
Abstract
In the USA, an interchangeability designation provides biosimilar sponsors with a pathway for achieving what is standard for small-molecule generics: pharmacy-level auto-substitution for an innovator. No other major health authority links interchangeability to automatic substitution, as all require the involvement of the prescriber or patient in a medication change. This editorial considers the clinical impact and practicality of auto-substitution. First, interchangeability is linked to non-medical switching (NMS), the practice of switching treatment in patients with stable disease for non-clinical reasons. NMS may generate negative sentiment in those unwilling or reluctant to switch, which can adversely impact treatment outcomes (i.e., nocebo effect). Indeed, in real-world studies of tumor necrosis factor inhibitors, discontinuation rates have been shown to be higher in patients switched to biosimilars for non-medical reasons than in historical cohorts maintained on innovators. Second, interchangeability may impede pharmacovigilance and traceability, as not all jurisdictions require innovators and biosimilars to have distinct biologic names. Third, an interchangeability designation from the US Food and Drug Administration only permits a biosimilar to be automatically substituted for its innovator, not other biosimilars (if available). Pharmacist education would be needed to avoid off-label, automatic substitution among biosimilars of a single innovator. Last, once granted, an interchangeability designation exists in perpetuity under current US federal law. However, the supply chains of innovators and biosimilars are maintained independently, with no requirement for reconfirmation of biosimilarity or interchangeability. We feel that additional guidance is needed for the auto-substitution of biosimilars and innovators to become a reality.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Definitions regarding the interchangeability of a biosimilar for its innovator biologic vary across jurisdictions, with some (e.g., the European Union) requiring the input of the prescriber and others (e.g., the USA) permitting automatic substitution at the pharmacy-level independent of physician direction |
Given potential issues with treatment persistence, pharmacovigilance, traceability, and manufacturing changes over time, additional data and guidance are needed to clarify the extent to which biosimilars should be considered interchangeable (if at all) and for how long |
Like many professional medical societies and patient advocacy groups, we believe that treatment decisions should remain under the purview of physicians in consultation with their patients |
Digital Features
This article is published with digital features, including a summary slide, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.14114252.
Introduction
The interchangeability of a biosimilar (Table 1) for its innovator (or vice versa) is a complex topic that has generated much discussion [1,2,3,4,5,6,7]. Interchangeability is closely linked to non-medical switching (NMS), the practice of switching treatment in patients with stable disease for non-clinical reasons, such as cost (Table 1) [8,9,10]. NMS can involve switching between molecules of the same class (e.g., tumor necrosis factor [TNF] inhibitors) or from an innovator to its biosimilar (or vice versa). Given the clinical and economic implications of interchangeability, this editorial seeks to educate on the different definitions of interchangeability, to outline considerations regarding the clinical impact and practicality of interchangeability, and to question the view, as advocated by others [1], that biosimilars should be, by default, considered interchangeable with their innovators. Because NMS often occurs with biologic products used long term, we will draw on experience with TNF inhibitors used in the management of immune-mediated inflammatory diseases (e.g., rheumatoid arthritis, Crohn’s disease, psoriasis). This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Definitions of Interchangeability
The definition of interchangeability varies by legal jurisdiction. In the USA, interchangeability is a designation endorsing the auto-substitution (Table 1) of an approved biosimilar with its innovator [11]. This means that an interchangeable biosimilar may be switched for the innovator by the pharmacist without the knowledge or intervention of the prescriber. Although there is no federal mandate, most states have enacted laws requiring the prescriber to at least be notified of a medication change, but the federal government provides no guidance on whether the pharmacist needs to inform the patient of any such change (Table 2). The US Food and Drug Administration (FDA) is the only major regulator with this designation, which places qualifying biosimilars—despite their complexity—on par with small-molecule generics. In contrast to biologic products, small-molecule generics, which are synthesized via chemical processes rather than generated in living systems, are considered bioequivalent to and, therefore, in most cases, automatically substitutable for their innovator (or, in US biologics parlance, interchangeable). (From a regulatory perspective, a biosimilar cannot be considered bioequivalent to its innovator because this term is applicable only to small molecules.) Biosimilars, on the other hand, are regarded in the US as similar to but not interchangeable (i.e., automatically substitutable) with innovators [12], unless a higher standard of evidence is provided [11]. Although auto-substitution is standard practice for small-molecule generics, no biosimilar approved in the US is currently approved as an interchangeable.
The FDA’s data requirements for demonstrating interchangeability are greater than those for biosimilarity. To receive an interchangeability designation in the US, a sponsor must show that (1) the biosimilar “can be expected to produce the same clinical result as the reference product in any given patient” and (2) for treatment administered more than once that “the risk in terms of safety or diminished efficacy of alternating or switching between use of the biological product and the reference product is not greater than the risk of using the reference product without such alteration or switch” [11]. However, product-dependent factors may impact the amount and types of data needed to support a demonstration of interchangeability. These include the complexity of the product (as this would affect the extent of comparative and functional characterizations) and the risk of immunogenicity.
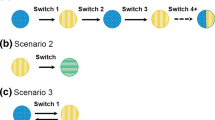
Currently, the FDA requires evidence of a single switch (Table 1) for the approval of a non-interchangeable biosimilar, but will generally require multiple-switch (Table 1) data from a biosimilar sponsor seeking an interchangeability designation [11]. Possible trial designs of single- and multi-switch studies are summarized in Fig. 1. The FDA advises that pharmacokinetics (and pharmacodynamics, if available) serve as the primary endpoint of studies examining multiple (≥ 3) switches of a proposed interchangeable biosimilar with its innovator, as these parameters “are generally more likely to be sensitive to detect changes in exposure and/or activity that may arise as a result of alternating or switching” than clinical endpoints [11]. In its guidance to industry on interchangeability, the FDA advises caution on the use of non-US-licensed products versus US-licensed innovators as comparators in multiple-switch studies of a proposed interchangeable biosimilar. This is due to the possibility of subtle differences in structural features, process-related impurities, and/or formulation between a US-licensed and non–US-licensed innovator [11]. There is potential for such differences to trigger an immune response that may become augmented following multiple exposures.

