
Abstract
Chronic granulomatous disease (CGD) is a primary immunodeficiency caused by defects in any of the five subunits of the NADPH oxidase complex responsible for the respiratory burst in phagocytic leukocytes. Patients with CGD are at increased risk of life-threatening infections with catalase-positive bacteria and fungi and inflammatory complications such as CGD colitis. The implementation of routine antimicrobial prophylaxis and the advent of azole antifungals has considerably improved overall survival. Nevertheless, life expectancy remains decreased compared to the general population. Inflammatory complications are a significant contributor to morbidity in CGD, and they are often refractory to standard therapies. At present, hematopoietic stem cell transplantation (HCT) is the only curative treatment, and transplantation outcomes have improved over the last few decades with overall survival rates now > 90% in children less than 14 years of age. However, there remains debate as to the optimal conditioning regimen, and there is question as to how to manage adolescent and adult patients. The current evidence suggests that myeloablative conditioning results is more durable myeloid engraftment but with increased toxicity and high rates of graft-versus-host disease. In recent years, gene therapy has been proposed as an alternative to HCT for patients without an HLA-matched donor. However, results to date have not been encouraging. with negligible long-term engraftment of gene-corrected hematopoietic stem cells and reports of myelodysplastic syndrome due to insertional mutagenesis. Multicenter trials are currently underway in the United States and Europe using a SIN-lentiviral vector under the control of a myeloid-specific promoter, and, should the trials be successful, gene therapy may be a viable option for patients with CGD in the future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
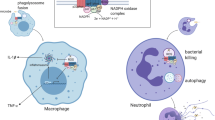
Chronic granulomatous disease (CGD) is an inherited primary immunodeficiency caused by functional impairment of the NADPH oxidase complex in neutrophilic granulocytes and monocytes and characterized by recurrent and severe infections, dysregulated inflammation, and autoimmunity. The NADPH oxidase complex is comprised of both membrane-bound and cytosolic proteins that function in concert upon phagocyte activation to produce reactive oxygen species (ROS) essential for the normal killing of bacteria and fungi [1]. The catalytic glycoprotein gp91phox and non-glycosylated protein p22phox are located in the cell membrane and together form the heterodimer cytochrome b 558. Upon phagocyte activation, the cytosolic proteins p47phox, p67phox, and p40phox translocate to cytochrome b 558 and recruit Rac1/2. This results in a conformational change in gp91phox, which enables cytosolic NADPH to donate an electron to molecular oxygen in the phagolysosome to form superoxide ions. Superoxide ions are then used to generate ROS such as hydrogen peroxide, hypochlorous acid, hydroxyl radicals, and secondary amines that are highly toxic to phagocytosed microorganisms.
Mutations in any of the five structural subunits of the NADPH oxidase complex result in defective ROS production and the syndrome of CGD. The transmembrane glycoprotein gp91phox is encoded by CYBB on the X chromosome and accounts for approximately two-thirds of cases of CGD. Autosomal recessive mutations in NCF1 (p47phox) account for about 20% of cases, and mutations in CYBA (p22phox) and NCF2 (p67phox) each account for about 5% of cases [2–6]. There has been one reported case of an NCF4 (p40phox) mutation resulting in CGD [7]. The incidence of CGD in the United States and Europe is around 1 in 200,000 to 1 in 250,000 live births [2, 5]. However, the incidence varies significantly worldwide, from 1:1 million in Italy to 1:70,000 in the Israeli Arab population [4, 8], and, in countries with high rates of consanguinity, the rate of autosomal recessive CGD exceeds that of X-linked CGD [5, 8–10].
CGD may present at any age from infancy to late adulthood; however, the vast majority of patients are diagnosed at less than 5 years of age [3–5]. In general, patients with X-linked CGD have a more severe disease course with earlier age at presentation and earlier age of death [2, 5]. Mechanistically, the survival of patients with CGD is strongly associated with residual superoxide production independent of the specific gene affected [6].
CGD was initially described as “a fatal granulomatous disease of childhood,” and. historically, most patients with CGD died by 10 years of age [11]. However, improved awareness of the disease and advances in management have led to a marked improvement in life expectancy. Before the introduction of oral antifungals, Winkelstein et al. reported a mortality rate of 5% per year for X-linked CGD and 2% per year for autosomal recessive CGD [2]. More recent studies now report a survival rate of approximately 90% at 10 years of age, which has been attributed to improved recognition and early diagnosis leading to earlier therapies, including more efficacious antimicrobial prophylaxis, use of interferon-gamma (IFN-γ) supplementation for infection prophylaxis, and use of hematopoietic stem cell transplantation (HCT) [6]. Nevertheless, the median age of death remains around 30–40 years, and patients tend to become increasingly debilitated with poor quality of life with advancing age [3–5, 12].
