
Abstract
Newly synthesized Pathway Preferential Estrogen-1 (PaPE-1) selectively activates membrane estrogen receptors (mERs), namely, mERα and mERβ, and has been shown to evoke neuroprotection; however, its effectiveness in protecting brain tissue against hypoxia and ischemia has not been verified in a posttreatment paradigm. This is the first study showing that a 6-h delayed posttreatment with PaPE-1 inhibited hypoxia/ischemia-induced neuronal death, as indicated by neutral red uptake in mouse primary cell cultures in vitro. The effect was accompanied by substantial decreases in neurotoxicity and neurodegeneration in terms of LDH release and Fluoro-Jade C staining of damaged cells, respectively. The mechanisms of the neuroprotective action of PaPE-1 also involved apoptosis inhibition demonstrated by normalization of both mitochondrial membrane potential and expression levels of apoptosis-related genes and proteins such as Fas, Fasl, Bcl2, FAS, FASL, BCL2, BAX, and GSK3β. Furthermore, PaPE-1-evoked neuroprotection was mediated through a reduction in ROS formation and restoration of cellular metabolic activity that had become dysregulated due to hypoxia and ischemia. These data provide evidence that targeting membrane non-GPER estrogen receptors with PaPE-1 is an effective therapy that protects brain neurons from hypoxic/ischemic damage, even when applied with a 6-h delay from injury onset.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acute stroke is a commonly used name for a cerebrovascular accident. Cerebrovascular accidents are the 2nd leading cause of death and a leading cause of disability worldwide. Research has been shown that up to 15 million people suffer from stroke annually, that more than 5 million of whom die, and that another 5 million of whom remain disabled for the rest of their lives (World Health Organisation (n.d.), Stroke, cerebrovascular accident). Approximately three-quarters of all strokes occur in persons aged ≥ 65 years, but strokes can occur at any age (Yousufuddin and Young 2019). The 3 main types of stroke are ischemic stroke, hemorrhagic stroke, and transient ischemic attack. Ischemic stroke accounts for 87% of all stroke cases and is caused by the blood clot resulting in the loss of blood circulation (Centers for Disease Control and Prevention. Types of stroke 2020). The gold standard for stroke treatment is an injection of recombinant tissue plasminogen activator (rtPA), which is also called alteplase. rtPA can be given up to 4.5 h after stroke symptoms start and has a long list of contraindications, such as intracranial hemorrhage and cerebral edema (Jilani and Siddiqui 2021). Consequently, only 5% of stroke patients can be treated with rtPA (Miller et al. 2011; Frendl and Csiba 2011).
Perinatal asphyxia is a condition characterized by fetal oxygen deprivation that leads to the death of approximately 1 million children and disability of another 1 million annually (Manandhar and Basnet 2019). It occurs during the antepartum, intrapartum, or perinatal period. First, a reduced oxygen supply (hypoxia) impairs neuronal cell function. Next, prolonged hypoxia leads to myocardial cell dysfunction, and cardiac failure causes ischemia in peripheral organs and the brain (Rainaldi and Perlman 2016). The current therapy for perinatal asphyxia must occur within 6 h and is based on therapeutic hypothermia (selective head or systemic cooling) and oxygen therapy (Zubcevic et al. 2015). Recent studies have shown that therapeutic hypothermia may increase the risk of persistent pulmonary hypertension in neonates (Vijverberg Joanna et al. 2021).
Several pathways are involved solely or in combination in hypoxia/ischemia injury, including apoptosis, necrosis, and oxidative stress. During the lack of blood supply due to hypoxia or ischemia, in addition to acute necrosis, apoptosis appears to determine the fate of cells (Wu et al. 2018). Apoptosis contributes to a significant proportion of neuronal death in the ischemic penumbra during blood flow restoration, which is also called reperfusion. Suppression of glucose and oxygen delivery causes activation of both prosurvival and proapoptotic signaling cascades (Chen et al. 2009). Moreover, reactive oxygen species (ROS) are strongly associated with brain injury after hypoxia and ischemia. Immediately after acute oxygen and/or glucose deprivation, ROS production rapidly increases as a consequence of an imbalance between pro-oxidants and antioxidants and overwhelmed antioxidant defenses. ROS can damage cell structures, including DNA, proteins, lipids, and membranes leading to apoptosis and necrosis. Moreover, during reperfusion, brain cell oxygenation increases and leads to a second burst of ROS generation (Li and Yang 2016).
Sex dimorphism seems to be an important factor in ischemic stroke outcomes. Women (45–74 years old) have a lower risk of stroke incidence and lower mortality than men of the same age (Jiang et al. 2020). Many studies have confirmed that estrogen receptors (ERs) have a neuroprotective capacity against ischemia reperfusion. However, therapy based on estrogen itself or on SERMs (selective ER modulators), e.g., tamoxifen and raloxifene, has several side effects (e.g., venous thromboembolic events and an increased risk of breast/uterine ER-dependent cancer). Sex dimorphism is caused by activation of the nuclear ERs responsible for regulating transcriptional processes and the expression of several genes in peripheral tissues in response to hormonal effects. In addition to the nuclear localization of classical nuclear ERs, receptors are also localized on cell membranes, i.e., mERα, mERβ, ER-X, GPER1, and GqmER, mainly in the nervous, metabolic, and cardiovascular systems. Stimulation of mERs has been shown to activate various neuronal survival-related rapid signaling pathways and prevent neuronal cell death through the kinases PI3K and ERK/MAPK and the tyrosine cascade as well as membrane-associated molecules such as ion channels and G-protein-coupled receptors (De Butte-Smith et al. 2012; Saczko et al. 2017). This action is called nonnuclear or membrane activity. In recent years, these differences have rendered membrane ERs ideal pharmacological targets devoid of the negative side effects associated with nuclear activity.