Designs of different single- and multi-switch studies [89]. Adapted from Moots R, et al. Curr Rheumatol Rep. 2017;19:37 https://s100.copyright.com/AppDispatchServlet?title=Switching%20Between%20Reference%20Biologics%20and%20Biosimilars%20for%20the%20Treatment%20of%20Rheumatology%2C%20Gastroenterology%2C%20and%20Dermatology%20Inflammatory%20Conditions%3A%20Considerations%20for%20the%20Clinician&author=Robert%20Moots%20et%20al&contentID=10.1007%2Fs11926-017-0658-4&publication=1523-3774&publicationDate=2017-06-16&publisherName=SpringerNature&orderBeanReset=true&oa=CC%20BY. “Blinded” indicates single- or double-blinded study phase. “Open-label” indicates open-label study phase. “No study treatment” indicates that at the time of study start, patients had not been receiving the biologic treatment that was the focus of the clinical trial
Although the FDA has the authority to grant an interchangeability designation, the decision of whether to actually implement pharmacy-level auto-substitution rests with individual states (i.e., there is no federal mandate) [5, 13, 14]. Of the 45 states currently with laws regarding the substitutability of a biosimilar, only one (Kansas) does not require a product to first have been designated as interchangeable by the FDA prior to allowing auto-substitution [15].
In contrast to the FDA, other major regulators do not have statutory/legal definitions of interchangeability. Although some have defined “interchangeability” in their guidance documents [16, 17], these definitions are not legally binding. Rather, in these jurisdictions, regulators approve biosimilars as “biosimilar,” a designation that is not linked to automatic substitution. Per the European Medicines Agency (EMA), interchangeability refers to the possibility of exchanging one medicine for another that is expected to have the same clinical effect. This may involve replacing an innovator with a biosimilar (or vice versa) or one biosimilar with another. Replacement can be mediated by either the physician (switching) or pharmacist (auto-substitution) (Table 2). With the latter, the EMA definition of interchangeability overlaps with that of the FDA, leading to confusion. Irrespective of whether switching or substitution occurs with or without the involvement of the prescriber, the EMA does not approve biosimilars as interchangeable; moreover, it has no explicit position on switching/auto-substitution/replacement, as the EMA views this as a matter of prescribing practice, the regulation of which is beyond its mandate. Rather, such decisions fall to the governing bodies of its member states [16]. Yet, most member states with positions on switching/auto-substitution/replacement leave treatment decisions with the prescribing physician [1, 18]; efforts are ongoing to change this [19]. Many member states, however, support the “interchangeability” of a biosimilar for its innovator under medical supervision [3]. Thus, in practice, interchangeability in the European Union (EU) has a different meaning relative to the US: it is not synonymous with auto-substitution and instead involves the prescriber.
In Australia, agents are approved by the Therapeutic Goods Administration (TGA). The TGA does not define or assess interchangeability. Instead, the TGA defers to the Pharmaceutical Benefits Advisory Committee (PBAC), a government-appointed body charged with recommending drugs for reimbursement and issuing policies related to the uptake of biosimilars. PBAC does not specifically use the term “interchangeability.” Rather, biosimilars that have been “a-flagged” by the PBAC can be replaced with an innovator (or vice versa) at the pharmacy-level in consultation with the patient rather than the physician (Table 2) [20]. However, even if a biosimilar has been a-flagged, the prescriber can indicate that “substitution is not permitted.” Under such circumstances, the pharmacist must dispense the drug prescribed or is otherwise legally bound to contact the prescriber [20, 21]. Yet, real-world scenarios could lead to situations in which biosimilars are dispensed, for example, when the innovator is out of stock. The sponsor of a proposed a-flag biosimilar must provide switch data. In addition to reviewing switch data, the PBAC assesses the practicality of switching at the pharmacy level [20].
The positions of health authorities in Canada, Brazil, and Japan on interchangeability are summarized in Table 2. Having a statutory/legal definition of interchangeability and linking it directly to auto-substitution are unique to the FDA, as other major health regulators require the involvement of the prescriber or patient.
Support for Automatic Substitution
Surveys have indicated that the majority of healthcare providers across jurisdictions/geographies are opposed to auto-substitution, irrespective of whether patients are treatment-naïve or on a stable treatment regimen [22,23,24]. Moreover, major US, Canadian, and European rheumatology and gastroenterology societies have released position statements advising against the practice of auto-substitution, stressing that there are insufficient data to support switching patients with stable disease on maintenance therapy [25] or that treatment decisions should remain under the purview of prescribing physicians in consultation with their patients [26,27,28,29,30]. The American College of Rheumatology (ACR) is opposed to mandated switching in the absence of evidence of interchangeability [31] and advocates that treatment decisions be made only by physicians [26]. However, in jurisdictions where auto-substitution is permitted, the ACR recommends that both patients and prescribers be informed immediately of any change in medication [26].
It must be stressed, however, that these societies are not opposed to the use of biosimilars, just the loss of physician input in the treatment dispensed. For example, according to the ACR, substitutions should only be made by the prescriber, and providers should retain the ability to mandate that prescriptions, including biologic products, be “dispensed as written” [26]. Of course, medical needs may change, warranting a switch in medication. In such situations, the prescriber (in consultation with the patient) may change treatments. However, if treatment with an innovator or biosimilar were to fail, the European League Against Rheumatism recommends that a biosimilar (if another is available) not be prescribed. Rather, treatment with another biologic product within the same class or a biologic product with a completely different mechanism of action should be initiated [32]. This is because, by definition, no difference in clinical effect is expected with a biosimilar relative to an innovator; thus, failure with one product will likely result in failure with the other. This is supported by immunogenicity data showing that anti-drug antibodies developed in response to innovators cross-react with biosimilars (and vice versa) [33,34,35].