This review aims to summarize the clinical phenotype of CGD, including infectious and inflammatory manifestations, and to update the current data on conventional management, HCT, and gene therapy. It also aims to identify questions that remain with regard to optimal management of patients with CGD, particularly with respect to HCT. This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Infections
Infections are primarily with a subset of catalase-positive microorganisms, and the most common sites of infection are the lungs, skin, lymph nodes, and liver. In North America and Europe, the most frequent pathogens are Aspergillus spp., Staphylococcus aureus, Burkholderia cepacia, Serratia marcescens, Nocardia spp., and Salmonella [1–5, 12]. In developing countries, Bacille Calmette-Guerin (BCG) and Mycobacterium tuberculosis are important pathogens [8, 13, 14]. There are a number of unusual bacteria that have been reported over the last few decades that are virtually pathognomonic for CGD. Chromcobacterium violaceum and Francisella philomiragia are found in brackish water and most frequently cause skin and deep tissue abscesses and sepsis in CGD [15–17]. Granulibacter bethesdensis is a Gram-negative rod that causes chronic necrotizing lymphadenitis and sepsis [18], and Burkholderia gladioli has been reported as a cause of osteomyelitis and sepsis [19, 20]. Infection with any of these microorganisms should prompt evaluation for CGD.
CGD has the highest prevalence of invasive fungal infections among all primary immunodeficiencies, affecting 20–40% of CGD patients, and invasive fungal infections remain an important contributor to morbidity and mortality. [12, 21–23]. The lungs and chest wall are the most common sites of infection, and Aspergillus fumigatus followed by A. nidulans are the most commonly isolated pathogens [12, 21–23]. A. fumigatus was previously the leading cause of mortality in CGD; however, with the advent of azole antifungal treatment, death from A. fumigatus is now uncommon [23]. Conversely, A. nidulans infections cause more severe, refractory, and invasive disease with high mortality rates [21–23]. Notably, the incidence of A. nidulans infections has increased since widespread implementation of itraconazole prophylaxis. Other Aspergillus spp, including A. viridinutans, A. tanneri, and Neosartorya udagawae also cause disease in CGD and are difficult to treat [24–26] After Aspergillus spp., Rhizopus spp. and Trichosporon spp. are the most commonly identified fungal pathogens in CGD [27]. Other rare fungi seen in patients with CGD include Paecilomyces variotii, Paecilomyces lilacinus, Phellinus tropicalis, and Geosmithia argillacea [28–32]. Mulch pneumonitis deserves special mention, as it is exclusive to CGD and is associated with a high rate of mortality if not identified early. Mulch pneumonitis is due to an exuberant inflammatory response to fungal elements in aerosolized decayed organic matter and should be considered in all cases of unexplained pneumonitis in previously well patients [33, 34]. Of note, dimorphic mold infections such as histoplasmosis and blastomycosis and the yeast infection cryptococcosis are not seen in CGD. Mucormycosis is also rare in CGD and only occurs in the setting of significant immunosuppression [35].
Inflammatory Complications
In addition to recurrent and severe infections, dysregulated inflammation is commonly seen in CGD patients. A recent study on a French cohort of 98 patients reported inflammatory manifestations in 69.4% of patients, and the most commonly affected organs were the GI tract (88.2% of patients), lungs (26.4%), urogenital tract (17.6%), and eyes (8.85%) [36]. About 10% of patients also had autoimmune complications. Patients with X-linked CGD had two times the rate of inflammatory complications compared to patients with autosomal recessive CGD.
GI tract manifestations are common, with a reported incidence ranging from 33% to 60% of patients with CGD [36, 37]. Symptom onset may be at any time, but most affected patients develop GI involvement in the first decade of life [37]. Importantly, GI manifestations may precede the diagnosis of CGD and the development of infectious complications. As such, CGD should be considered in all patients who present with early onset inflammatory bowel disease. GI symptoms are generally non-specific and include abdominal pain, noninfectious diarrhea, oral aphthae, nausea and vomiting, and failure to thrive [36–38]. The colon is the most frequently affected site, and patients with CGD are particularly prone to developing perianal disease with high rates of anal fistulae and perirectal abscesses [37–40].
