
Abstract
Background
Hirschsprung’s disease (HSCR) is one of the most common congenital digestive tract malformations and can cause stubborn constipation or gastrointestinal obstruction after birth, causing great physical and mental pain to patients and their families. Studies have shown that more than 20 genes are involved in HSCR, and most cases of HSCR are sporadic. However, the overall rate of familial recurrence in 4331 cases of HSCR is about 7.6%. Furthermore, familial HSCR patients show incomplete dominance. We still do not know the penetrance and genetic characteristics of these known risk genes due to the rarity of HSCR families.
Methods
To find published references, we used the title/abstract terms “Hirschsprung” and “familial” in the PubMed database and the MeSH terms “Hirschsprung” and “familial” in Web of Science. Finally, we summarized 129 HSCR families over the last 40 years.
Results
The male-to-female ratio and the percentage of short segment-HSCR in familial HSCR are much lower than in sporadic HSCR. The primary gene factors in the syndromic families are ret proto-oncogene (RET) and endothelin B receptor gene (EDNRB). Most families show incomplete dominance and are relevant to RET, and the RET mutation has 56% penetrance in familial HSCR. When one of the parents is a RET mutation carrier in an HSCR family, the offspring’s recurrence risk is 28%, and the incidence of the offspring does not depend on whether the parent suffers from HSCR.
Conclusion
Our findings will help HSCR patients obtain better genetic counseling, calculate the risk of recurrence, and provide new insights for future pedigree studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hirschsprung’s disease (HSCR) is one of the most common congenital digestive tract malformations, with an incidence of 1 in 5000 and a male‒female ratio of 4:1. Due to the loss of enteric neurons in the distal colon, HSCR can cause stubborn constipation or gastrointestinal obstruction after birth due to loss of enteric neurons in the distal colon, bringing great physical and mental pain to patients and their families [1]. With the development of sequencing technology and bioinformatics analysis, more than 20 genes [ret proto-oncogene (RET), endothelin B receptor gene (EDNRB), paired-like homeobox 2B gene (PHOX2B), etc.] have been linked to HSCR [2,3,4,5].
Although most cases of HSCR are sporadic, some families with two or more HSCR family members are classified as HSCR families. Three studies reported a series of HSCR families [6,7,8], implying that HSCR has a genetic predisposition. Mc Laughlin and Puri reported a 7.6% overall rate of familial recurrence in 4331 HSCR index cases [9]. Familial HSCR is also due to a list of pathogenic genes, such as RET [10], EDNRB [11], and PHOX2B [12]. However, familial HSCR does not follow Mendelian inheritance, and RET and PHOX2B show incomplete penetrance in members of familial HSCR [10, 12]. Because HSCR families are uncommon, current gene studies are based on a summary of a few families. The penetrance and genetic characteristics of these known risk genes in familial HSCR, particularly the major pathogenic gene RET, which occurs in 50% of familial HSCR and 35% of sporadic HSCR, remain unknown [13].
In this study, we summarized 129 HSCR families reported in 53 references to analyze the penetrance, recurrence risk, and genetic characteristics of familial HSCR. Our study will elucidate the genetic characteristics of familial HSCR, provide preferable genetic counseling for HSCR patients, help in calculating the risk of recurrence, and provide new insights for future pedigree studies.
Methods
We used the title/abstract terms “Hirschsprung” and “familial” in the PubMed database and the MeSH terms “Hirschsprung” and “familial” in the Web of Science to search published references. At least two family members with HSCR were required for inclusion. Studies of multicenter investigated data were excluded to avoid including duplicate families in the analysis. Finally, we confirmed and analyzed 53 references containing 129 families (Supplemental Table 1) [6,7,8, 10,11,12, 14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60].