Several patient societies have expressed opposition to auto-substitution or have commented on the data needed to increase confidence in auto-substitution. The Alliance for Patient Access, for example, contends that patients who are clinically stable should be allowed to continue the use of their medication and that both patients and physicians should be informed in writing prior to a switch in medication for non-medical reasons [36]. The Crohn’s and Colitis Foundation “urges the FDA, when considering interchangeability, to provide reasonable proof that switching from the originator to the biosimilar would not incur immunogenicity or loss of response to the originator (and vice versa)” [37]. We will now discuss the main reasons associated with the lack of support for auto-substitution: NMS, traceability, and manufacturing.
Issues with Non-medical Switching
Prior to the arrival of biosimilars, NMS among TNF inhibitors involved switching between innovators within the same class. Studies demonstrated that this practice was associated with loss of tolerance and efficacy [38,39,40] and increased healthcare costs [41,42,43], prompting recommendations that patients with stable disease continue current therapy and not switch among innovators. Once biosimilars became available, researchers began to investigate the impact of NMS between two versions of a single TNF inhibitor (i.e., biosimilar and innovator). Although data from randomized controlled trials were generally positive [44,45,46,47,48,49], real-world data indicated that discontinuation rates were higher in patients switched to a biosimilar for non-medical reasons than in historical cohorts maintained on innovator TNF inhibitors [8, 50,51,52,53,54]. Limited real-world data exist on NMS from a biosimilar to an innovator [55], but as a biosimilar would have been approved only if it was shown not to yield clinically meaningful differences relative to its innovator, it is reasonable to assume that the act of switching in and of itself—rather than the direction of the switch (innovator to biosimilar or vice versa)—is responsible for the increased discontinuation rates reported in the real-world setting.
Several reasons are possible for the discrepant discontinuation rates observed in real-world studies versus randomized controlled trials, including the open-label nature, broad patient populations examined, patient selection process (or rather the lack thereof), and inadequate education and monitoring, all inherent to the former. Patient perceptions have also been shown to play a role in treatment persistence. Switching between an innovator and a biosimilar for non-medical reasons may generate negative sentiment in those unwilling or reluctant to switch, which, in turn, can adversely impact expectations of treatment safety and efficacy—a phenomenon known as the nocebo effect (Table 1) [56,57,58,59]. This has been described in multiple cohorts, most notably in the BIO-SWITCH study, a Dutch registry of patients with long-term (median of 7 years), stable disease on innovator infliximab switched to biosimilar CT-P13 following a government mandate [52]. The discontinuation rate at month 6 was 24%. Of the adverse events reported, 78% were due to events that could be considered subjective (e.g., mood disorders, fatigue). Moreover, among those who discontinued CT-P13 due to a perceived lack of efficacy, scores on subjective measures of disease (tender joint count and global disease activity) worsened relative to baseline, whereas objective measures (swollen joint count and C-reactive protein) were stable. Interestingly, the majority (79%) of patients in BIO-SWITCH who discontinued CT-P13 resumed treatment with innovator infliximab (outcomes not provided).
The increased discontinuation rates and effects of patient perceptions of treatment safety/efficacy following NMS are supported by a systematic literature review examining nocebo effects in patients switched from an innovator TNF inhibitor to a biosimilar for non-medical reasons [8]. In the absence of a validated measure of nocebo effects, the authors used biosimilar discontinuation rate and switchback (i.e., rate of reversion to the innovator following biosimilar discontinuation) as proxies [8]. Discontinuation rates were generally similar between the switched and control groups of randomized controlled studies. However, rates of discontinuation varied among real-world studies that included a control arm. Across the 12 studies reporting switchback data, 50–100% of patients who discontinued a biosimilar successfully resumed treatment with the innovator. Of note, the median rates of discontinuation following NMS were numerically higher in studies in which the switch to the biosimilar was mandatory. Similarly, the median rates of switchback to the innovator were higher when the switch was mandated. These findings have now been found across biosimilars of infliximab, etanercept, adalimumab, and rituximab [8, 60,61,62,63].
Although the higher rates of discontinuation observed in real-world studies of NMS may reflect only a minority of patients (2–18% additional patients above an expected discontinuation rate of 10% per year) [54], they suggest the consideration of mitigation measures. The available data indicate that some patients may be more susceptible to nocebo effects (and thus more likely to “fail” treatment following NMS). If it were possible to identify such patients a priori, then the risks/benefits of switching treatment for non-medical reasons could be reevaluated. If no other option exists, then such patients may need to be closely monitored and/or educated to maximize the likelihood of treatment persistence with the biosimilar. Indeed, mitigation measures, such as patient education, selection (e.g., based on disease stability and willingness to switch), and follow-up, have been associated with improved outcomes following switching for non-medical reasons [53, 64,65,66].
Although nocebo effects factor into the decreased persistence rates seen in real-world studies of NMS, they do not explain the phenomenon fully. Differences in delivery device and product formulation can also have an impact. The latter is likely responsible for the objective events, such as serum sickness, skin reactions, and loss of response, leading to biosimilar discontinuation following NMS [60, 67, 68].
Of note, the development of anti-drug antibodies, an objective adverse effect that is a key concern with biologic products, has not been shown to increase in randomized controlled trials or real-world studies of a single switch [2, 44, 46, 48, 52, 69, 70] or in randomized controlled trials of multiple switches [49, 71] (although evaluation of multiple switches in the real-world setting remains understudied) [72]. This result is perhaps unsurprising, as the differences in immunogenicity between a biosimilar and its innovator would have been demonstrated to be minimal during the regulatory approval process [3, 73].