In addition to inflammatory bowel disease, liver involvement is frequent and can be significant. Patients with CGD may develop nodular regenerative hyperplasia, non-cirrhotic portal hypertension, hepatosplenomegaly, and splenic sequestration [41]. Liver manifestations are often progressive, and, notably, the development of thrombocytopenia secondary to splenic sequestration is a strong predictor of mortality [42]. Genitourinary tract manifestations are common and include bladder granulomata, ureteral obstruction, and urinary tract infections, especially in patients with gp91phox and p22phox deficiency [43]. Eosinophilic cystitis has also been reported in children with CGD [44, 45]. Pulmonary manifestations may include granulomatous lung disease and interstitial pulmonary fibrosis [36, 38]. Ocular involvement with chorioretinitis, uveitis, and ocular granulomata has been reported [36]. Of note, macrophage activation syndrome has also been reported in CGD patients and is a potentially life-threatening inflammatory complication [46, 47].
X-Linked Carriers
Female carriers of X-linked CGD have a dual phagocyte population due to lyonization, and cases of severe skewing of X-chromosome inactivation have been reported in which patients are at risk for CGD-type infections. Reports of female carriers with discoid lupus erythematosus, photosensitivity rashes, and other autoimmune phenomena have been published [48, 49]. Inflammatory bowel disease has also been reported in women with skewed X-inactivation [50].
A recent UK survey of 94 female carriers of X-linked CGD demonstrated that these individuals may be more symptomatic than previously thought [51]. Cutaneous symptoms were reported by 63 (79%) women, most frequently photosensitivity but also malar-like lupus rash and eczema; skin abscesses were reported by 14 (17%) women; gastrointestinal symptoms were reported by 40 (42%) women; and 24 (26%) women met criteria for systemic lupus erythematosus. Female carriers with skin abscesses and chronic diarrhea were found to have a significantly lower neutrophil respiratory oxidative burst than unaffected carriers. Interestingly, there seemed to be no relationship between autoimmunity and neutrophil respiratory oxidative burst.
Another recent study from the NIH of 162 female carriers of X-linked CGD again showed greater symptomatology than previously recognized, although not to the same extent as the UK survey [52]. In the NIH study, 25% of women had cutaneous symptoms, and 19% had autoimmunity. Fourteen women (15%) had a history of severe infection caused by typical CGD pathogens. There was a clear correlation between history of severe infection and percent of neutrophils with normal oxidative capacity. Women with less that 20% normal neutrophil oxidative capacity had increased infections, and less than 10% was highly associated with severe infection. As in the UK survey, there was no association between neutrophil oxidative capacity and symptomatic autoimmunity.
These findings create questions regarding the long-term health of female carriers and their suitability for consideration as donors for HCT in their affected family members, since the degree of lyonization can change over time.
Conventional Management
Conventional management is predominantly with lifelong antibiotic and antifungal prophylaxis. Trimethoprim-sulfamethoxazole has been shown to reduce the incidence of bacterial infections from 15.8 to 6.9 infections per 100 patient-months in patients with X-linked CGD and from 7.1 to 2.4 per 100 patient-months in patients with autosomal recessive CGD [53]. Itraconazole prophylaxis was shown to be well tolerated in a trial of 39 patients randomized to receive either placebo or itraconazole. Only one patient receiving itraconazole had a serious fungal infection compared to seven in the placebo group [54]. As such, lifelong prophylaxis with trimethroprim-sulfamethoxazole (5 mg/kg/d div BID up to 320 mg trimethoprim a day) and itraconazole (5 mg/kg/d up to 200 mg daily) is recommended. Dicloxacillin and ciprofloxacin are options for patients with sulfamethoxazole allergy or G6PD deficiency. For those unable to tolerate itraconazole, posaconazole has been shown to be safe and effective [55]. Patients with CGD should receive all routine childhood immunizations except for the BCG vaccine.
The prophylactic use of IFN-γ remains variable. A large randomized, double-blind, placebo-controlled study of 128 patients with CGD showed a clear benefit of IFN-γ prophylaxis with a decrease in both the number and severity of infections (14/63 patients assigned to IFN-γ developed serious infections during the study period versus 30/65 patients assigned to placebo) regardless of inheritance pattern, sex, or use of antibiotic prophylaxis [56]. Long-term follow-up of 9 years demonstrated sustained benefit [57]. Conversely, a prospective Italian study showed that long-term prophylaxis with IFN-γ did not significantly change the rate of total infection per patient-year compared to control, and the group determined there was no evidence to justify long-term prophylaxis with IFN-γ [4]. At our institution, the decision for IFN-γ prophylaxis is made on a case-by-case basis, and is particularly encouraged for patients experiencing increased infections.