The group-divisible designs are as follows: (1) HSCR subtypes: short segment-HSCR (S-HSCR), long segment-HSCR (L-HSCR), total colonic aganglionosis (TCA), total bowel aganglionosis, not available (NA); (2) syndromic HSCR and non-syndromic HSCR/not mentioned, syndromic HSCR refers to HSCR patients suffering other syndromic symptoms, such as familial medullary thyroid cancer, multiple endocrine neoplasia type 2a (MEN2A) and Waardenburg syndrome; (3) male and female; (4) familial genetic characteristics: “parent to child, lineal 3 generation” means determined HSCR occurs in both the proband’s parents (father, mother, or both) and grandparents (grandfather, grandmother, or both), “parent to child, lineal 2 generation” means determined HSCR occurs in the proband’s parents (father, mother, or both), “siblings” means determined HSCR occurs in the proband’s brothers or sisters, “collateral relatives” means determined HSCR occurs in the proband’s cousins or relatives; (5) syndromic symptoms: familial medullary thyroid cancer (FMTC), MEN2A, Waardenburg syndrome, Bardet–Biedl syndrome, special physical characteristics, respiratory symptoms, external auditory canal agenesis, anisocoria, multiple sclerosis, congenital central hypoventilation syndrome, Currarino syndrome, neuroblastoma, congenital heart disease, trisomy 21, meningocele and intellectual disability; (6) genetic patterns or pathogenic mechanisms: dominant inheritance, recessive inheritance, incomplete dominance, compound heterozygous inheritance, epistasis; (7) transmission pattern: affected father to affected son, affected father to unaffected son, affected father to affected daughter, affected father to unaffected daughter, unaffected father to affected son, unaffected father to unaffected son, unaffected father to affected daughter, unaffected father to unaffected daughter, affected mother to affected son, affected mother to unaffected son, affected mother to affected daughter, affected mother to unaffected daughter, unaffected mother to affected son, unaffected mother to unaffected son, unaffected mother to affected daughter, unaffected mother to unaffected daughter (Supplementary Table 2).
We analyzed the penetrance and transmission patterns of the RET gene. Inclusion criteria are as follows: (1) families with equal to or more than two members diagnosed with HSCR; (2) the RET mutation was linked to families; (3) detailed RET mutation sites are available; and (4) there is only one variation of RET. The exclusion criteria were as follows: (1) consanguineous marriage; (2) unavailable RET mutation information or de novo variants; and (3) equal to or more than two variations of RET. Finally, we summarized 110 RET carriers from 21 references (Supplementary Table 3) [10, 16,17,18,19, 21, 24, 27, 30, 31, 36,37,38,39,40, 43, 44, 47, 50, 52, 54].
Results
Sex ratio of HSCR subtypes and syndromic HSCR in familial HSCR
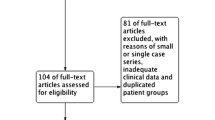
We finally discovered 53 references (Fig. 1), including 129 families with 416 HSCR cases (Tables 1 and 2). The ratio of males to females in S-HSCR was 1.11 (39/35); the ratio of males to females in L-HSCR was 1.48 (43/29); the ratio of males to females in TCA/total bowel aganglionosis was 0.78 (7/9); and the ratio of males to females in all familial HSCR cases was 1.51 (250/166) (Table 1), which is much lower than the sporadic HSCR ratio (4:1) [61]. The male-to-female ratio in syndromic HSCR was 1.11 (39/35), and the male-to-female ratio in non-syndromic HSCR/not mentioned was 1.61 (211/131) (Table 2). The percentages of S-HSCR, L-HSCR, TCA and total bowel ananglionos in familial HSCR cases are 46% (74/162), 44% (72/162), 8% (13/162) and 2% (3/162), respectively, while the reported percentage of S-HSCR reported in sporadic HSCR cases is 80% [61].

Preferred Reporting Items for Systematic Review and Meta-Analysis 2020 flow diagram for new systematic reviews that included searches of databases and registers only. HSCR Hirschsprung’s Disease
Syndromic HSCR families exist mainly in sibling families
We examined the 129 families (Table 3). Syndromic HSCR families accounted for 30% (39/129); parent to child (lineal 2 generation) families accounted for 32% (41/129); and sibling families accounted for 53% (68/129). The ratio of syndromic HSCR families to non-syndromic/not mentioned HSCR families in sibling families is 0.48 (22/46); in parent to child (lineal 2 generation) families, the ratio is 0.28 (9/32); and in all families, the ratio of syndromic HSCR families to non-syndromic/not mentioned HSCR families is 0.43 (39/90). Sibling families account for the majority of syndromic HSCR families (22/39, 56%); sibling and parent to child (lineal 2 generation) families are the most common familial HSCR characteristics.