Traceability of Biologic Products
Pharmacovigilance, the practice of identifying adverse events during routine clinical use of a drug, is essential to ensuring patient safety [74, 75]. It is needed not only to identify events too rare to be observed in the randomized controlled setting, but also to identify issues related to a particular batch or lot of a product (i.e., manufacturing- or storage-related issues). Unique identifiers, such as non-proprietary/brand names and batch numbers, are key to traceability. In major markets, approved innovators and their biosimilars have distinct brand names. However, the convention for non-proprietary names differs between geographies. For example, in the US, the non-proprietary name is appended with a unique, randomly generated, four-letter sequence, but in the EU, all products (innovator and biosimilar[s]) with the same active ingredient share the same non-proprietary name (i.e., no four-letter suffix) [76]. To improve traceability in the EU, the brand name is used preferentially, and when the non-proprietary name is referenced, it is often (but not always) accompanied by the name of the marketing authorization holder.
Without a unique, non-proprietary name for each approved biologic, including biosimilars, it can be difficult to unambiguously attribute adverse events to a specific agent (innovator or biosimilar). For example, in Australia, innovator and biosimilar versions of infliximab and filgrastim share the same biological name, whereas innovator and biosimilar epoetin have distinct biological names owing to differences in glycosylation [77]. In an analysis of an adverse event database maintained by the Australian TGA, the rates of ambiguous adverse event reporting were higher for infliximab (25%) and filgrastim (36%) than for epoetin (3%), which the authors attributed to the absence of distinct Australian Biological Names for biosimilars of infliximab and filgrastim [77]. In the EU, the reporting of suspected adverse drug reactions (ADRs) requires the inclusion of the biologic product’s brand name, as well as its batch number [74], but only 5% of ADRs have been shown to include both the brand name and batch number [78]. The lack and/or omission of traceable information can lead to delays in the identification of safety issues with a particular product, batch, or lot [74, 79]. This may be exacerbated by the auto-substitution of biosimilars and their innovators.
Manufacturing
In addition to strengthening adverse event reporting, another reason for tracking biologic products relates to the complexity of the manufacturing process itself. Although the amino acid sequence of a biosimilar and its innovator is identical, the cell line used for the innovator, as well as manufacturing processes and quality specifications, is proprietary. Consequently, biosimilar sponsors use reverse engineering techniques to generate their product, such that the cell line used, manufacturing process, and quality specifications are all developed and maintained completely independently of the innovator sponsor.
However, the sponsor of a biologic—whether innovator or biosimilar—may elect to implement changes to the manufacturing process post-approval. Because these changes can introduce product variability, the sponsor of an agent undergoing manufacturing changes undertakes comparability assessments throughout the manufacturing process, including product intermediates, ensuring that any variability introduced by manufacturing changes falls within pre-specified limits. Since the molecule generated before and after the manufacturing change is produced by the same sponsor in the same cell line [75], with impurities, inactive ingredients, and microheterogeneity (Table 1) kept within regulatory-approved specifications, and since any manufacturing change must be approved by health authorities, the consistency of the biologic product is maintained over time. In contrast, biosimilarity exercises for a biosimilar can occur only with the commercially available version of the innovator (i.e., the final product) [12], not with intermediates (as would occur in a single-supply chain). Of note, biosimilarity and interchangeability assessments are undertaken only once, as part of the submission for regulatory approval; there is no re-evaluation of either designation following the introduction of manufacturing changes in one or both supply chains.
Need for a Better Understanding of Interchangeability
The FDA has committed to increasing the availability of biosimilars and interchangeable biosimilars, despite requiring more rigorous data for the latter. Although we support the FDA’s position that interchangeability requires a higher evidence standard than biosimilarity, we argue that additional data and guidance beyond this are needed to make fully informed decisions regarding auto-substitution. First, the FDA recommends that sponsors seeking an interchangeability designation provide data on ≥ 3 switches between the innovator and biosimilar [11]. Although data on multiple switches between a biosimilar and its innovator have been collected in the randomized controlled setting [49, 71] and—consistent with single-switch data—do not indicate a loss of efficacy or an increase in adverse events [44, 46, 48], limited data exist on multiple switches in the real-world setting [72]. As such, the increased discontinuation rates seen with a single switch in real-world studies may be exacerbated following multiple switches. Since anti-drug antibodies to TNF inhibitors are more likely to develop over time [80] and the two multi-switch randomized controlled trials published to date followed patients for only 1 year [49, 71], the absence of evidence should not be interpreted as proof that there are no immunogenicity issues with multiple switches over time. Moreover, the aforementioned causes (e.g., nocebo effects) of increased discontinuation rates discussed in the context of real-world, single-switch studies would apply to multiple switches. Are post-marketing commitments needed to evaluate these potential issues?
Second, switching between biosimilars has not been evaluated in randomized controlled trials. If a biosimilar were to receive an interchangeability designation from the FDA, it is important to note that that biosimilar can only be automatically substituted for its innovator, not other biosimilars (if available) of the innovator. Thus, do the sponsors of late-to-market biosimilars need to demonstrate interchangeability not only with the innovator, but with all regulatory-approved biosimilars with the same active ingredient? This is neither practical nor required by the FDA, but its absence means that physician and pharmacist education, as well as robust tracking procedures, will be needed to avoid prescription errors.
Last, once a biosimilar has been deemed interchangeable with an innovator, that designation exists in perpetuity under current US federal law, since there is no requirement for it to be repeated. Given the independent supply chains of innovators and biosimilars and the fact that one or both may be subject to manufacturing changes over time [81,82,83], should the “interchangeability” status of a biosimilar be periodically reconfirmed?
Conclusions
Some have advocated that all biosimilars approved by major regulators be considered interchangeable [1], but the lack of a singular definition of “interchangeability” has led to confusion. If “interchangeability” means that the decision to switch treatment is made by the physician in consultation with the patient (akin to the EMA definition), then this practice should be adopted universally. However, if “interchangeability” is equivalent to automatic substitution (akin to the US definition), then global acceptance of this practice is inadvisable in light of the points raised above. This opinion is aligned with the major regulatory authorities covered in this editorial, as upon approval of a biosimilar, either the physician must prescribe or the patient must be informed of—if not consent to—pharmacy-level substitution. Like many physicians, professional medical societies, and patient advocacy groups, we believe that treatment decisions should remain under the purview of physicians in consultation with their patients.