Several large studies have reported rates of infection of around 0.3 per year despite appropriate antimicrobial prophylaxis [4, 12]. Infections should be treated early and aggressively, and initial antibiotic therapy should provide strong coverage for both S. aureus and Gram-negative bacteria, including B. cepacia (e.g., combination of vancomycin/clindamycin/oxacillin and ceftazidime/carbapenem depending on local resistance patterns). Treatment strength dosing of trimethoprim-sulfamethoxazole to cover ceftazidime-resistant B. cepacia and Nocardia should also be considered as part of the initial empiric therapy. If patients do not improve within 24–48 h, then more aggressive diagnostic procedures to identify the responsible pathogen should be considered.
There should always be a high index of suspicion for invasive fungal infection in patients with CGD. Invasive fungal infections most commonly affect the lungs and chest wall. In patients with pulmonary symptoms and/or fever of uncertain origin, antifungal therapy should be initiated as part of the initial empiric therapy. Of note, Aspergillus serological tests (e.g., Aspergillus galactomannan) and bronchial alveolar lavage have particularly low sensitivity in patients with CGD and should not be relied upon for diagnosis [23]. Similarly, the efficacy of 1,3-β-d-glucan testing is unclear in CGD. Voriconazole is recommend as first-line therapy for its activity against Aspergillus spp. For infections refractory to voriconazole, liposomal amphotericin B, caspofungin, posaconazole, or some combination thereof may be considered. Surgical intervention is often necessary, and patients generally require prolonged treatment courses. Rescue HCT and/or gene therapy have been proposed as viable options for life-threatening infections resistant to antifungal treatment.
Corticosteroids have traditionally been avoided in patients with CGD and active infection; however, a number of reports indicate that steroids may be used in conjunction with appropriate antimicrobials to treat hyperactive inflammation. Liver abscesses affect about one-third of patients with CGD, and they are often recurrent [12, 58]. Liver abscesses are dense, caseous, and difficult to drain and frequently require surgical intervention. However, in a case series of nine patients at the NIH with Staphylococcal liver abscesses refractory to conventional therapy, the addition of corticosteroids led to the successful resolution of liver abscesses without need for surgical intervention [59]. Corticosteroids have also been shown to play a role in the treatment of respiratory infections, including Nocardia pneumonia and mulch pneumonitis [33, 60, 61].
Treatment of CGD colitis is often long-term and difficult. Patients typically respond to steroids, but relapse is common [37]. Treatment with infliximab also leads to rapid improvement; however, it is associated with increased infections and death in patients with CGD [62]. As such, TNF-α inhibitors should be strictly avoided. Steroid-sparing agents used with varying degrees of success include salicylic acid derivatives, antimetabolites such as azathioprine, and 6-mercaptopurine. IL-1R blockade using Anakinra resulted in rapid and sustained improvement of colitis in two patients with CGD [63]. Of note, hematopoietic stem cell transplantation is curative, and most patients have complete resolution of colitis following transplantation.
Hematopoietic Stem Cell Transplantation
Allogeneic HCT is the only curative treatment for CGD and may reverse both infectious and inflammatory complications. However, patients with CGD are prone to graft failure, and prior infections and organ dysfunction may increase transplant-related complications. Early reports showed that HCT was possible, but outcomes were poorer than reported for other primary immunodeficiencies with a high rate of mortality, graft failure, and low donor chimerism [64]. These complications were at least partly attributed to the use of reduced intensity conditioning (RIC), and it was proposed that more myeloablative conditioning (MAC) may be necessary for stable engraftment. However, higher intensity conditioning typically results in more prolonged immunosuppression leading to an increased risk of infection and requirement for blood/platelet transfusions. HCT is also associated with risk of developing graft-versus-host disease (GVHD).
In 2002, Seger et al. published results from a cohort of 27 European patients ranging from 3 to 38.7 years of age who underwent HCT for CGD between 1985 and 2000 [65]. Twenty-five of the 27 patients received grafts from an HLA-identical sibling, and all but four patients received MAC with busulfan and cyclophosphamide. The four patients who received RIC were severely debilitated and not candidates for MAC. Overall survival was reported at 85% with an event-free survival of 81%. Notably, all patients without severe active infection or inflammation did extremely well (18/18 patients survived). Conversely, all four patients with active fungal infections died, and those with active inflammation had high rates of GVHD. Also of note, only two of the four patients who received RIC had stable myeloid engraftment. Ultimately, this study showed that HCT with MAC was a viable option for patients with CGD and an HLA-identical donor.