In 2016, Madak-Erdogan et al. designed Pathway Preferential Estrogen-1 (PaPE-1), which provides beneficial metabolic and vascular effects without stimulating reproductive tissues. PaPE-1 ((S)-5-(4-hydroxy-3,5-dimethyl-phenyl)-indan-1-ol) preferably interacts with the mER signaling pathway (50,000 times less bound to nuclear receptors); specifically, PaPE-1 selectively interacts with mERα and mERβ without activating the nuclear signaling pathway of the controversial GPER1 receptor. Another important feature of PaPE-1 is the short lifetime of ER-PaPE-1 complexes; the half-life of the ER-E2 (17β-estradiol) complex is nearly 30 h, whereas that of the ER-PaPE-1 complex is less than 1 min. This short time is sufficient to activate kinase cascades and at the same time is insufficient to promote transcription via nuclear receptors. Therefore, PaPE-1 does not induce ERα or ERK2 recruitment to gene enhancers or stimulate the expression of proliferation-associated genes, as observed with E2. However, similar to E2, PaPE-1 strongly activates the MAPK and mTOR pathways. The neuroprotective capacity of PaPE-1 against Amyloid β (Aβ)-induced neurotoxicity and apoptosis has already been evidenced by our group (Wnuk et al. 2020a). We showed that following 24 h of exposure, PaPE-1 inhibited Aβ-evoked effects, as shown by reduced parameters of neurotoxicity, oxidative stress, and apoptosis. Moreover, PaPE-1 normalized the Aβ-induced loss of mitochondrial membrane potential and restored the BAX/BCL2 ratio, suggesting that the anti-Alzheimer’s disease (AD) capacity of PaPE-1 particularly relies on inhibition of the mitochondrial apoptotic pathway.
Since a posttreatment paradigm is the most relevant approach to improve hypoxia-/ischemia-based injuries, the present study aims to identify the neuroprotective potential and mechanisms of action of PaPE-1 as a posttreatment therapy in cellular models of ischemic stroke and perinatal asphyxia.
Materials and Methods
Materials
Phosphate-buffered saline (PBS) was purchased from Biomed Lublin (Lublin, Poland). B27 and neurobasal media were obtained from Gibco (Grand Island, NY, USA). The ROS-Glo™ H2O2 assay was obtained from Promega (Madison, WI, USA). The cytotoxicity detection kit and neutral red dye were purchased from Roche Diagnostics GmbH (Mannheim, Germany). ELISA kits for FAS, FASL, BAX, BCL2, and GSK3B were purchased from Bioassay Technology Laboratory (Shanghai, China). Culture plates were obtained from TPP Techno Plastic Products AG (Trasadingen, Switzerland). L-Glutamine, fetal bovine serum (FBS), dimethyl sulfoxide (DMSO), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate hydrate (CHAPS), ammonium persulfate, N,N,N′,N′-tetramethylethane-1,2-diamine (TEMED), 2-amino-2-(hydroxymethyl)-1,3-propanediol (Trizma base), DL-dithiothreitol, sodium deoxycholate, protease inhibitor (ethylenediaminetetraacetic acid-free), bromophenol blue, radioimmunoprecipitation assay buffer (RIPA) buffer, Fluoro-Jade C, PaPE-1 ((S)-5-(4-hydroxy-3,5-dimethyl-phenyl)-indan-1-ol, 0.01–10 μM), thiazolyl blue tetrazolium bromide, protease inhibitor cocktail for mammalian tissues, and polyornithine were obtained from Sigma-Aldrich (St. Louis, MO, USA). JC-10 was purchased from Abcam (Cambridge, UK). The RNeasy Mini Kit was obtained from Qiagen (Hilden, Germany). The RNA quality (integrity) was analyzed using PrimePCR™ RNA Quality Probe Assay, Mouse (Bio-Rad, USA). A High-Capacity cDNA-Reverse Transcription Kit, TaqMan Gene Expression Master Mix, and TaqMan probes for specific genes encoding Hprt, β-actin, Gapdh, Fas, Fasl, Bax, Bcl2, and Gsk3b were obtained from Thermo Fisher Scientific (Waltham, MA, USA).
Methods
Primary Neuronal Cell Culture
Primary neocortical cultures were prepared from CD-1® IGS Swiss mouse embryos at 15 days of gestation (Charles River, Germany) as previously described (Kajta et al. 2006; Wnuk et al. 2020a, b). The cortices were mechanically and then enzymatically fragmented with 0.1% trypsin for 15 min at 37 °C. Next, the cells were centrifuged for 5 min at 1,500 × g and plated in medium containing 10% fetal bovine serum. The cells were cultured on plates (TPP Techno Plastic Products AG, Switzerland) covered by poly-L-ornithine in neurobasal medium (Thermo Fisher Scientific, USA) with L-glutamine (Sigma–Aldrich, USA), B27 (Thermo Fisher Scientific, USA), and penicillin–streptomycin antibiotics (Sigma-Aldrich, USA) at 37 °C in a humidified atmosphere containing 5% CO2 for 7 days in vitro (DIV). The density of the cells was 2.0 × 105 cells per cm2. All animals used in the research were maintained according to the principles of the Three Rs in compliance with European Union Legislation (Directive 2010/63/EU, amended by Regulation (EU) 2019/1010).
Experimental Models of Hypoxia and Ischemia
To induce hypoxic conditions, the cell medium was replaced with standard medium, whereas to induce ischemia, the cell medium was replaced with medium without glucose. Then, the procedure was the same for both experimental models. The cells were placed in a prewarmed and humidified hypoxia modular incubator chamber (Billups-Rothenberg, Inc., CA, USA) with 95% N2/5% CO2 for 6 h. The O2 level was measured with an oxygen analyzer (Greisinger, Germany) and reached less than 0.5%. After 6 h of hypoxic/ischemic conditions, i.e., at the reoxygenation period, the culture medium was replaced immediately with standard medium for 18 h (Wnuk, Przepiórska et al. 2021).
Treatment
Cell cultures were treated with PaPE-1 ((S)-5-(4-hydroxy-3,5-dimethyl-phenyl)-indan-1-ol) at a concentration of 0.01–10 μM. During reoxygenation, cells were cultured in a humidified incubator (New Brunswick Scientific, NJ, USA). PaPE-1 was dissolved in DMSO, not exceeding a concentration of 0.1% in the culture medium, as previously described (Wnuk et al. 2020a).
Assessment of Lactate Dehydrogenase Release
The cytotoxicity in neuronal cell cultures was assessed by measuring lactate dehydrogenase (LDH) release into the cell culture medium as previously described (Kajta et al. 2019). After experiments, the supernatant was collected and incubated with the relevant reaction mixture for 60 min at room temperature. The absorbance was measured after 30 and 60 min at 490 nm with the use of an Infinite M200 PRO microplate reader (Tecan Mannedorf, Switzerland), and the results were analyzed by i-control software. Data were normalized to the vehicle-treated cells (DMSO 0.1%) and presented as a percentage of the control ± SEM. The intensity of the red color was proportional to LDH release from damaged cells.