The primary genetic factors of syndromic familial HSCR are RET and EDNRB
There were 74 HSCR patients with syndromic symptoms in 39 families (Table 4). FMTC, MEN2A, and Waardenburg syndrome families accounted for 41% (16/39) and 31% (12/39), respectively, of the total; FMTC/MEN2A and Waardenburg syndrome patients accounted for 39% (29/74) and 31% (29/73) of the total, respectively. FMTC/MEN2A, Waardenburg syndrome, and intellectual disability are all linked to RET mutations; EDNRB mutations are linked to Waardenburg syndrome, special physical characteristics, and multiple sclerosis; and PHOX2B mutations can cause respiratory symptoms, anisocoria, congenital central hypoventilation syndrome, and congenital heart disease.
Familial HSCR has complicated genetic patterns
There were 62 families with detailed gene information in this analysis (Table 5). Twenty-seven percent (17/62) of families show dominant inheritance, and 47% (29/62) of families show incomplete dominance. RET-associated families accounted for 65% (40/62). In 40 RET-associated families, 30% (12/40) showed dominant inheritance, and 58% (23/40) showed incomplete dominance.
The penetrance of the RET mutation and recurrence risk in familial HSCR
There are 110 RET mutation carriers in 21 familial HSCR references. The number of affected carriers (HSCR and the RET mutation) is 62, implying that the RET mutation is 56% (62/110) penetrant in familial HSCR (“carriers” means RET gene mutation members; “affected carriers” means HSCR along with RET mutation). As a result, when one of the parents is a RET mutation carrier in an HSCR family, the offspring’s recurrence risk is 28% (1/2 of 56%). We counted the number of different transmission patterns (Fig. 2): unaffected RET-carrying parents transmitted the variation to children, and the percentage of affected children was 73% (16/22); affected RET-carrying parents transmitted the variation to children, and the percentage of affected children was 78% (7/9). They seem to have a similar HSCR rate, meaning that the incidence of HSCR in offspring does not depend on whether the parents suffer from it.

Number of different transmission patterns in RET mutation carriers. RET ret proto-oncogene
Discussion
Previous studies only looked at a few HSCR families due to their rarity. They usually focus on one of these pathogenic genes and study its biological function. However, there is a lack of systematic research on the genetic characteristics and gene penetrance of familial HSCR. We summarized all 129 families reported in this study over the last 40 years, concluded that the genetic feature of familial HSCR exists, provided preferable genetic counseling for HSCR patients, assisted in calculating the risk of recurrence, and provided new insights for future pedigree studies.
We determined that 65% of HSCR families were associated with RET in our study. RET shows complex genetic patterns, including dominant inheritance (30%) and incomplete dominance (58%). We determined the penetrance of the RET mutation to be 56% in familial HSCR. That is, when a member of a RET-associated family is diagnosed with HSCR, other carriers with the RET mutation have a 56% chance of developing HSCR. When one of the parents is a RET mutation carrier in an HSCR family, the offspring’s recurrence risk is 28%.
A few risk genes are highly associated with familial HSCR and show multiple genetic characteristics. The function of RET in HSCR is complicated and varied. Previous research has revealed the dominant inheritance and incomplete dominance of RET. However, some scholars have also proposed dosage-dependent penetrance and epistasis to explain the phenomenon of incomplete dominance and different subtypes of HSCR [30, 33]. For other risk genes, EDNRB shows dominant inheritance, recessive inheritance and incomplete dominance, and PHOX2B shows dominant inheritance and incomplete dominance. Thirty percent of families are syndromic, and 56% (22/39) of syndromic HSCR families are sibling groups. Syndromic symptoms (FMTC/MEN2A and Waardenburg syndrome) are mainly caused by RET and EDNRB. Syndrome symptoms are common and varied, so it is important to focus on other complications in HSCR patients and pay attention to risk genes, such as RET, EDNRB and PHOX2B.
Twelve percent (15/129) of the families had consanguineous marriages. Consanguineous marriage appears to be a risk factor for HSCR, and it is recommended that consanguineous marriage be avoided. We also observed 11 pairs of twins (six pairs of monozygotic twins and five pairs of dizygotic twins). In six pairs of monozygotic twins, four pairs (six males and two females) were diagnosed with HSCR, while there was only one case diagnosed with HSCR in the remaining two pairs. Two pairs of dizygotic twins were diagnosed with HSCR, while the remaining pairs had only one HSCR patient. Thus, it seems that the risk of HSCR was independent in both monozygotic and dizygotic twins due to the incomplete dominance of HSCR.