In the past 15 years, several more series have been published with encouraging results (Table 1). Transplantation outcomes for pediatric patients less than 14 years of age have been excellent with reported survival rates now consistently > 90% although long-term follow-up is limited. Importantly, several studies have demonstrated that outcomes with 10/10 matched unrelated donors (MUD) are comparable to those with matched sibling donors (MSD) [66–69]. Tewari et al. reported six patients who received unrelated umbilical cord blood transplantation with MAC [70]. All patients survived, and while two of the six patients experienced graft failure, they were both successfully re-transplanted using umbilical cord blood. There have also been a handful of reports of successful haplo-identical transplantation for CGD in recent years [71–74]. The probability of finding an unaffected MSD in most populations is less than 25%. The issue of X-linked carriers as potential MSD is controversial due to increasing evidence of CGD disease burden in carriers that can increase with age, as well as the need to ensure higher engraftment levels to achieve symptomatic cure than may be needed with an unaffected donor.
Patients with intractable infection or active inflammation at time of transplantation and adolescents and young adults have remained difficult to transplant. Adolescents and young adults (14 years of age or older) have historically had increased transplant-related mortality rates between 28% and 50% [64–67]. Unfortunately, efforts to reduce toxicity by utilizing reduced intensity conditioning regimens have generally been complicated by high rates of graft failure. In 2014, Gungor et al. published a large prospective, multicenter study that included 56 patients aged 0–40 years (median 13 years) who underwent HCT with RIC using low-dose busulfan, fludarabine, and serotherapy with either ATG or alemtuzumab [78]. Importantly, 42 of the 56 patients were considered high risk due to active infection and/or autoinflammation. The group reported an impressive 2-year overall survival of 96% and an event-free survival of 91%. The cumulative incidence of grade III–IV acute GVHD was low at 4%, and chronic GVHD was 7%. Graft failure occurred in only 3 of 56 (5%) patients, and stable (≥ 90%) myeloid chimerism was found in 93% of surviving patients. However, subsequent studies have reported high rates of graft failure and mixed chimerism. Khandelwal et al. recently compared a cohort of 14 patients who received MAC with busulfan, cyclophosphamide, and ATG versus four patients who received RIC with fludarabine, melphalan, and alemtuzumab [80]. All four patients who received RIC survived, but three patients developed mixed chimerism, and two required stem cell boosts to maintain donor chimerism. The results at our own institution using RIC have also not been successful. Three patients who underwent HCT with RIC all had poor engraftment and needed further intervention with either donor lymphocyte infusions or repeat HCT [81]. As such, we currently use MAC in all patients with CGD at our institution.
As with all HCT recipients, there is concern regarding the potential for development of late effects and durability of immune reconstitution in CGD patients following HCT. This is of particular concern as many CGD patients may be treated early in life, leading to early exposure to the toxicities of conditioning agents. Further study specifically aimed to address late effects and durability of immune reconstitution are needed.
Cole et al. found that pediatric patients who underwent HCT had fewer infections (0.15 episodes of infection/admission/surgery per year vs. 0.71 episodes infection/admission/surgery per year) and improved growth parameters (height and BMT) compared to those treated conventionally [75]. The same group also demonstrated that quality of life (QOL) was significantly higher in transplanted children versus non-transplanted children [76]. In fact, parent-reported and patient-reported QOL in transplanted children were comparable to levels reported from healthy children. A recent Swedish study directly compared outcomes of 14 X-linked CGD patents who underwent HCT with 13 patients who received conventional management [77]. Thirteen of 14 (92%) men aged 1–35 years of age survived HCT at median follow-up of 7 years (range 1–16) and were cured of disease. Contrarily, 7 of 13 (54%) men treated conventionally died at a mean age of 19 years, and all others suffered from life-threatening infections.
Gene Therapy for CGD
Gene therapy is an attractive alternative to HCT and would provide an option for patients without an HLA-identical donor. Autologous HCT also eliminates the risk of GVHD and abrogates the need for long-term immunosuppressive therapy. Furthermore, full engraftment is likely unnecessary for CGD, as data from healthy female carriers of X-linked CGD indicate that as low as 10–20% functional neutrophils is adequate to protect against severe infection [53, 54].
The first gene therapy trials for CGD took place at the NIH in the 1990s [82]. Five patients with autosomal recessive p47phox deficiency received gene-corrected CD34 + hematopoietic stem cells (HSCs) without conditioning using a recombinant γ-retroviral vector. Functionally corrected granulocytes were detected in all 5 patients but with a peak of only 0.004–0.05% total circulating granulocytes at 3–6 weeks, and no gene-corrected neutrophils were present at 1 year post-gene therapy. Protocol modifications to enhance mobilization of CD34+ cells and to improve retroviral transduction efficiency failed to improve long-term engraftment [83]. These initial studies demonstrated the necessity of at least some degree of conditioning with gene therapy for CGD.