The FDA is the only major health authority to explicitly distinguish between biosimilarity and interchangeability. With its interchangeability designation, the FDA provides biosimilar sponsors with a pathway for achieving what is otherwise standard for small-molecule generics: pharmacy-level auto-substitution for a biologic. Given the complexity of biologic products, the FDA recognizes that additional data beyond those required for demonstrating biosimilarity are needed for an interchangeability designation. Although the acquisition of such data inflates biosimilar development costs, they are necessary to ensure patient safety. These costs may not be a deterrent, as several pharmaceutical companies have undertaken clinical trials to support an interchangeability designation for their candidate biosimilars [84, 85].
While the FDA’s evidence requirements for interchangeability are commendable, we feel that they could be strengthened, given real-world data on the impact of NMS on treatment persistence and the challenges of traceability and possible manufacturing changes on the integrity of any agent being considered automatically substitutable indefinitely. If the auto-substitution of biosimilars for their innovators (or vice versa) were to become a reality, then pharmacists would need to remain up to date on which biosimilars can be substituted (one biosimilar for one innovator or all biosimilars of a single innovator) and tracking procedures would need to be strengthened for pharmacovigilance. Moreover, even with the FDA’s requirements, there might not be sufficient data at the time of approval to understand the impact of multiple switches among interchangeable agents in the clinical practice setting; real-world studies showed that even a single switch for non-medical reasons can increase rates of treatment discontinuation, which could destabilize disease control. Post-marketing evaluation is needed to ascertain whether multiple switches have similar effects. Lastly, we believe that consideration should be given to evaluating and reconfirming the interchangeability status of a biosimilar periodically. If the climate were to change and support for auto-substitution were to become universal, then only biosimilars with robust, supportive evidence should receive an interchangeability designation.
References
Ebbers HC, Schellekens H. Are we ready to close the discussion on the interchangeability of biosimilars? Drug Discov Today. 2019;24:1963–7.
McKinnon RA, Cook M, Liauw W, et al. Biosimilarity and interchangeability: principles and evidence: a systematic review. BioDrugs. 2018;32:27–52.
Kurki P, van Aerts L, Wolff-Holz E, Giezen T, Skibeli V, Weise M. Interchangeability of biosimilars: a European perspective. BioDrugs. 2017;31:83–91.
de Mora F, Balsa A, Cornide-Santos M, et al. Biosimilar and interchangeable: inseparable scientific concepts? Br J Clin Pharmacol. 2019;85:2460–3.
McKinley L, Kelton JM, Popovian R. Sowing confusion in the field: the interchangeable use of biosimilar terminology. Curr Med Res Opin. 2019;35:619–21.
Trifirò G, Marcianò I, Ingrasciotta Y. Interchangeability of biosimilar and biological reference product: updated regulatory positions and pre- and post-marketing evidence. Expert Opin Biol Ther. 2018;18:309–15.
O’Callaghan J, Barry SP, Bermingham M, Morris JM, Griffin BT. Regulation of biosimilar medicines and current perspectives on interchangeability and policy. Eur J Clin Pharmacol. 2019;75:1–11.
Fleischmann R, Jairath V, Mysler E, Nicholls D, Declerck P. Nonmedical switching from originators to biosimilars: does the nocebo effect explain treatment failures and adverse events in rheumatology and gastroenterology? Rheumatol Ther. 2020;7:35–64.
Dolinar R, Kohn CG, Lavernia F, Nguyen E. The non-medical switching of prescription medications. Postgrad Med. 2019;131:335–41.
Benucci M, Cantini F. Non-medical switching: save today and pay tomorrow. J Med Econ. 2019;22:1160–1.
United States Food and Drug Administration. Considerations in demonstrating interchangeability with a reference product; guidance for industry. 2019. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/considerations-demonstrating-interchangeability-reference-product-guidance-industry. Accessed 14 Sept 2020.
Declerk PJ. Biologicals and biosimilars: a review of the science and its implications. GaBI J. 2012;1:13–6.
Derbyshire M. Update on US state legislation on biosimilars substitution. GaBI J. 2015;4:95–7.
Derbyshire M. Interchangeability of biosimilars in the US and around the world. GaBI J. 2017;6:97–8.
National Conference of State Legislatures. State Laws and Legislation Related to Biologic Medications and Substitution of Biosimilars. 2019. https://www.ncsl.org/research/health/state-laws-and-legislation-related-to-biologic-medications-and-substitution-of-biosimilars.aspx. Accessed 14 Sept 2020.
European Medicines Agency and European Commission. Biosimilars in the EU; information guide for healthcare professionals. 2019. https://www.ema.europa.eu/en/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_en.pdf. Accessed 14 Sept 2020.
Medicines & Healthcare Products Regulatory Agency. Consultation document: MHRA guidance on the licensing of biosimilar products. 2020. https://www.gov.uk/government/consultations/mhra-draft-guidance-on-the-licensing-of-biosimilar-products/consultation-document-mhra-guidance-on-the-licensing-of-biosimilar-products. Accessed 21 Jan 2021.
GaBI Journal Editor. Biosimilar substitution in Europe. 2017. http://www.gabionline.net/Reports/Biosimilar-substitution-in-Europe. Accessed 14 Sept 2020.
APM HealthEurope. Germany’s patients and industry warn against ‘cheapest drug’ approach in biosimilar substitution. 2020. https://www.apmhealtheurope.com/freestory/0/70168/germany-s-patients-and-industry-warn-against--cheapest-drug--approach-in-biosimilar-substitution. Accessed 21 Jan 2021.
Australian Government Department of Health. Who chooses whether the biosimilar medicine or the reference biological medicine is used? 2019. https://www1.health.gov.au/internet/main/publishing.nsf/Content/biosimilar-hp-who-chooses-whether-biosimilar-medicine-or-reference-biological-medicine-is-used. Accessed 14 Sept 2020
Australian Rheumatology Association. ARA position statement on the introduction of biosimilars for the treatment of rheumatic diseases. November 2016. https://rheumatology.org.au/gps/documents/ARAbiosimilarsPANov16-updateJun17.pdf Accessed 9 Sept 2020.