The mutated genes or loci, especially the gene RET, reported in the families in the references we included conform to the law of genetic coseparation and are predicted or proven to be highly pathogenic. Based on this, all reported mutated genes or loci were included and analyzed. We performed a large familial HSCR study and determined a series of ratios and percentages. However, there may be some statistical bias, as the reported HSCR families are typical and characteristic.
In conclusion, the male-to-female ratio in familial HSCR is close to one. Most families show incomplete dominance and are relevant to RET, and the RET mutation has 56% penetrance in familial HSCR. The incidence of HSCR in the offspring does not depend on whether the parent suffers from HSCR. Overall, our findings will enhance the comprehensive characterization of the genetic landscape for familial HSCR and help HSCR patients obtain better genetic counseling.
Data availability
All data generated or analyzed during this study are included in this published article (and its supplementary information files).
References
Kyrklund K, Sloots CEJ, de Blaauw I, Bjørnland K, Rolle U, Cavalieri D, et al. ERNICA guidelines for the management of rectosigmoid Hirschsprung’s disease. Orphanet J Rare Dis. 2020;15:164.
Romeo G, Ronchetto P, Luo Y, Barone V, Seri M, Ceccherini I, et al. Point mutations affecting the tyrosine kinase domain of the RET proto-oncogene in Hirschsprung’s disease. Nature. 1994;367:377–8.
Hosoda K, Hammer RE, Richardson JA, Baynash AG, Cheung JC, Giaid A, et al. Targeted and natural (piebald-lethal) mutations of endothelin-B receptor gene produce megacolon associated with spotted coat color in mice. Cell. 1994;79:1267–76.
Garcia-Barceló M, Sham MH, Lui VC, Chen BL, Ott J, Tam PK. Association study of PHOX2B as a candidate gene for Hirschsprung’s disease. Gut. 2003;52:563–7.
Tilghman JM, Ling AY, Turner TN, Sosa MX, Krumm N, Chatterjee S, et al. Molecular genetic anatomy and risk profile of Hirschsprung’s disease. N Engl J Med. 2019;380:1421–32.
Bolk S, Pelet A, Hofstra RM, Angrist M, Salomon R, Croaker D, et al. A human model for multigenic inheritance: phenotypic expression in Hirschsprung disease requires both the RET gene and a new 9q31 locus. Proc Natl Acad Sci U S A. 2000;97:268–73.
Russell MB, Russell CA, Fenger K, Niebuhr E. Familial occurrence of Hirschsprung’s disease. Clin Genet. 1994;45:231–5.
Edery P, Pelet A, Mulligan LM, Abel L, Attié T, Dow E, et al. Long segment and short segment familial Hirschsprung’s disease: variable clinical expression at the RET locus. J Med Genet. 1994;31:602–6.
Mc Laughlin D, Puri P. Familial Hirschsprung’s disease: a systematic review. Pediatr Surg Int. 2015;31:695–700.
Kawano T, Hosomichi K, Inoue I, Shimono R, Onishi S, Nakame K, et al. Identification of a novel variant of the RET proto-oncogene in a novel family with Hirschsprung’s disease. Pediatr Surg Int. 2017;33:1041–6.
Granström AL, Markljung E, Fink K, Nordenskjöld E, Nilsson D, Wester T, et al. A novel stop mutation in the EDNRB gene in a family with Hirschsprung’s disease associated with multiple sclerosis. J Pediatr Surg. 2014;49:622–5.
Pace NP, Pace Bardon M, Borg I. A respiratory/Hirschsprung phenotype in a three-generation family associated with a novel pathogenic PHOX2B splice donor mutation. Mol Genet Genomic Med. 2020;8:e1528.
Attié T, Pelet A, Edery P, Eng C, Mulligan LM, Amiel J, et al. Diversity of RET proto-oncogene mutations in familial and sporadic Hirschsprung disease. Hum Mol Genet. 1995;4:1381–6.
Le TL, Galmiche L, Levy J, Suwannarat P, Hellebrekers DM, Morarach K, et al. Dysregulation of the NRG1/ERBB pathway causes a developmental disorder with gastrointestinal dysmotility in humans. J Clin Invest. 2021;131:e145837.
Lombardo RC, Kramer E, Cnota JF, Sawnani H, Hopkin RJ. Variable phenotype in a novel mutation in PHOX2B. Am J Med Genet A. 2017;173:1705–9.