Caspase-3 Activity Measurement
Caspase-3 activity was measured in neocortical cell cultures 18 h after treatment with PaPE-1 (Kajta et al. 2009; Rzemieniec et al. 2019). To initiate cell lysis, caspase assay buffer (containing 50 mM HEPES, pH 7.4, 100 mM NaCl, 0.1% CHAPS, 1 mM EDTA, 10% glycerol, and 10 mM DTT) was added to each well. After this step, cell lysates were incubated with acetyl-Asp-Glu-Val-Asp p-nitroanilide (Ac-DEVD-pNA) for 60 min at 37 °C. The reaction was based on the hydrolysis of Ac-DEVD-pNA by caspase-3 and the release of p-nitroanilide detected at 405 nm. Absorbance measurements were performed using an Infinite M200 PRO microplate reader (Tecan, Mannedorf, Switzerland). The results were analyzed by i-control software, normalized to the absorbance of vehicle-treated cells (DMSO 0.1%), and presented as a percentage of the control ± SEM. The amount of the reaction product was proportional to caspase-3 activity.
Measurement of Neutral Red Uptake
To assess the viability of neuronal cell cultures after hypoxia/ischemia, neutral red dye was used. This dye which is able to bind to the lysosomes of viable cells, as previously described (Szychowski et al. 2019). First, a 10% neutral red solution was prepared, filtered, and heated to 37 °C. Then, the solution was added to the cells and was followed by 2 h of incubation at 37 °C. Then, the neuronal cultures were washed with PBS and incubated with acidified ethanol solution (50% ethanol, 1% acetic acid, 49% H2O) for 10 min. The absorbance was measured at a wavelength of 540 nm using an Infinite M200 PRO microplate reader (Tecan, Mannedorf, Switzerland) and i-control software. Data were normalized to vehicle-treated cells and presented as a percentage of the control ± SEM. The extracted dye was proportional to the number of living cells.
Assessment of MTT Reduction
This colorimetric assay is based on reducing yellow 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to purple formazan through oxidoreductase enzymes. The purple color intensity of dissolved formazan correlates with the mitochondrial activity of neuronal cells. After the experiment, MTT solution was added to the cell cultures for incubation at 37 °C for 1 h. Next, the cell medium was replaced with DMSO to dissolve formazan crystals. The absorbance was measured at 570 nm with the use of an Infinite M200 PRO microplate reader (Tecan Mannedorf, Switzerland), and the data were analyzed with i-control software. The results were normalized to vehicle-treated cell cultures and presented as the percentage of the control ± SEM.
Measurement of Degenerating Neurons
To determine the degree of neurodegeneration under hypoxic/ischemic conditions and after PaPE-1 treatment, Fluoro-Jade C (FJ-C) staining was used. This novel method enables in vitro detection of cytotoxicity using fluorochromatic dyes. The solution was prepared by mixing FJ-C with distilled water, and the culture medium was replaced with a mixture. After 1 h of incubation, the fluorescence was measured at Ex/Em = 490/525 by an Infinite M200 PRO microplate reader (Tecan Mannedorf, Switzerland). The results analyzed by i-control software were normalized to data from vehicle-treated cells and presented as the percentage of the control ± SEM.
Measurement of ROS Formation
The ROS-Glo™ H2O2 Assay (Promega, Madison, WI, USA) was used to assess the level of reactive oxygen species (ROS) in neocortical cells exposed to hypoxia/ischemia and PaPE-1 treatment. ROS-Glo™ H2O2 substrate was added to cell cultures at the end of the treatment and reacted with H2O2 present in our samples. The reaction product, a luciferin precursor, was converted to luciferin by adding ROS-Glo™ Detection Solution. The bioluminescence was measured with the use of a GloMax® Navigator Microplate Luminometer (Promega, Madison, WI, USA), and the light signal was proportional to the amount of H2O2 in cultured cells. The data were normalized to the bioluminescent signal intensity of vehicle-treated cells and expressed as a percentage of the control ± SEM.
Assessment of the JC-10 Concentration
JC-10 is a widely used dye that aggregates in mitochondria and changes its color from green to orange, which is detected as a membrane potential increase. To monitor mitochondrial membrane potential changes after hypoxic/ischemic episodes and PaPE-1 treatment, the JC-10 Mitochondrial Membrane Potential Assay Kit (Abcam, Cambridge, UK) was used. JC-10 dye-loading solution was added to each well and incubated with cell cultures for 1 h at 37 °C. After adding Assay Buffer B, the fluorescence was monitored at Ex/Em = 490/525 and Ex/Em = 540/590 nm with an Infinite M200 PRO microplate reader (Tecan Mannedorf, Switzerland). The fluorescence intensity was used for the ratio analysis, and the results are presented as a percentage of the control ± SEM. Data were normalized to the fluorescence intensity of vehicle-treated cells.
qPCR Analysis of mRNA-Specific for Genes Encoding Apoptosis-Related Factors
To extract total RNA from neocortical cells, a Qiagen RNeasy Mini Kit (Hilden, Germany) was used as previously described (Wnuk et al. 2019). The quantity of RNA was spectrophotometrically determined at 260 nm and 260/280 nm (ND/1000 UV/Vis; Thermo Fisher NanoDrop, Waltham, MA, USA). Two-step qPCR consisting of reverse transcription and qPCR was performed using the CFX96 Real-Time System (Bio-Rad, Hercules, CA, USA). Total RNA was reverse transcribed with a High-Capacity cDNA Reverse Transcription Assay (Thermo Fisher Scientific, Waltham, MA, USA). The collected cDNA was then amplified with TaqMan probes of the specific genes encoding Hprt, β-actin, Gapdh, Fas, Fasl, Bax, Bcl2, and Gsk3b. Twenty microliters of the final amplification mixture consisted of 10 µl of FastStart Universal Probe Master Mix (Roche, Basel, Switzerland), 8 µl of RNAse-free water, 1 µl of template cDNA, and 1 µl of the TaqMan probe. The qPCR procedure was performed as follows: 2 min at 50 °C, 10 min at 95 °C followed by 40 cycles of 15 s at 95 °C and 1 min at 60 °C. The obtained data were analyzed according to the delta Ct method. To choose the reference gene, RefFinder was used (Xie et al. 2012), which led to the selection of Hprt as a reference gene.