Danese S, Fiorino G, Michetti P. Changes in biosimilar knowledge among European Crohn’s Colitis Organization [ECCO] members: an updated survey. J Crohns Colitis. 2016;10:1362–5.
Aladul MI, Fitzpatrick RW, Chapman SR. Healthcare professionals’ perceptions and perspectives on biosimilar medicines and the barriers and facilitators to their prescribing in UK: a qualitative study. BMJ Open. 2018;8:e023603.
Teeple A, Ellis LA, Huff L, et al. Physician attitudes about non-medical switching to biosimilars: results from an online physician survey in the United States. Curr Med Res Opin. 2019;35:611–7.
Lichtenstein GR, Loftus EV, Isaacs KL, Regueiro MD, Gerson LB, Sands BE. ACG Clinical guideline: management of Crohn’s disease in adults. Am J Gastroenterol. 2018;113:481–517.
American College of Rheumatology. American College of Rheumatology position statement. Biosimilars. 2018. https://www.rheumatology.org/Portals/0/Files/Biosimilars-Position-Statement.pdf. Accessed 14 Sept 2020.
European League Against Rheumatism (EULAR) Standing Committee of People with Arthritis/Rheumatism in Europe (PARE). Biosimilars—position paper. https://www.eular.org/myUploadData/files/biosimilars_paper_updated_2018_09_14_dw.pdf. Accessed 14 Sept 2020.
Burri E, Juillerat P, Maillard MH, on behalf of IBDnet, et al. Position statement on the use of biosimilars in inflammatory bowel disease. Swiss Med Wkly. 2019;149:w20148.
Danese S, Fiorino G, Raine T, et al. ECCO position statement on the use of biosimilars for inflammatory bowel disease—an update. J Crohns Colitis. 2017;11:26–34.
Moayyedi P, Benchimol EI, Armstrong D, Yuan C, Fernandes A, Leontiadis GI. Joint Canadian Association of Gastroenterology and Crohn’s and Colitis Canada position statement on biosimilars for the treatment of inflammatory bowel disease. J Can Assoc Gastroenterol. 2020;3:e1-9.
Ward MM, Deodhar A, Gensler LS, et al. 2019 Update of the American College of Rheumatology/Spondylitis Association of America/spondyloarthritis research and treatment network recommendations for the treatment of ankylosing spondylitis and nonradiographic axial spondyloarthritis. Arthritis Rheumatol. 2019;71:1599–613.
Smolen JS, Landewé R, Bijlsma J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;76:960–77.
Ruiz-Argüello MB, Maguregui A, Ruiz Del Agua A, et al. Antibodies to infliximab in Remicade-treated rheumatic patients show identical reactivity towards biosimilars. Ann Rheum Dis. 2016;75:1693–6.
Ben-Horin S, Yavzori M, Benhar I, et al. Cross-immunogenicity: antibodies to infliximab in Remicade-treated patients with IBD similarly recognise the biosimilar Remsima. Gut. 2016;65:1132–8.
Reinisch W, Jahnsen J, Schreiber S, et al. Evaluation of the cross-reactivity of antidrug antibodies to CT-P13 and infliximab reference product (Remicade): an analysis using immunoassays tagged with both agents. BioDrugs. 2017;31:223–37.
Alliance for Patient Access. Non-medical switching: a position statement. 2019. https://admin.allianceforpatientaccess.org/wp-content/uploads/2020/02/AfPA-Position-Statement-Non-Medical_Switching_July-2019.pdf. Accessed 12 Mar 2021.
Crohn’s and Colitis Organization. Biosimilars position statement. https://www.crohnscolitisfoundation.org/sites/default/files/2019-06/biosimilars-statement-needs_0.pdf. Accessed 14 Sept 2020.
Van Assche G, Vermeire S, Ballet V, et al. Switch to adalimumab in patients with Crohn’s disease controlled by maintenance infliximab: prospective randomised SWITCH trial. Gut. 2012;61:229–34.
Hoentjen F, Hanauer SB, Hart J, Rubin DT. Long-term treatment of patients with a history of ulcerative colitis who develop gastritis and pan-enteritis after colectomy. J Clin Gastroenterol. 2013;47:52–7.
Boktor M, Motlis A, Aravantagi A, et al. Substitution with alternative anti-TNFα therapy (SAVANT)-outcomes of a Crohn’s disease cohort undergoing substitution therapy with certolizumab. Inflamm Bowel Dis. 2016;22:1353–61.
Liu Y, Skup M, Lin J, Chao J. Impact of non-medical switching on healthcare costs: a claims database analysis. Value Health. 2015;18:A252.
Gibofsky A, Skup M, Mittal M, et al. Effects of non-medical switching on outcomes among patients prescribed tumor necrosis factor inhibitors. Curr Med Res Opin. 2017;33:1945–53.
Wolf D, Skup M, Yang H, et al. Clinical outcomes associated with switching or discontinuation from anti-TNF inhibitors for nonmedical reasons. Clin Ther. 2017;39:849–62.
Alten R, Batko B, Hala T, et al. Randomised, double-blind, phase III study comparing the infliximab biosimilar, PF-06438179/GP1111, with reference infliximab: efficacy, safety and immunogenicity from week 30 to week 54. RMD Open. 2019;5:e000876.
Odinet JS, Day CE, Cruz JL, Heindel GA. The biosimilar nocebo effect? A systematic review of double-blinded versus open-label studies. J Manag Care Spec Pharm. 2018;24:952–9.
Smolen JS, Choe JY, Prodanovic N, et al. Safety, immunogenicity and efficacy after switching from reference infliximab to biosimilar SB2 compared with continuing reference infliximab and SB2 in patients with rheumatoid arthritis: results of a randomised, double-blind, phase III transition study. Ann Rheum Dis. 2018;77:234–40.