Jiang Q, Wang Y, Gao Y, Wang H, Zhang Z, Li Q, et al. RET compound inheritance in Chinese patients with Hirschsprung disease: lack of penetrance from insufficient gene dysfunction. Hum Genet. 2021;140:813–25.
Jiang Q, Wang Y, Li Q, Zhang Z, Xiao P, Wang H, et al. Sequence characterization of RET in 117 Chinese Hirschsprung disease families identifies a large burden of de novo and parental mosaic mutations. Orphanet J Rare Dis. 2019;14:237.
Yang W, Chen SC, Lai JY, Ming YC, Chen JC, Chen PL. Distinctive genetic variation of long-segment Hirschsprung’s disease in Taiwan. Neurogastroenterol Motil. 2019;31:e13665.
Wu W, Lu L, Xu W, Liu J, Sun J, Zheng L, et al. Whole exome sequencing identifies a novel pathogenic RET variant in Hirschsprung disease. Front Genet. 2018;9:752.
Shen Q, Zhang H, Su Y, Wen Z, Zhu Z, Chen G, et al. Identification of two novel PCDHA9 mutations associated with Hirschsprung’s disease. Gene. 2018;658:96–104.
Sribudiani Y, Chauhan RK, Alves MM, Petrova L, Brosens E, Harrison C, et al. Identification of variants in RET and IHH pathway members in a large family with history of Hirschsprung disease. Gastroenterology. 2018;155:118–29.e6.
Luzón-Toro B, Gui H, Ruiz-Ferrer M, Sze-Man Tang C, Fernández RM, Sham PC, et al. Exome sequencing reveals a high genetic heterogeneity on familial Hirschsprung disease. Sci Rep. 2015;5:16473.
Doubaj Y, Pingault V, Elalaoui SC, Ratbi I, Azouz M, Zerhouni H, et al. A novel mutation in the endothelin B receptor gene in a moroccan family with shah-waardenburg syndrome. Mol Syndromol. 2015;6:44–9.
Igarashi T, Okamura R, Jikuzono T, Uchino S, Sugitani I, Shimizu K. An extended family with familial medullary thyroid carcinoma and Hirschsprung’s disease. J Nippon Med Sch. 2014;81:64–9.
Bygarski E, Paterson M, Lemire EG. Extreme intra-familial variability of congenital central hypoventilation syndrome: a case series. J Med Case Rep. 2013;7:117.
Van Camp G, Van Thienen MN, Handig I, Van Roy B, Rao VS, Milunsky A, et al. Chromosome 13q deletion with Waardenburg syndrome: further evidence for a gene involved in neural crest function on 13q. J Med Genet. 1995;32:531–6.
Sánchez-Mejías A, Fernández RM, López-Alonso M, Antiñolo G, Borrego S. Contribution of RET, NTRK3 and EDN3 to the expression of Hirschsprung disease in a multiplex family. J Med Genet. 2009;46:862–4.
Cui L, Wong EH, Cheng G, Firmato de Almeida M, So MT, Sham PC, et al. Genetic analyses of a three generation family segregating Hirschsprung disease and iris heterochromia. PLoS One. 2013;8:e66631.
Ohno K, Nakamura T, Azuma T, Nakaoka T, Takama Y, Hayashi H, et al. Familial Currarino syndrome associated with Hirschsprung disease: two cases of a mother and daughter. J Pediatr Surg. 2013;48:233–8.
de Pontual L, Zaghloul NA, Thomas S, Davis EE, McGaughey DM, Dollfus H, et al. Epistasis between RET and BBS mutations modulates enteric innervation and causes syndromic Hirschsprung disease. Proc Natl Acad Sci U S A. 2009;106:13921–6.
Moore SW, Zaahl M. Familial associations in medullary thyroid carcinoma with Hirschsprung disease: the role of the RET-C620 “Janus” genetic variation. J Pediatr Surg. 2010;45:393–6.
Cherian MP, Al-Sanna’a NA, Al-Mulhim SI. Hirschsprung’s disease in Arab siblings with Bardet-Biedl syndrome. J Pediatr Surg. 2008;43:1213–7.
Basel-Vanagaite L, Pelet A, Steiner Z, Munnich A, Rozenbach Y, Shohat M, et al. Allele dosage-dependent penetrance of RET proto-oncogene in an Israeli-Arab inbred family segregating Hirschsprung disease. Eur J Hum Genet. 2007;15:242–5.