Several small trials subsequently performed gene therapy for X-linked CGD using γ-retroviral vectors and non-myeloablative conditioning. Two adult men aged 25 and 26 years underwent gene therapy in Germany using a γ-retroviral vector expressing gp91phox under control of the spleen focus-forming virus promoter and low-dose busulfan at 8 mg/kg [84, 85]. Approximately 15% of peripheral blood neutrophils were found to express gp91phox within the first 5 months after transplantation, and both patients experienced clinical benefit with resolution of bacterial and fungal infections. However, gene marking increased with time due to insertional activation of the proto-oncogene MDS1-EVI1, while methylation of the viral promoter resulted in transgene silencing and loss of clinical benefit. Both patients ultimately went on to develop myelodysplastic syndrome (MDS); one patient died at 27 months post-gene therapy from septic shock, and the other patient went on to receive a MUD HCT at 45 months [85]. The same vector and protocol were used to treat two children in Switzerland with therapy-resistant A. nidulans infections with resolution of infection [86, 87]. However, the same clonal expansion was observed in both children, and both were rescued with allogeneic HCT [87].
Three patients, aged 28, 28, and 19 years, with X-linked CGD and severe infection not responsive to conventional management underwent gene therapy at the NIH using a murine Moloney retrovirus-derived vector to introduce gp91phox cDNA into CD34+ HSCs and low-dose busulfan at 5 mg/kg/day for 2 days [88]. The team achieved early marking of 26, 5, and 4% of neutrophils, but there was sustained long-term marking of only 1.1% and 0.03% of neutrophils in patients 1 and 3, respectively. Nevertheless, both patients had full or partial resolution of infection. Gene-marked neutrophils had sustained correction of oxidase activity, indicating that silencing of the transgene was not a problem with this vector, and gene marking was polyclonal. Unfortunately, patient 2 had no detectable corrected neutrophils at 4 weeks, and he died at 6 months post-gene therapy from a pre-existing fungal infection.
In total, five phase I/II clinical trials were performed in Germany, London, the NIH, and Seoul with 12 patients transplanted using γ-retroviral vectors and RIC [83, 89]. All the trials demonstrated initial engraftment of transduced neutrophils at 10–30% of total neutrophils and clinical benefit with resolution of pre-existing and life-threatening infections. However, cell engraftment progressively decreased with time in all patients, and several patients developed MDS. Corrected HSCs do not have a selective growth advantage compared to NADPH oxidase-deficient cells, contrary to what is seen with some other primary immunodeficiencies. This suggests that more myeloablative conditioning may be needed for sustained engraftment. It has also been suggested that constitutive expression of gp91phox in hematopoietic progenitor cells may lead to the inappropriate production of ROS with resultant toxicity and loss of gene-corrected cells over time.
In response to the high incidence of MDS seen with the γ-retroviral vectors and concerns related to stem cell toxicity mentioned above, codon-optimized self-inactivating (SIN) lentiviral vectors have been developed whereby transgene expression is limited to the myeloid lineage. Santilli et al. developed a SIN-lentiviral vector with gp91phox cDNA under the control of a chimeric promotor that contains binding sites for the myeloid transcription factors CAAT box enhancer-biding family proteins (C/EBPs) and PU.1, which are highly expressed during granulocyte differentiation [90]. This allows for high levels of gp91phox expression in terminally differentiated neutrophils while de-targeting expression in HSC’s. Chiriaco et al. took this one step further and developed a dual-regulated lentiviral vector employing a myeloid-specific promoter and microRNA to post-transcriptionally regulate transgene expression [91]. Preclinical trials in mice demonstrated high levels of gp91phox expression in myeloid cells with sparing of the CD34 + HSC compartment. Multicenter clinical trials using the Santilli et al. vector and low-dose busulfan are currently underway in the United States and Europe. So far, three patients have undergone gene therapy in the United States, and early results are promising, with resolution of CGD phenotype and sustained neutrophil gene marking with time (unpublished data).
Finally, targeted genome editing approaches using zinc-finger nucleases (ZFN), transcription activator-like effector nucleases, or the clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 system have been proposed to allow gene correction in situ such that gene expression remains under the control of the gene’s own cell-specific promoters. However, many CGD patients have unique mutations, and these approaches would require engineering unique systems for each individual patient. Another approach is to target specific “safe harbors” in the genome such as the AAVS1 locus (the common integration site of adeno-associated virus), the disruption of which does not result in genomic instability. However, the advantage of gene correction in situ is lost using this technique. Thus far, preclinical experiments using ZFNs and the CRISPR-Cas9 system have demonstrated successful transgene expression in CD34+ HSCs and induced pluripotent stem cells from CGD patients without off-target effects [92–94]. Nevertheless, gene editing approaches remain limited by low efficiency and hematopoietic stem cell toxicity through the process of transfection, expansion, and selection.