Ye BD, Pesegova M, Alexeeva O, et al. Efficacy and safety of biosimilar CT-P13 compared with originator infliximab in patients with active Crohn’s disease: an international, randomised, double-blind, phase 3 non-inferiority study. Lancet. 2019;393:1699–707.
Jørgensen KK, Olsen IC, Goll GL, NOR-SWITCH Study Group, et al. Switching from originator infliximab to biosimilar CT-P13 compared with maintained treatment with originator infliximab (NOR-SWITCH): a 52-week, randomised, double-blind, non-inferiority trial. Lancet. 2017;389:2304–16.
Griffiths CEM, Thaçi D, Gerdes S, et al. The EGALITY study: a confirmatory, randomized, double-blind study comparing the efficacy, safety and immunogenicity of GP2015, a proposed etanercept biosimilar, vs. the originator product in patients with moderate-to-severe chronic plaque-type psoriasis. Br J Dermatol. 2017;176:928–38.
Glintborg B, Sørensen IJ, Loft AG, et al. A nationwide non-medical switch from originator infliximab to biosimilar CT-P13 in 802 patients with inflammatory arthritis: 1-year clinical outcomes from the DANBIO registry. Ann Rheum Dis. 2017;76:1426–31.
Glintborg B, Loft AG, Omerovic E, et al. To switch or not to switch: results of a nationwide guideline of mandatory switching from originator to biosimilar etanercept. One-year treatment outcomes in 2061 patients with inflammatory arthritis from the DANBIO registry. Ann Rheum Dis. 2019;78:192–200.
Tweehuysen L, van den Bemt BJF, van Ingen IL, de Jong AJL, van der Laan WH, van den Hoogen FHJ, den Broeder AA. Subjective complaints as the main reason for biosimilar discontinuation after open-label transition from reference infliximab to biosimilar infliximab. Arthritis Rheumatol. 2018;70:60–8.
Tweehuysen L, Huiskes VJB, van den Bemt BJF, et al. Open-label, non-mandatory transitioning from originator etanercept to biosimilar SB4: six-month results from a controlled cohort study. Arthritis Rheumatol. 2018;70:1408–18.
Bakalos G, Zintzaras E. Drug discontinuation in studies including a switch from an originator to a biosimilar monoclonal antibody: a systematic literature review. Clin Ther. 2019;41(155–73):e13.
Ilias A, Szanto K, Gonczi L, et al. Outcomes of patients with inflammatory bowel diseases switched from maintenance therapy with a biosimilar to Remicade. Clin Gastroenterol Hepatol. 2019;17(2506–13):e2.
Planès S, Villier C, Mallaret M. The nocebo effect of drugs. Pharmacol Res Perspect. 2016;4:e00208.
Kristensen LE, Alten R, Puig L, et al. Non-pharmacological effects in switching medication: the nocebo effect in switching from originator to biosimilar agent. BioDrugs. 2018;32:397–404.
Boone NW, Liu L, Romberg-Camps MJ, et al. The nocebo effect challenges the non-medical infliximab switch in practice. Eur J Clin Pharmacol. 2018;74:655–61.
Kravvariti E, Kitas GD, Mitsikostas DD, Sfikakis PP. Nocebos in rheumatology: emerging concepts and their implications for clinical practice. Nat Rev Rheumatol. 2018;14:727–40.
Nisar MK. Rituximab biosimilar non-medical switch—does it work? Ann Rheum Dis. 2019;78(suppl_2):A722.
Barberio B, Melatti P, Zingone F, et al. Switching from adalimumab originator to ABP 501 biosimilar: a multicentre North Italian study. Presented at: European Crohn's and Colitis Organisation 2020; February 12–15, 2020; Vienna, Austria.
Dragoni G, Pieraccini A, Bagnoli S, et al. Maintenance of clinical remission with SB5 biosimilar after switch from adalimumab originator: Real-life experience of a tertiary referral centre. Presented at: European Crohn’s and Colitis Organisation 2020; February 12–15, 2020; Vienna, Austria.
Tansley S, Smith S, Bond D, et al. Apparent high frequency of disease flare following switch from reference biologic rituximab (Mabthera) to a biosimilar (Truxima) in patients with vasculitis. Presented at: British Society of Rheumatology 2019; April 30–May 2, 2019; Birmingham, England.
Bhat S, Altajar S, Shankar D, et al. Process and clinical outcomes of a biosimilar adoption program with infliximab-dyyb. J Manag Care Spec Pharm. 2020;26:410–6.
Ho SL, Niu F, Pola S, Velayos FS, Ning X, Hui RL. Effectiveness of switching from reference product infliximab to infliximab-dyyb in patients with inflammatory bowel disease in an integrated healthcare system in the united states: a retrospective, propensity score-matched, non-inferiority cohort study. BioDrugs. 2020;34:395–404.
Plevris N, Jones GR, Jenkinson PW, et al. Implementation of CT-P13 via a managed switch programme in Crohn’s disease: 12-month real-world outcomes. Dig Dis Sci. 2019;64:1660–7.
Mahmmod S, Schultheiss JPD, van Bodegraven AA, et al. Outcome of reverse switching from CT-P13 to originator infliximab in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2021:izaa364. https://doi.org/10.1093/ibd/izaa364.
Scherlinger M, Germain V, Labadie C, et al. Switching from originator infliximab to biosimilar CT-P13 in real-life: the weight of patient acceptance. Jt Bone Spine. 2018;85(5):561–7.
Cohen HP, Blauvelt A, Rifkin RM, Danese S, Gokhale SB, Woollett G. Switching reference medicines to biosimilars: a systematic literature review of clinical outcomes. Drugs. 2018;78:463–78.
Glintborg B, Kringelbach T, Bolstad N, et al. Drug concentrations and anti-drug antibodies during treatment with biosimilar infliximab (CT-P13) in routine care. Scand J Rheumatol. 2018;47:418–21.