Enzyme-Linked Immunosorbent Assays for Apoptosis-Related Factors
The expression of apoptosis-related factors, i.e., FAS, FASL, BAX, BCL2, and GSK3β, was assessed by enzyme-linked immunosorbent assay (ELISA) after hypoxia/ischemia and PaPE-1 treatment as previously described (Kajta et al. 2020). Cells were lysed with ice-cold RIPA lysis buffer and protease inhibitor cocktail. Next, cell lysates were sonicated and centrifuged at 15,000 × g for 20 min at 4 °C. The protein concentration in the collected supernatant was measured using Bradford reagent and bovine serum albumin as a standard. Antigens from our samples were attached to the precoated wells with mouse antibody surfaces. Biotin-conjugated polyclonal antibodies specific for FAS, FASL, BAX, BCL2, and GSK3β were added to each well. After every step, the plates were washed to remove any nonspecifically bound proteins and antibodies. Then, streptavidin-HRP attached to biotinylated antibodies and the addition of substrate solution caused a color change from blue to yellow. The absorbance was measured with an Infinite M200 PRO microplate reader (Tecan Mannedorf, Switzerland), and the results were expressed as a percentage of the control value ± SEM and in terms of picograms per milligram of total protein. The intensity of yellow color was proportional to the amount of a specific protein.
Statistical Analysis of the Data
Statistical tests were performed on raw data. The results are expressed as the mean absorbance, fluorescence intensity (in arbitrary units) and luminescence signal per well containing 50,000 cells for analyses of LDH release, caspase-3 activity, Fluoro-Jade C, ROS activity, neutral red staining, MTT, and mitochondrial membrane potential as fluorescence units per 1.5 million cells for qPCR or as the picograms per miligram of total protein for the ELISAs. One-way analysis of variance (ANOVA) was preceded by Levene’s test of homogeneity of variances and was used to determine overall significance. Differences between the control and experimental groups were assessed with a post hoc Newman–Keuls test. Significant differences were indicated as follows: *p < 0.05, **p < 0.01, ***p < 0.001 compared to the normoxic control; #p < 0.05, ##p < 0.01, ###p < 0.001 compared to the cell cultures exposed to hypoxia; and ^p < 0.05, ^^p < 0.01, and ^^^p < 0.001 compared to the cell cultures exposed to ischemia. The results are expressed as the mean ± SEM of 3 independent experiments. The number of replicates ranged from 6 to 12.
Results
PaPE-1 Inhibited Lactate Dehydrogenase (LDH) Release and Caspase-3 Activity in Neocortical Cell Cultures Exposed to Hypoxia/Ischemia
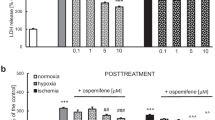
In this study, after 6 h of hypoxia/ischemia, 18 h of reoxygenation was applied. Hypoxia and ischemia conditions induced LDH release to 263% and 430% of the normoxic value, respectively (Fig. 1a). Caspase-3 activity remained unchanged under hypoxic/ischemic conditions (Fig. 1b). In this study, PaPE-1 was added during the reoxygenation period as a posttreatment therapy 6 h after the initial injury. Under hypoxic and ischemic conditions, PaPE-1 (1–10 μM) inhibited LDH release to 72–93% of the hypoxia/ischemia value, whereas concentrations of 0.01 μM and 0.1 μM did not change the LDH release level (Fig. 1c). For caspase-3 activity, only 5 and 10 μM PaPE-1 reduced the value to 88–96% of the control value (Fig. 1d). Under normoxic conditions, PaPE-1 used at concentrations ranging from 0.01 to 10 μM did not change LDH release or caspase-3 activity.

PaPE-1 inhibited LDH release and caspase-3 activity in neocortical cell cultures exposed to hypoxia/ischemia injury. The results are presented as a percentage of the control. Each bar represents the mean ± SEM of 3 independent experiments, with 7 to 10 replicates per group. ***p < 0.001 compared to the normoxic control; #p < 0.05, ##p < 0.01, ###p < 0.001 compared to the cell cultures exposed to hypoxia; ^p < 0.05, ^^p < 0.01, ^^^p < 0.001 compared to the cell cultures exposed to ischemia
Since the most promising concentrations were 5 and 10 µM PaPE-1, these concentrations were used in the next experiments.
PaPE-1 Partially Reversed Hypoxia/Ischemia-Evoked Neurodegeneration
The degree of neurodegeneration was measured using Fluoro-Jade C staining. Six hours of hypoxic or ischemic conditions followed by 18 h of reoxygenation elevated the degree of neurodegeneration to 130% and 147% of the normoxic value, respectively. Administration of PaPE-1 at concentrations of 5 and 10 μM reversed neurodegeneration by decreasing the parameter values to 110 and 108%, respectively, under hypoxia and 130 and 132%, respectively, under ischemia. Under normoxic conditions, PaPE-1 at concentrations of 5 and 10 μM did not change the degree of degenerating neurons (Fig. 2).

PaPE-1 (5 and 10 μM) partially reversed hypoxia/ischemia-evoked neurodegeneration measured using Fluoro-Jade C staining in 7 DIV neocortical cultures. The results are presented as a percentage of the control. Each bar represents the mean ± SEM of 3 independent experiments, with 8–12 replicates per group. ***p < 0.001 compared to the normoxic control; ###p < 0.001 compared to the cell cultures exposed to hypoxia; ^^^p < 0.001 compared to the cell cultures exposed to ischemia
PaPE-1 Inhibited Hypoxia/Ischemia-Evoked Loss of Mitochondrial Membrane Potential
JC-10 labeling showed that hypoxia and ischemia resulted in a decrease in mitochondrial membrane potential, reaching 84 and 61% of the control value, respectively. Under hypoxic conditions, PaPE-1 administration (5 and 10 μM) normalized the mitochondrial membrane potential to 94–96% of the control value. In the ischemic model, PaPE-1 (5 and 10 μM) partially inhibited ischemia-evoked loss of mitochondrial membrane potential to 75% of the normoxic value. During normoxia, PaPE-1 at concentrations of 5 μM and 10 μM did not change this parameter (Fig. 3).