Moore SW, Zaahl MG. A review of genetic mutation in familial Hirschsprung’s disease in South Africa: towards genetic counseling. J Pediatr Surg. 2008;43:325–9.
Fialkowski EA, DeBenedetti MK, Moley JF, Bachrach B. RET proto-oncogene testing in infants presenting with Hirschsprung disease identifies 2 new multiple endocrine neoplasia 2A kindreds. J Pediatr Surg. 2008;43:188–90.
Lui VC, Leon TY, Garcia-Barceló MM, Ganster RW, Chen BL, Hutson JM, et al. Novel RET mutation produces a truncated RET receptor lacking the intracellular signaling domain in a 3-generation family with Hirschsprung disease. Clin Chem. 2005;51:1552–4.
Gilchrist DM, Morrish DW, Bridge PJ, Brown JL. Cost analysis of DNA-based testing in a large Canadian family with multiple endocrine neoplasia type 2. Clin Genet. 2004;66:349–52.
Fernández RM, Antiñolo G, Eng C, Borrego S. The RET C620S mutation causes multiple endocrine neoplasia type 2A (MEN2A) but not Hirschsprung disease (HSCR) in a family cosegregating both phenotypes. Hum Mutat. 2003;22:412–5.
Fitze G, Cramer J, Serra A, Schreiber M, Roesner D, Schackert HK. Within-gene interaction between c.135 G/A genotypes and RET proto-oncogene germline mutations in HSCR families. Eur J Pediatr Surg. 2003;13:152–7.
Munnes M, Fanaei S, Schmitz B, Muiznieks I, Holschneider AM, Doerfler W. Familial form of hirschsprung disease: nucleotide sequence studies reveal point mutations in the RET proto-oncogene in two of six families but not in other candidate genes. Am J Med Genet. 2000;94:19–27.
Brooks AS, Breuning MH, Osinga J, vd Smagt JJ, Catsman CE, Buys CH, et al. A consanguineous family with Hirschsprung disease, microcephaly, and mental retardation (Goldberg-Shprintzen syndrome). J Med Genet. 1999;36:485–9.
Decker RA, Peacock ML, Watson P. Hirschsprung disease in MEN 2A: increased spectrum of RET exon 10 genotypes and strong genotype-phenotype correlation. Hum Mol Genet. 1998;7:129–34.
Romeo G, Ceccherini I, Celli J, Priolo M, Betsos N, Bonardi G, et al. Association of multiple endocrine neoplasia type 2 and Hirschsprung disease. J Intern Med. 1998;243:515–20.
Borrego S, Eng C, Sánchez B, Sáez ME, Navarro E, Antiñolo G. Molecular analysis of the ret and GDNF genes in a family with multiple endocrine neoplasia type 2A and Hirschsprung disease. J Clin Endocrinol Metab. 1998;83:3361–4.
Peretz H, Luboshitsky R, Baron E, Biton A, Gershoni R, Usher S, et al. Cys 618 Arg mutation in the RET proto-oncogene associated with familial medullary thyroid carcinoma and maternally transmitted Hirschsprung’s disease suggesting a role for imprinting. Hum Mutat. 1997;10:155–9.
Maris JM, Chatten J, Meadows AT, Biegel JA, Brodeur GM. Familial neuroblastoma: a three-generation pedigree and a further association with Hirschsprung disease. Med Pediatr Oncol. 1997;28:1–5.
Salomon R, Attié T, Pelet A, Bidaud C, Eng C, Amiel J, et al. Germline mutations of the RET ligand GDNF are not sufficient to cause Hirschsprung disease. Nat Genet. 1996;14:345–7.
Attié T, Till M, Pelet A, Edery P, Bonnet JP, Munnich A, et al. Exclusion of RET and Pax 3 loci in Waardenburg-Hirschsprung disease. J Med Genet. 1995;32:312–3.
Hofstra RM, Osinga J, Tan-Sindhunata G, Wu Y, Kamsteeg EJ, Stulp RP, et al. A homozygous mutation in the endothelin-3 gene associated with a combined Waardenburg type 2 and Hirschsprung phenotype (Shah-Waardenburg syndrome). Nat Genet. 1996;12:445–7.
Angrist M, Bolk S, Thiel B, Puffenberger EG, Hofstra RM, Buys CH, et al. Mutation analysis of the RET receptor tyrosine kinase in Hirschsprung disease. Hum Mol Genet. 1995;4:821–30.