Blauvelt A, Lacour JP, Fowler JF Jr, et al. Phase III randomized study of the proposed adalimumab biosimilar GP2017 in psoriasis: impact of multiple switches. Br J Dermatol. 2018;179:623–31.
Lauret A, Moltó A, Abitbol V, et al. Effects of successive switches to different biosimilars infliximab on immunogenicity in chronic inflammatory diseases in daily clinical practice. Semin Arthritis Rheum. 2020;S0049–0172(20):30030–5.
McCamish M, Woollett G. Worldwide experience with biosimilar development. MAbs. 2011;3:209–17.
Felix T, Jordan JB, Akers C, Patel B, Drago D. Current state of biologic pharmacovigilance in the European Union: improvements are needed. Expert Opin Drug Saf. 2019;18:231–40.
Zharkov A, Barton B, Heinzmann D, Bakalos G, Schreitmüller T. Development pathways for subcutaneous formulations of biologics versus biosimilar development. Expert Rev Precis Med Drug Dev. 2019:1–8. https://doi.org/10.1080/23808993.2019.1585806.
Felix T, Patel B, Bradbury BD, Grampp G. Pharmacovigilance of biosimilars: global experience and perspective. In: Gutka H, Yang H, Kakar S, editors. Biosimilars. Springer: Cham; 2018. p. 631–52.
Sarshad M, Campbell R, Pitts PJ, Vanderpuye-Orgle J. The need for distinct nomenclature for originator and biosimilar products. GaBI J. 2018;7:152–7.
Klein K, Scholl JH, Vermeer NS, et al. Traceability of biologics in the Netherlands: an analysis of information-recording systems in clinical practice and spontaneous ADR reports. Drug Saf. 2016;39(2):185–92.
Vermeer NS, Ebbers HC, Straus SM, Leufkens HG, Egberts TC, De Bruin ML. The effect of exposure misclassification in spontaneous ADR reports on the time to detection of product-specific risks for biologicals: a simulation study. Pharmacoepidemiol Drug Saf. 2016;25(3):297–306.
Quistrebert J, Hässler S, Bachelet D, et al; ABIRISK Consortium. Incidence and risk factors for adalimumab and infliximab anti-drug antibodies in rheumatoid arthritis: a European retrospective multicohort analysis. Semin Arthritis Rheum. 2019;48:967–75.
Lamanna WC, Holzmann J, Cohen HP, et al. Maintaining consistent quality and clinical performance of biopharmaceuticals. Expert Opin Biol Ther. 2018;18:369–79.
Kim S, Song J, Park S, et al. Drifts in ADCC-related quality attributes of Herceptin®: impact on development of a trastuzumab biosimilar. MAbs. 2017;9:704–14.
Schiestl M, Stangler T, Torella C, Cepeljnik T, Toll H, Grau R. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol. 2011;29:310–2.
Pharmacy Times. Interchangeability clinical study between adalimumab, biosimilar candidate enrolls first patient. https://www.pharmacytimes.com/news/interchangeability-clinical-study-between-adalimumab-biosimilar-candidate-enrolls-first-patient. Accessed 14 Sept 2020.
The Center for Biosimilars. Eye on Pharma: Nichi-Iko seeks interchangeable designation for NI-071. https://www.centerforbiosimilars.com/news/eye-on-pharma-nichiiko-seeks-interchangeable-designation-for-ni071. Accessed 14 Sept 2020.
Health Canada. Biosimilar biologic drugs in Canada: fact sheet. 2019. https://www.canada.ca/content/dam/hc-sc/migration/hc-sc/dhp-mps/alt_formats/pdf/brgtherap/applic-demande/guides/Fact-Sheet-EN-2019-08-23.pdf. Accessed 14 Sept 2020.
de Assis MR, Pinto V. Strengths and weaknesses of the Brazilian regulation on biosimilars: a critical view of the regulatory requirements for biosimilars in Brazil. Ther Adv Musculoskelet Dis. 2018;10:253–9.
GaBI Journal Editor. Naming and interchangeability for biosimilars in Japan. 2016. http://www.gabionline.net/Reports/Naming-and-interchangeability-for-biosimilars-in-Japan. Accessed 14 Sept 2020.
Moots R, Azevedo V, Coindreau JL, et al. Switching between reference biologics and biosimilars for the treatment of rheumatology, gastroenterology, and dermatology inflammatory conditions: considerations for the clinician. Curr Rheumatol Rep. 2017;19:37.
Acknowledgements
All authors meet ICMJE criteria for authorship and no deserving authors have been omitted. All individuals who did not meet authorship criteria are within the Acknowledgment section.
Funding
Janssen additionally provided funding for the journal’s rapid service fee.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authorship Contributions
All authors were involved in the development of the content and direction of this manuscript; revised it critically for important intellectual content; approved the final version; and agree to be accountable for all aspects of the manuscript in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Medical Writing and Editorial Assistance
Editorial and medical writing support for this manuscript were provided by Tiffany DeSimone, PhD, of Ashfield MedComms, an Ashfield Health company, and were funded by Janssen.
Disclosures
Anita Afzali has served as a consultant to or speaker for Janssen, Abbvie, Takeda, and Pfizer and on advisory boards for Janssen, Abbvie, Takeda, and Celgene/Bristol-Myers Squibb. She has also received research grant/clinical trials support from Janssen, Abbvie, Takeda, and Bristol-Myers Squibb. Daniel Furtner is an employee of Johnson & Johnson Pte Ltd and may own stock/options. Richard Melsheimer is an employee of Janssen Pharmaceuticals, a division of Johnson & Johnson, and may own stock and/or options in Johnson & Johnson. Philip J. Molloy has nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Afzali, A., Furtner, D., Melsheimer, R. et al. The Automatic Substitution of Biosimilars: Definitions of Interchangeability are not Interchangeable. Adv Ther 38, 2077–2093 (2021). https://doi.org/10.1007/s12325-021-01688-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-021-01688-9