PaPE-1 (5 and 10 μM) inhibited hypoxia/ischemia-evoked loss of mitochondrial membrane potential in 7 DIV neocortical cultures. The results are presented as a percentage of the control. Each bar represents the mean ± SEM of 3 independent experiments, with 10–12 replicates per group. ***p < 0.001 compared to the normoxic control; ###p < 0.001 compared to the cell cultures exposed to hypoxia; ^^^p < 0.001 compared to the cell cultures exposed to ischemia
PaPE-1 Partially Reversed the Hypoxia/Ischemia-Evoked Decrease in Cell Metabolic Activity
MTT staining revealed that hypoxia and ischemia led to a loss of cell metabolic activity. As a consequence of hypoxia, a decrease to 79% of the normoxic value was observed. Comparably, ischemia resulted in 75% of the normoxic cells’ metabolic activity. PaPE-1 (5 and 10 μM) applied for the reoxygenation period increased the metabolic activity of cells subjected to hypoxia and ischemia. When administered after hypoxic insult, the compound normalized the tested parameter to control values, i.e., 96–98% of the control. Under ischemic conditions, 5 μM PaPE-1 increased cell metabolic activity to 87%, and 10 μM PaPE-1 improved it to 92% of the normoxic value. Under normoxic conditions, PaPE-1 did not induce changes in the MTT parameter (Fig. 4).

PaPE-1 (5 and 10 μM) partially reversed the hypoxia/ischemia-evoked decrease in cell metabolic activity measured using MTT staining in 7 DIV neocortical cultures. The results are presented as a percentage of the control. Each bar represents the mean ± SEM of 3 independent experiments, with 10–12 replicates per group. ***p < 0.001 compared to the normoxic control; ###p < 0.001 compared to the cell cultures exposed to hypoxia; ^^^p < 0.001 compared to the cell cultures exposed to ischemia
PaPE-1 Inhibited Hypoxia-/Ischemia-Evoked Cell Viability Decrease
In this study, neutral red staining revealed that 6 h of hypoxia/ischemia followed by 18 h of reoxygenation decreased cell viability to 78 and 41% of the normoxic value, respectively. PaPE-1 applied at the beginning of reoxygenation improved the viability of neurons subjected to both hypoxic and ischemic conditions. In the hypoxia model, a 5-μM concentration of the tested compound increased the measured parameter to 88% (10% increase), while treatment with a 10-μM concentration resulted in a rise to 91% (13% increase). PaPE-1 also protected cortical neurons after ischemic episodes by increasing cell viability to 51% (10% increase) after treatment with both 5 and 10 μM PaPE-1. Under normoxic conditions, neither 5 nor 10 μM PaPE-1 changed cell viability (Fig. 5).

PaPE-1 (5 and 10 μM) inhibited the hypoxia-/ischemia-evoked decrease in cell viability measured using neutral red uptake in neocortical cultures at 7 DIV. The results are presented as a percentage of the control. Each bar represents the mean ± SEM of 3 independent experiments, with 12 replicates per group. ***p < 0.001 compared to the normoxic control; ###p < 0.001 compared to the cell cultures exposed to hypoxia; ^^^p < 0.001 compared to the cell cultures exposed to ischemia
PaPE-1 Partially Reversed Hypoxia/Ischemia-Evoked ROS Formation
In neocortical cell cultures subjected to hypoxia or ischemia, increased ROS formation was observed. In both models, ROS formation doubled in response to oxygen or oxygen and glucose deprivation (212% of the normoxic value under hypoxia and 225% of the normoxic value under ischemia). Under hypoxia, treatment with PaPE-1 at concentrations of 5 and 10 μM substantially reduced the parameter to 145–167%. Similarly, under ischemic conditions, PaPE-1 also lowered the ROS formation, resulting in a decrease to 137–162%. Moreover, under normoxic conditions, neither concentration of PaPE-1 altered ROS activity in neocortical cells (Fig. 6).

PaPE-1 (5 and 10 μM) partially reversed hypoxia/ischemia-evoked ROS formation in neocortical cultures at 7 DIV. The results are presented as a percentage of the control. Each bar represents the mean ± SEM of 3 independent experiments, with 10–12 replicates per group. ***p < 0.001 compared to the normoxic control; ###p < 0.001 compared to the cell cultures exposed to hypoxia; ^^^p < 0.001 compared to the cell cultures exposed to ischemia
PaPE-1 Normalized the Hypoxia/Ischemia-Dysregulated Expression Levels of Apoptosis-Related Genes and Proteins
In our study, exposure to hypoxia enhanced the mRNA expression levels of apoptosis-related genes, i.e., Fas (0.64-fold increase) and Bcl2 (0.36-fold increase), although the expression of Fasl, Bax, and Gsk3b remained unchanged. Treatment with PaPE-1 (5 μM) decreased only the expression of Fas to 1.2-fold. In ischemia-exposed cells, the expression of apoptosis-related genes substantially increased, i.e., Fas (7.82-fold increase), Fasl (1.25-fold increase), Bax (0.85-fold increase), Bcl2 (0.81-fold increase), and Gsk3b (0.23-fold increase). After PaPE-1 (5 μM) administration, the expression of Fasl decreased to 1.66-fold, and the expression of Fas and Bcl2 increased to 11.6-fold and 2.35-fold, respectively. PaPE-1 did not change the mRNA expression levels of Bax and Gsk3b. In normoxic cells treated with PaPE-1, the expression of all tested apoptosis-related genes did not change (Fig. 7a).

PaPE-1 (5 μM) affected the hypoxia/ischemia-induced increased expression levels of apoptosis-related genes and proteins in mouse neocortical cells at 7 DIV. Receptor levels were measured using qPCR (a) and specific ELISAs (b). Each result is presented as a fold change, a percentage of the control or in terms of pg per mg of total protein. Each bar represents the mean ± SEM of 3 independent experiments, with 6 replicates per group. *p < 0.05, **p < 0.01, ***p < 0.001 compared to the normoxic control; ##p < 0.01, ###p < 0.001 compared to the cell cultures exposed to hypoxia; ^p < 0.05, ^^^p < 0.001 compared to the cell cultures exposed to ischemia
ELISA analyses showed that the protein levels of FAS, FASL, BAX, BCL2, and GSK3β in the control cells (normoxia) reached 0.06, 134.43, 19.07, 1.47, and 2.17 pg per mg of total protein, respectively. Hypoxia increased the expression of all analyzed proteins by 46–140%, and PaPE-1 (5 μM) treatment partially inhibited the excessive expression of FAS (108% decrease), FASL (60% decrease), BAX (42% decrease), and GSK3β (71% decrease); however, the expression of BCL2 increased by 360% in response to PaPE-1 treatment. Similar to those under hypoxia, the protein levels of FAS, FASL, BAX, BCL2, and GSK3β significantly increased by 44–262% in response to ischemic conditions. Exposure to PaPE-1 (5 μM) decreased the protein expression of FAS (102% decrease), FASL (50% decrease), BAX (65% decrease), and GSK3β (127% decrease), whereas the expression of BCL2 increased by 111% in response to PaPE-1 treatment. Normoxic cells treated with PaPE-1 did not exhibit changes in the expression of apoptosis-related proteins (Fig. 7b).