Pierpont JW, St Jacques D, Seaver LH, Erickson RP. A family with unusual Waardenburg syndrome type I (WSI), cleft lip (palate), and Hirschsprung disease is not linked to PAX 3. Clin Genet. 1995;47:139–43.
Attie T, Pelet A, Sarda P, Eng C, Edery P, Mulligan LM, et al. A 7 bp deletion of the RET proto-oncogene in familial Hirschsprung’s disease. Hum Mol Genet. 1994;3:1439–40.
Bonnet JP, Till M, Edery P, Attie T, Lyonnet S. Waardenburg-Hirschsprung disease in two sisters: a possible clue to the genetics of this association? Eur J Pediatr Surg. 1996;6:245–8.
Mulligan LM, Eng C, Attié T, Lyonnet S, Marsh DJ, Hyland VJ, et al. Diverse phenotypes associated with exon 10 mutations of the RET proto-oncogene. Hum Mol Genet. 1994;3:2163–7.
Dow E, Cross S, Wolgemuth DJ, Lyonnet S, Mulligan LM, Mascari M, et al. Second locus for Hirschsprung disease/Waardenburg syndrome in a large Mennonite kindred. Am J Med Genet. 1994;53:75–80.
Angrist M, Kauffman E, Slaugenhaupt SA, Matise TC, Puffenberger EG, Washington SS, et al. A gene for Hirschsprung disease (megacolon) in the pericentromeric region of human chromosome 10. Nat Genet. 1993;4:351–6.
Luo Y, Ceccherini I, Pasini B, Matera I, Bicocchi MP, Barone V, et al. Close linkage with the RET protooncogene and boundaries of deletion mutations in autosomal dominant Hirschsprung disease. Hum Mol Genet. 1993;2:1803–8.
Stannard VA, Fowler C, Robinson L, Besner G, Glick PL, Allen JE, et al. Familial Hirschsprung’s disease: report of autosomal dominant and probable recessive X-linked kindreds. J Pediatr Surg. 1991;26:591–4.
Schiller M, Levy P, Shawa RA, Abu-Dalu K, Gorenstein A, Katz S. Familial Hirschsprung’s disease-a report of 22 affected siblings in four families. J Pediatr Surg. 1990;25:322–5.
Verdy M, Weber AM, Roy CC, Morin CL, Cadotte M, Brochu P. Hirschsprung’s disease in a family with multiple endocrine neoplasia type 2. J Pediatr Gastroenterol Nutr. 1982;1:603–7.
Amiel J, Sproat-Emison E, Garcia-Barcelo M, Lantieri F, Burzynski G, Borrego S, et al. Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet. 2008;45:1–14.
Funding
This work was supported by National Natural Science Foundation of China (82071685 to FJX), Clinical Research Pilot Project of Tongji Hospital (2019YBKY026 to FJX), Provincial Key Research and Development Program (2020BCB008 to FJX), Science and Technology Innovation Base Platform (2020DCD006 to FJX), and Project of Shenzhen San Ming (SZSM201812055 to FJX).
Author information
Authors and Affiliations
Contributions
FJX and MXY contributed to conceptualization, data curation, formal analysis, funding acquisition, and writing of the original draft. XJ contributed to conceptualization, data curation, project administration, writing of the original draft, and reviewing and editing. HLW, WJ and YXS contributed to investigation, methodology, project administration, and resources. YJY, LZJ and MHD contributed to resources, software, supervision, validation, and visualization. All authors approved the final manuscript as submitted and agreed to be accountable for all aspects of the work. The first author is XJ. FJX and MXY contributed equally to the work and are co-corresponding authors.
Corresponding authors
Ethics declarations
Ethical approval
Not needed.
Conflict of interest
No financial or non-financial benefits have been received or will be received from any party related directly or indirectly to the subject of this article. Author Jie-Xiong Feng is a member of the Editorial Board for World Journal of Pediatrics. The paper was handled by the other Editor and has undergone rigorous peer review process. Author Jie-Xiong Feng was not involved in the journal's review of, or decisions related to, this manuscript. The authors have no conflict of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xiao, J., Hao, LW., Wang, J. et al. Comprehensive characterization of the genetic landscape of familial Hirschsprung’s disease. World J Pediatr 19, 644–651 (2023). https://doi.org/10.1007/s12519-023-00686-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12519-023-00686-x