Conclusions
Life expectancy of CGD patients has increased more than three-fold over the last few decades due to increased recognition of the disease, the advent of azole antifungals, and improved management of infectious and inflammatory complications. Nevertheless, the incidence of triazole-resistant Aspergillus is increasing, and the management of inflammatory complications remains difficult. Survival following HCT has increased from approximately 85% before 2000 to greater than 90% in recent reports, and outcomes have been encouraging regardless of the donor source. Children who undergo HCT are also healthier with better QoL than those managed conservatively. As such, HCT should be considered for all patients with CGD regardless of sex, genetic mutation, and clinical manifestations. MAC appears to reduce the risk of graft failure and increase the likelihood of long-term myeloid engraftment. It is preferable to perform HCT as early as possible, but definitive cure can also be considered for adolescent and young adult patients, including those with a history of severe infection and autoinflammation. Ultimately, as HCT becomes more widely available and better tolerated, we expect overall life expectancy for patients with CGD to increase substantially over the next several years. Furthermore, with the new SIN-lentiviral vectors and optimization of conditioning regimens, gene therapy may become a viable alternative to allogeneic HCT for those without an HLA-matched donor.
References
Segal BH, Leto TL, Gallin JI, et al. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine (Baltimore). 2000;79(3):170–200.
Winkelstein JA, Marino MC, Johnston RB, et al. Chronic granulomatous disease. Report on a national registry of 368 Patients. Medicine (Baltimore). 2000;79(3):155–69.
Jones LB, McGrogan P, Flood TJ, et al. Special article: chronic granulomatous disease in the United Kingdom and Ireland: a comprehensive national patient-based registry. Clin Exp Immunol. 2008;152(2):211–8.
Martire B, Rondelli R, Soresina A, et al. Clinical features, long-term follow-up and outcome of a large cohort of patients with Chronic Granulomatous Disease: an Italian multicenter study. Clin Immunol. 2008;126(2):155–64.
van den Berg JM, van Koppen E, Ahlin A, et al. Chronic granulomatous disease: the European experience. PLoS ONE. 2009;4(4):e5234.
Kuhns DB, Alvord WG, Heller T, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363(27):2600–10.
Matute JD, Arias AA, Wright NA, et al. A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40phox and selective defects in neutrophil NADPH oxidase activity. Blood. 2009;114(15):3309–15.
Wolach B, Gavrieli R, de Boer M, et al. Chronic granulomatous disease: clinical, functional, molecular, and genetic studies. The Israeli experience with 84 patients. Am J Hematol. 2017;92(1):28–36.
Fattahi F, Badalzadeh M, Sedighipour L, et al. Inheritance pattern and clinical aspects of 93 Iranian patients with chronic granulomatous disease. J Clin Immunol. 2011;31(5):792–801.
Köker MY, Camcioglu Y, van Leeuwen K, et al. Clinical, functional, and genetic characterization of chronic granulomatous disease in 89 Turkish patients. J Allergy Clin Immunol. 2013;132(5):1156–63.
Mouy R, Fischer A, Vilmer E, et al. Incidence, severity, and prevention of infections in chronic granulomatous disease. J Pediatr. 1989;114(4):555–60.
Marciano BE, Spalding C, Fitzgerald A, et al. Common severe infections in chronic granulomatous disease. Clin Infect Dis. 2015;60(8):1176–83.
Lee PP, Chan KW, Jiang L, et al. Susceptibility to mycobacterial infections in children with X-linked chronic granulomatous disease: a review of 17 patients living in a region endemic for tuberculosis. Pediatr Infect Dis J. 2008;27(3):224–30.
Conti F, Lugo-Reyes SO, Blancas Galicia L, et al. Mycobacterial disease in patients with chronic granulomatous disease: a retrospective analysis of 71 cases. J Allergy Clin Immunol. 2016;138(1):241–8.
Sirinavin S, Techasaensiri C, Benjaponpitak S, et al. Invasive Chromobacterium violaceum infection in children: case report and review. Pediatr Infect Dis J. 2005;24(6):559–61.
Meher-Homji Z, Mangalore RP, DR Johnson P, Y L Chua K. Chromobacterium violaceum infection in chronic granulomatous disease: a case report and review of the literature. JMM Case Rep. 2017;4(1):005084.