Discussion
Newly synthesized PaPE-1 selectively activates membrane (nonnuclear/extranuclear) estrogen receptors (ERs), namely, mERα, and mERβ, and has been shown to evoke neuroprotection in a cellular model of AD and in a mouse model of stroke, specifically transient middle cerebral artery occlusion (tMCAO). PaPE-1 appeared to inhibit both Aβ-induced neurotoxicity in mouse primary neurons in in vitro cultures and tMCAO-evoked infarction and leukocyte infiltration into the ischemic mouse brain (Wnuk et al. 2020a; Selvaraj et al. 2018). However, until recently, PaPE-1’s effectiveness in protecting brain tissue against hypoxia/ischemia has been verified only in a pretreatment paradigm, namely, PaPE-1 was administered 24 h before inducing brain lesions via tMCAO.
This is the first study showing that 6-h delayed posttreatment with PaPE-1 inhibited hypoxia-/ischemia-induced neuronal death, as indicated by neutral red uptake in mouse primary neocortical cell cultures in vitro. The effect was accompanied by substantial decreases in neurotoxicity and neurodegeneration measured by LDH release and Fluoro-Jade C staining of damaged cells, respectively. Previously, we used the same cellular models of perinatal asphyxia and stroke, i.e., 6-h hypoxia or ischemia followed by 18-h reoxygenation, which reflected the major features of brain pathologies in vivo (Wnuk, Przepiórska et al. 2021), including oxidative stress due to enhanced ROS production, oxidative DNA damage, neurotoxicity, and neurodegeneration caused by elevated LDH release and Fluoro-Jade C staining, and neuronal cell death due to decreased MTT activity.
Since PaPE-1 has only been recently synthesized, no relevant reports are available to compare the results of our present studies. The beneficial effects of ER agonists, particularly estrogens, on brain tissue undergoing hypoxia and/or ischemia have been extensively reported; however, only a few research reports have focused on membrane ERs, mainly GPER1 (previously GPR30). In the mouse brain challenged with MCAO-induced ischemic injury, 17β-estradiol (E2)-activated GPR30 was shown to inhibit Toll-like receptor 4 (TLR4)-mediated microglial inflammation as well as infarction volume and neuronal damage (Zhang et al. 2018). Similarly, GPER1 appeared to mediate E2-exerted neuroprotection in mouse hippocampal neurons exposed to oxygen and glucose deprivation (OGD) (Zhao et al. 2016). Activation of GPER1 by the selective agonist G1 exerted anti-inflammatory effects and preserved cognitive function in rats subjected to global cerebral ischemia (Bai et al. 2020). Intriguingly, in female mice subjected to cerebral ischemia, G1 appeared to reduce infarct volume and neurological deficits, whereas in male mice, G1 increased infarct volume and worsened functional outcome (Broughton et al. 2014).
Much less is known about the involvement of non-GPER membrane ERs in neuroprotection against hypoxic/ischemic injury. GPER1 mainly involves PKA, PKB, and ERK signaling cascades and phosphoinositide-3-kinase (PI3K), in addition to G-proteins (Gαs, Gαi); in contrast, non-GPER membrane ERs act preferably via Ca2+ liberation from intracellular stores, PLC, PKC, Src/ERK, PI3K/AKT, p38/MAPK, JAK/STAT, Pak1, and FAK (Acconcia et al. 2021). A recently identified membrane-based estrogen receptor, ER-α36, that is expressed in human and rodent brains exhibited neuroprotective properties in animal and cellular models of ischemic stroke (Zou et al. 2015). In particular, ER-α36 was activated by E2 in OGD-exposed PC12 cells, which confirmed its participation in rescuing the cells from ischemic damage. In rat brains subjected to global hypoxia, Cicer microphyllum seed supplementation caused neuroprotection that was mediated through ERβ-dependent extranuclear activation of ERK1/2 (Sharma et al. 2017), which is partially in line with the mERα/mERβ-mediated neuroprotection observed in our present study after treatment with PaPE-1.
In this study, the mechanisms of neuroprotective action of PaPE-1 against hypoxia-/ischemia-induced injury also involved an inhibition of apoptosis that was evidenced by normalization of both mitochondrial membrane potential and expression levels of apoptosis-related genes, i.e., Fas, Fasl, and Bcl2, and proteins such as FAS, FASL, BCL2, BAX, and GSK3β. Furthermore, in the present study, PaPE-1-evoked neuroprotection was mediated through a reduction in ROS formation and enhancement of cellular metabolic activity that was dysregulated in the course of hypoxia and ischemia. Previously, we showed that PaPE-1 has the ability to reduce ROS formation and to prevent apoptosis through the restoration of the mitochondrial membrane potential and BAX/BCL2 ratio as well as the downregulation of Fas/FAS expression (Wnuk et al. 2020a), which concurs with the results of the present research. Interestingly, estrogen deficiency appeared to predispose brain neurons to apoptosis following cerebral ischemia (Guo et al. 2017), which supports essential roles of estrogen-based compounds such as PaPE-1 in pharmacotherapy of stroke and perinatal asphyxia.
Conclusion
These data provide evidence that targeting membrane non-GPER estrogen receptors with PaPE-1 is an effective therapy that protects brain neurons from hypoxic/ischemic damage by inhibiting neurotoxicity, neurodegeneration, and apoptosis and by reducing oxidative stress and enhancing cellular metabolic activity, even when applied with a 6-h delay from injury onset. Hence, investigating the molecular mechanisms by which PaPE-1 regulates hypoxia-/ischemia-related processes may lead to the development of a novel posttreatment therapy that targets the membrane-associated non-GPER ERs and opens up new therapeutic perspectives for stroke and perinatal asphyxia.
