression of E-cadherin. Cancer Res 67(17):7972–7976
Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E et al (2005) MicroRNA gene expression deregulation in human breast cancer. Cancer Res 65(16):7065–7070
Kanellopoulou C, Muljo SA, Kung AL, Ganesan S, Drapkin R, Jenuwein T, Livingston DM, Rajewsky K (2005) Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev 19(4):489–501
Kawase-Koga Y, Low R, Otaegi G, Pollock A, Deng H, Eisenhaber F, Maurer-Stroh S, Sun T (2010) RNAase-III enzyme Dicer maintains signaling pathways for differentiation and survival in mouse cortical neural stem cells. J Cell Sci 123(Pt 4):586–594
Kedde M, Strasser MJ, Boldajipour B, Oude Vrielink JA, Slanchev K, le Sage C, Nagel R et al (2007) RNA-binding protein Dnd1 inhibits microRNA access to target mRNA. Cell 131(7):1273–1286
Kim HH, Kuwano Y, Srikantan S, Lee EK, Martindale JL, Gorospe M (2009) HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev 23(15):1743–1748
Klinakis A, Szabolcs M, Chen G, Xuan S, Hibshoosh H, Efstratiadis A (2009) Igf1r as a therapeutic target in a mouse model of basal-like breast cancer. Proc Natl Acad Sci USA 106(7):2359–2364
Klinge CM (2009) Estrogen regulation of microRNA expression. Curr Genomics 10(3):169–183. doi:10.2174/138920209788185289
Kobayashi S (2008) Basal-like subtype of breast cancer: a review of its unique characteristics and their clinical significance. Breast Cancer 15(2):153–158
Korpal M, Lee ES, Hu G, Kang Y (2008) The miR-200 family inhibits epithelial–mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem 283(22):14910–14914
Kuehbacher A, Urbich C, Zeiher AM, Dimmeler S (2007) Role of Dicer and Drosha for endothelial microRNA expression and angiogenesis. Circ Res 101(1):59–68
Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T (2007) Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet 39(5):673–677
Lambertz I, Nittner D, Mestdagh P, Denecker G, Vandesompele J, Dyer MA, Marine JC (2010) Monoallelic but not biallelic loss of Dicer1 promotes tumorigenesis in vivo. Cell Death Differ 17(4):633–641
Law JH, Habibi G, Hu K, Masoudi H, Wang MY, Stratford AL, Park E et al (2008) Phosphorylated insulin-like growth factor-i/insulin receptor is present in all breast cancer subtypes and is related to poor survival. Cancer Res 68(24):10238–10246
Lee EJ, Baek M, Gusev Y, Brackett DJ, Nuovo GJ, Schmittgen TD (2008) Systematic evaluation of microRNA processing patterns in tissues, cell lines, and tumors. RNA 14(1):35–42
Leivonen SK, Makela R, Ostling P, Kohonen P, Haapa-Paananen S, Kleivi K, Enerly E et al (2009) Protein lysate microarray analysis to identify microRNAs regulating estrogen receptor signaling in breast cancer cell lines. Oncogene 28(44):3926–3936
Li X, Carthew RW (2005) A microRNA mediates EGF receptor signaling and promotes photoreceptor differentiation in the Drosophila eye. Cell 123(7):1267–1277
Lowery AJ, Miller N, Devaney A, McNeill RE, Davoren PA, Lemetre C, Benes V et al (2009) MicroRNA signatures predict oestrogen receptor, progesterone receptor and HER2/neu receptor status in breast cancer. Breast Cancer Res 11(3):R27
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A et al (2005) MicroRNA expression profiles classify human cancers. Nature 435(7043):834–838
Martello G, Rosato A, Ferrari F, Manfrin A, Cordenonsi M, Dupont S, Enzo E et al (2010) A MicroRNA targeting dicer for metastasis control. Cell 141(7):1195–1207
Mattie MD, Benz CC, Bowers J, Sensinger K, Wong L, Scott GK, Fedele V, Ginzinger D, Getts R, Haqq C (2006) Optimized high-throughput microRNA expression profiling provides novel biomarker assessment of clinical prostate and breast cancer biopsies. Mol Cancer 5:24
Mayr C, Bartel DP (2009) Widespread shortening of 3′ UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138(4):673–684
Michael MZ, O’ Connor SM, van Holst Pellekaan NG, Young GP, James RJ (2003) Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res 1(12):882–891
Miller TE, Ghoshal K, Ramaswamy B, Roy S, Datta J, Shapiro CL, Jacob S, Majumder S (2008) MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27Kip1. J Biol Chem 283(44):29897–29903
Nagle JA, Ma Z, Byrne MA, White MF, Shaw LM (2004) Involvement of insulin receptor substrate 2 in mammary tumor metastasis. Mol Cell Biol 24(22):9726–9735
Nicoloso MS, Sun H, Spizzo R, Kim H, Wickramasinghe P, Shimizu M, Wojcik SE et al (2010) Single-nucleotide polymorphisms inside microRNA target sites influence tumor susceptibility. Cancer Res 70(7):2789–2798
Olive V, Jiang I, He L (2010) miR-17-92, a cluster of miRNAs in the midst of the cancer network. Int J Biochem Cell Biol 42(8):1348–1354
Pandey DP, Picard D (2009) miR-22 inhibits estrogen signaling by directly targeting the estrogen receptor alpha mRNA. Mol Cell Biol 29(13):3783–3790
Pankratz SL, Tan EY, Fine Y, Mercurio AM, Shaw LM (2009) Insulin receptor substrate-2 regulates aerobic glycolysis in mouse mammary tumor cells via glucose transporter 1. J Biol Chem 284(4):2031–2037
Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP (2010) A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 465(7301):1033–1038