Abbreviations
- AD :
-
Alzheimer’s disease
- Aβ:
-
Amyloid β
- E2:
-
17β-Estradiol
- ERs:
-
Estrogen receptors
- ERα/β:
-
Estrogen receptor α/β
- FJ-C:
-
Fluoro-Jade C
- LDH:
-
Lactate dehydrogenase
- MCAO:
-
Middle cerebral artery occlusion
- mERs:
-
Membrane estrogen receptors
- MTT:
-
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- OGD:
-
Oxygen and glucose deprivation
- PaPE-1:
-
Pathway preferential estrogen-1
- ROS:
-
Reactive oxygen species
- rtPA:
-
Recombinant tissue plasminogen activator
- tMCAO:
-
Transient middle cerebral artery occlusion
References
Acconcia F, Fiocchetti M, Busonero C, Fernandez VS, Montalesi E, Cipolletti M, Pallottini V, Marino M (2021) The extranuclear interactome of the estrogen receptors: implications for physiological functions. Mol Cell Endocrinol 538:111452. https://doi.org/10.1016/j.mce.2021.111452
Bai N, Zhang Q, Zhang W, Liu B, Yang F, Brann D, Wang R (2020) G-protein-coupled estrogen receptor activation upregulates interleukin-1 receptor antagonist in the hippocampus after global cerebral ischemia: implications for neuronal self-defense. J Neuroinflammation 17(1):45. https://doi.org/10.1186/s12974-020-1715-x
Broughton BR, Brait VH, Kim HA, Lee S, Chu HX, Gardiner-Mann CV, Guida E, Evans MA, Miller AA, Arumugam TV, Drummond GR, Sobey CG (2014) Sex-dependent effects of G protein-coupled estrogen receptor activity on outcome after ischemic stroke. Stroke 45(3):835–841. https://doi.org/10.1161/STROKEAHA.113.001499
Centers for Disease Control and Prevention. Types of stroke (2020) cdc.gov https://www.cdc.gov/stroke/types_of_stroke.htm Accessed 3 Nov 2021
Chen W, Ostrowski RP, Obenaus A, Zhang JH (2009) Prodeath or prosurvival: two facets of hypoxia inducible factor-1 in perinatal brain injury. Exp Neurol 216(1):7–15. https://doi.org/10.1016/j.expneurol.2008.10.016
De Butte-Smith M, Zukin RS, Etgen AM (2012) Effects of global ischemia and estradiol pretreatment on phosphorylation of Akt, CREB and STAT3 in hippocampal CA1 of young and middle-aged female rats. Brain Res 1471:118–128. https://doi.org/10.1016/j.brainres.2012.06.036
Frendl A, Csiba L (2011) Pharmacological and non-pharmacological recanalization strategies in acute ischemic stroke. Front Neurol 2:32. https://doi.org/10.3389/fneur.2011.00032
Guo JM, Shu H, Wang L, Xu JJ, Niu XC, Zhang L (2017) SIRT1-dependent AMPK pathway in the protection of estrogen against ischemic brain injury. CNS Neurosci Ther 23(4):360–369. https://doi.org/10.1111/cns.12686
Jiang M, Ma C, Li H, Shen H, Li X, Sun Q, Chen G (2020) Sex dimorphisms in ischemic stroke: from experimental studies to clinic. Front Neurol 11:504. https://doi.org/10.3389/fneur.2020.00504
Jilani TN, Siddiqui AH (2021) Tissue plasminogen activator. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan–. PMID: 29939694
Kajta M, Wójtowicz AK, Maćkowiak M, Lasoń W (2009) Aryl hydrocarbon receptor-mediated apoptosis of neuronal cells: a possible interaction with estrogen receptor signaling. Neuroscience 158(2):811–822. https://doi.org/10.1016/j.neuroscience.2008.10.045
Kajta M, Rzemieniec J, Wnuk A, Lasoń W (2020) Triclocarban impairs autophagy in neuronal cells and disrupts estrogen receptor signaling via hypermethylation of specific genes. The Science of the Total Environment 701:134818. https://doi.org/10.1016/j.scitotenv.2019.134818
Kajta M, Trotter A, Lasoń W, Beyer C (2006) Impact of 17beta-estradiol on cytokine-mediated apoptotic effects in primary hippocampal and neocortical cell cultures. Brain Res 1116(1):64–74. https://doi.org/10.1016/j.brainres.2006.07.105
Kajta M, Wnuk A, Rzemieniec J, Lason W, Mackowiak M, Chwastek E, Staniszewska M, Nehring I, Wojtowicz AK (2019) Triclocarban disrupts the epigenetic status of neuronal cells and induces AHR/CAR-mediated apoptosis. Mol Neurobiol 56(5):3113–3131. https://doi.org/10.1007/s12035-018-1285-4
Li W, Yang S (2016) Targeting oxidative stress for the treatment of ischemic stroke: upstream and downstream therapeutic strategies. Brain Circ Oct-Dec 2(4):153–163. https://doi.org/10.4103/2394-8108.195279
Madak-Erdogan Z, Kim SH, Gong P, Zhao YC, Zhang H, Chambliss KL, Carlson KE, Mayne CG, Shaul PW, Korach KS, Katzenellenbogen JA, Katzenellenbogen BS (2016) Design of pathway preferential estrogens that provide beneficial metabolic and vascular effects without stimulating reproductive tissues. Sci Signal 9(429):ra53. https://doi.org/10.1126/scisignal.aad8170
Manandhar SR, Basnet R (2019) Prevalence of perinatal asphyxia in neonates at a tertiary care hospital: a descriptive cross-sectional study. JNMA J Nepal Med Assoc 57(219):287–292. https://doi.org/10.31729/jnma.4550
Miller DJ, Simpson JR, Silver B (2011) Safety of thrombolysis in acute ischemic stroke: a review of complications, risk factors, and newer technologies. The Neurohospitalist 1(3):138–147. https://doi.org/10.1177/1941875211408731
Rainaldi MA, Perlman JM (2016) Pathophysiology of birth asphyxia. Clin Perinatol 43(3):409–22. https://doi.org/10.1016/j.clp.2016.04.002
Rzemieniec J, Wnuk A, Lasoń W, Bilecki W, Kajta M (2019) The neuroprotective action of 3,3’-diindolylmethane against ischemia involves an inhibition of apoptosis and autophagy that depends on HDAC and AhR/CYP1A1 but not ERα/CYP19A1 signaling. Apoptosis : an International Journal on Programmed Cell Death 24(5–6):435–452. https://doi.org/10.1007/s10495-019-01522-2
Saczko J, Michel O, Chwiłkowska A, Sawicka E, Mączyńska J, Kulbacka J (2017) Estrogen receptors in cell membranes: regulation and signaling. Adv Anat Embryol Cell Biol 227:93–105. https://doi.org/10.1007/978-3-319-56895-9_6
Selvaraj UM, Zuurbier KR, Whoolery CW, Plautz EJ, Chambliss KL, Kong X, Zhang S, Kim SH, Katzenellenbogen BS, Katzenellenbogen JA, Mineo C, Shaul PW, Stowe AM (2018) Selective nonnuclear estrogen receptor activation decreases stroke severity and promotes functional recovery in female mice. Endocrinology 159(11):3848–3859. https://doi.org/10.1210/en.2018-00600
Sharma D, Biswal SN, Kumar K, Bhardwaj P, Barhwal KK, Kumar A, Hota SK, Chaurasia OP (2017) Estrogen receptor β mediated neuroprotective efficacy of Cicer microphyllum seed extract in global hypoxia. Neurochem Res 42(12):3474–3489. https://doi.org/10.1007/s11064-017-2395-5
Szychowski KA, Wnuk A, Rzemieniec J, Kajta M, Leszczyńska T, Wójtowicz AK (2019) Triclosan-evoked neurotoxicity involves NMDAR subunits with the specific role of GluN2A in caspase-3-dependent apoptosis. Mol Neurobiol 56(1):1–12. https://doi.org/10.1007/s12035-018-1083-z
Vijverberg Joanna RG, Lopriore E, Te Pas AB, Rijken M, van Zwet EW, de Bruine FT, Steggerda SJ (2021) Persistent pulmonary hypertension in neonates with perinatal asphyxia and therapeutic hypothermia: a frequent and perilous combination. The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal Obstetricians, 1–7. Advance online publication. https://doi.org/10.1080/14767058.2021.1873941
Wu MY, Yiang GT, Liao WT, Tsai AP, Cheng YL, Cheng PW, Li CY, Li CJ (2018) Current mechanistic concepts in ischemia and reperfusion injury. Cell Physiol Biochem 46(4):1650–1667. https://doi.org/10.1159/000489241
Wnuk A, Przepiórska K, Pietrzak BA, Kajta M (2021) Post-treatment with Amorfrutin B evokes PPARγ-mediated neuroprotection against hypoxia and ischemia. Biomedicines 9(8):854. https://doi.org/10.3390/biomedicines9080854
Wnuk A, Przepiórska K, Rzemieniec J, Pietrzak B, Kajta M (2020a) Selective targeting of non-nuclear estrogen receptors with PaPE-1 as a new treatment strategy for Alzheimer’s disease. Neurotox Res 38(4):957–966. https://doi.org/10.1007/s12640-020-00289-8
Wnuk A, Rzemieniec J, Przepiórska K, Wesołowska J, Wójtowicz AK, Kajta M (2020b) Autophagy-related neurotoxicity is mediated via AHR and CAR in mouse neurons exposed to DDE. The Science of the Total Environment 742:140599. https://doi.org/10.1016/j.scitotenv.2020.140599
Wnuk A, Rzemieniec J, Staroń J, Litwa E, Lasoń W, Bojarski A, Kajta M (2019) Prenatal exposure to benzophenone-3 impairs autophagy, disrupts RXRs/PPARγ signaling, and alters epigenetic and post-translational statuses in brain neurons. Mol Neurobiol 56(7):4820–4837. https://doi.org/10.1007/s12035-018-1401-5
World Health Organisation (n.d.) Stroke, cerebrovascular accident. Retrieved from: https://www.emro.who.int/health-topics/stroke-cerebrovascular-accident/index.html (accessed on 2 Oct 2021)
Xie F, Xiao P, Chen D, Xu L, Zhang B (2012) miRDeepFinder: a miRNA analysis tool for deep sequencing of plant small RNAs. Plant Molecular Biology, Advance Online Publication. https://doi.org/10.1007/s11103-012-9885-2
Yousufuddin M, Young N (2019) Aging and ischemic stroke. Aging May 1;11(9):2542–2544. https://doi.org/10.18632/aging.101931
Zhang Z, Qin P, Deng Y, Ma Z, Guo H, Guo H, Hou Y, Wang S, Zou W, Sun Y, Ma Y, Hou W (2018) The novel estrogenic receptor GPR30 alleviates ischemic injury by inhibiting TLR4-mediated microglial inflammation. J Neuroinflammation 15(1):206. https://doi.org/10.1186/s12974-018-1246-x
Zhao TZ, Shi F, Hu J, He SM, Ding Q, Ma LT (2016) GPER1 mediates estrogen-induced neuroprotection against oxygen-glucose deprivation in the primary hippocampal neurons. Neuroscience 328:117–126. https://doi.org/10.1016/j.neuroscience.2016.04.026
Zou W, Fang C, Ji X, Liang X, Liu Y, Han C, Huang L, Zhang Q, Li H, Zhang Y, Liu J, Liu J (2015) Estrogen receptor (ER)-α36 is involved in estrogen- and tamoxifen-induced neuroprotective effects in ischemic stroke models. PLoS One 10(10):e0140660. https://doi.org/10.1371/journal.pone.0140660
Zubcevic S, Heljic S, Catibusic F, Uzicanin S, Sadikovic M, Krdzalic B (2015) Neurodevelopmental follow up after therapeutic hypothermia for perinatal asphyxia. Medical Archives (sarajevo, Bosnia and Herzegovina) 69(6):362–366. https://doi.org/10.5455/medarh.2015.69.362-366
Acknowledgements
The manuscript was edited for proper English language, grammar, punctuation, spelling, and overall style by one or more of the highly qualified native English-speaking editors at American Journal Experts—79ED-CE2B-7F94-70E0-D8B9.
Funding
This research was funded by the National Science Centre of Poland, grant number 2018/31/B/NZ7/01815, and statutory fund of the Maj Institute of Pharmacology, Polish Academy of Sciences in Krakow, Poland.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wnuk, A., Przepiórska, K., Pietrzak, B.A. et al. Posttreatment Strategy Against Hypoxia and Ischemia Based on Selective Targeting of Nonnuclear Estrogen Receptors with PaPE-1. Neurotox Res 39, 2029–2041 (2021). https://doi.org/10.1007/s12640-021-00441-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-021-00441-y