Ratner E, Lu L, Boeke M, Barnett R, Nallur S, Chin LJ, Pelletier C et al (2010) A KRAS-variant in ovarian cancer acts as a genetic marker of cancer risk. Cancer Res
Sachdev D, Li SL, Hartell JS, Fujita-Yamaguchi Y, Miller JS, Yee D (2003) A chimeric humanized single-chain antibody against the type I insulin-like growth factor (IGF) receptor renders breast cancer cells refractory to the mitogenic effects of IGF-I. Cancer Res 63(3):627–635
Saetrom P, Heale BS, Snove O Jr, Aagaard L, Alluin J, Rossi JJ (2007) Distance constraints between microRNA target sites dictate efficacy and cooperativity. Nucleic Acids Res 35(7):2333–2342
Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, Diehn M et al (2009) Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 138(3):592–603
Thomson JM, Newman M, Parker JS, Morin-Kensicki EM, Wright T, Hammond SM (2006) Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev 20(16):2202–2207
Tokumaru S, Suzuki M, Yamada H, Nagino M, Takahashi T (2008) Let-7 regulates Dicer expression and constitutes a negative feedback loop. Carcinogenesis 29(11):2073–2077
Wang J, Liu X, Wu H, Ni P, Gu Z, Qiao Y, Chen N, Sun F, Fan Q (2010) CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids Res
Webster RJ, Giles KM, Price KJ, Zhang PM, Mattick JS, Leedman PJ (2009) Regulation of epidermal growth factor receptor signaling in human cancer cells by microRNA-7. J Biol Chem 284(9):5731–5741
Zehir A, Hua LL, Maska EL, Morikawa Y, Cserjesi P (2010) Dicer is required for survival of differentiating neural crest cells. Dev Biol 340(2):459–467
Zhang L, Zhang B, Valdez JM, Wang F, Ittmann M, Xin L (2010) Dicer ablation impairs prostate stem cell activity and causes prostate atrophy. Stem Cells. doi:10.1002/stem.455
Zhao JJ, Lin J, Yang H, Kong W, He L, Ma X, Coppola D, Cheng JQ (2008) MicroRNA-221/222 negatively regulates ERalpha and associates with tamoxifen resistance in breast cancer. J Biol Chem 83(45):31079–31086
Acknowledgements
This work was supported by Department of Defense Breast Cancer Research Program Idea Award BC084162, Susan G Komen Foundation KG090415 (J. Richer), and the National Institutes of Health R01CA74285 (D. Yee).We thank Aik-Choon Tan (University of Colorado, Division of Medical Oncology) for bioinformatics and Dorraya El-Ashry and Phil Miller (University of Miami, Miller School of Medicine) for help with bioinformatics and thoughtful comments on the manuscript. We also thank the University of Colorado Cancer Center Cores for DNA Sequencing and Analysis, Tissue Procurement and Pathology, supported by the NIH/National Cancer Institute Cancer Core Support Grant P30 CA046934.
Conflict of Interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Supplemental Figure 1
Analysis of microarray data comparing Dicer1 expression in ESR1+ and ESR1− breast cancer patients. Four breast cancer patients microarray gene expression data sets and their corresponding ESR1 status were downloaded from GEO database (GSE2034 (PMID: 15721472), GSE2603 (PMID: 16049480), and GSE3494 (PMID: 16141321)), and MD Anderson Cancer Center (MDACC) website (http://bioinformatics.mdanderson.org/pubdata.html PMID: 16896004). Raw signals from the gene arrays were extracted using Affymetrix Power Tools (APT) with MAS 5.0 and normalized by the Robust Multiarray Average (RMA) (PMID: 12925520) method as previously described. Multiple probe sets representing the same gene were collapsed based on maximum values. The Dicer1 gene expressions were compared between ESR1+ and ESR1− cohorts. Welch two-sample t test was performed and p < 0.05 was considered as statistically significant results. The box portion of the plot includes 50% of the data, the bold line representing the median and the borders of the box represent the lower and upper quartiles. The upper and lower whiskers represent the maximum and minimum Dicer1 expression. Welch two-sample t test was performed on these comparisons (GIF 60 kb)
Supplemental Figure 2
MiR-222 is expressed in ESR1− cells, but not in ESR1+ cells. In situ hybridization for miR-222 in MCF7 and MDA-231 cells. Positive staining is purple. (×200 magnification) (GIF 46 kb)
Supplemental Figure 3
Real time PCR for miR-29a (a), miR-221 (b), and miR-222 (c) in T47D cells that have been mock transfected, transfected with a scrambled negative control, or miRNA mimics. Values are normalized to the mock-transfected sample. Error bars represent standard error of the mean (GIF 37 kb)
Supplemental Figure 4
a Immunoblot for Dicer in MDA-MB-231 cells stably expressing shRNA targeting ZEB1 or shRNA targeting luciferase (as a negative control). b Real time PCR for miR-200c in MDA-MB-231 cells stably expressing shRNA targeting ZEB1 or shRNA targeting luciferase. Values are normalized to the shLuciferase, and error bars represent the standard error of the mean (GIF 84 kb)
Rights and permissions
About this article
Cite this article
Cochrane, D.R., Cittelly, D.M., Howe, E.N. et al. MicroRNAs Link Estrogen Receptor Alpha Status and Dicer Levels in Breast Cancer. HORM CANC 1, 306–319 (2010). https://doi.org/10.1007/s12672-010-0043-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-010-0043-5