Mailman TL, Schmidt MH. Fancisella philomiragia adenitis and pulmonary nodules in a child with chronic granulomatous disease. Can J Infect Dis Med Microbiol. 2005;16(4):245–8.
Greenberg DE, Shoffner AR, Zelazny AM, et al. Recurrent Granulibacter bethesdensis infections and chronic granulomatous disease. Emerg Infect Dis. 2010;16(9):1341–8.
Ross JP, Holland SM, Gill VJ, et al. Severe Burkholderia (Pseudomonas) gladioli infection in chronic granulomatous disease: report of two successfully treated cases. Clin Infect Dis. 1995;21(5):1291–3.
Boyanton BL, Noroski LM, Reddy H, et al. Burkholderia gladioli osteomyelitis in association with chronic granulomatous disease: as case report and review. Pediatr Infect Dis J. 2005;24(9):837–9.
Falcone EL, Holland SM. Invasive fungal infection in chronic granulomatous disease: insights into pathogenesis and management. Curr Opin Infect Dis. 2012;25(6):658–69.
Beauté J, Obenga G, Le Mignot L, et al. Epidemiology and outcome of invasive fungal diseases in patients with chronic granulomatous disease: a multicenter study in France. Pediatr Infect Dis J. 2011;30(1):57–62.
Blumental S, Mouy R, Mahlaoui N, et al. Invasive mold infections in chronic granulomatous disease: a 25-year retrospective survey. Clin Infectious Dis. 2011;53(12):e159–69.
Vinh DC, Shea YR, Jones PA, et al. Chronic invasive aspergillosis caused by Aspergillus viridinutans. Emerg Infect Dis. 2009;15(8):1292–4.
Sigui JA, Peterson SW, Clark LP, et al. Aspergillus tanneri sp. nov., a new pathogen that causes invasive disease refractory to antifungal therapy. J Clin Microbiol. 2012;50(10):3309–17.
Vinh DC, Shea YR, Sugui JA, et al. Invasive aspergillosis due to Neosartorya udagawae. Clin Infect Dis. 2009;49(1):102–11.
Dotis J, Pana ZD, Roilides E. Non-Aspergillus fungal infections in chronic granulomatous disease. Mycoses. 2013;56(4):449–62.
Wang SM, Shieh CC, Liu CC. Successful treatment of Paecilomyces variotii splenic abscesses: a rare complication in a previously unrecognized chronic granulomatous disease child. Diagn Microbiol Infect Dis. 2005;53(2):149–52.
Silliman CC, Lawellin DW, Lohr JA, et al. Paecilomyces lilacinus infection in a child with chronic granulomatous disease. J Infect. 1992;24(2):191–5.
Ramesh M, Resnick E, Hui Y, et al. Phellinus tropicalis abscesses in a patient with chronic granulomatous disease. J Clin Immunol. 2014;34(2):130–3.
De Ravin SS, Challipalli M, Anderson V, et al. Geosmithia argillacea: an emerging cause of invasive mycosis in human chronic granulomatous disease. Clin Infect Dis. 2011;52(6):e136–43.
Haidar G, Zerbe CS, Cheng M, et al. Phellinus species: an emerging cause of refractory fungal infections in patients with X-linked chronic granulomatous disease. Mycoses. 2017;60(3):155–60.
Siddiqui S, Anderson VL, Hilligoss DM, et al. Fulminant mulch pneumonitis: an emergency presentation of chronic granulomatous disease. Clin Infect Dis. 2007;45(6):673–81.
Holland SM. Chronic granulomatous disease. Hematol Oncol Clin North Am. 2013;27(1):89–99.
Vinh DC, Freeman AF, Shea YR, et al. Mucormycosis in chronic granulomatous disease: association with iatrogenic immunosuppression. J Allergy Clin Immunol. 2009;123(6):1411–3.
Magnani A, Brosselin P, Beauté J, et al. Inflammatory manifestations in a single-center cohort of patients with chronic granulomatous disease. J Allergy Clin Immunol. 2014;134(3):655–62.
Marciano BE, Rosenzweig SD, Kleiner DE, et al. Gastrointestinal involvement in chronic granulomatous disease. Pediatrics. 2004;114(2):462–8.
Damen GM, van Krieken JH, Hoppenreijs E, et al. Overlap, common features, and essential differences in pediatric granulomatous inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 2010;51(6):690–7.
Alimchandani M, Lai JP, Aung PP, et al. Gastrointestinal histopathology in chronic granulomatous disease: a study of 87 patients. Am J Surg Pathol. 2013;37(9):1365–72.
Marks DJ, Miyagi K, Rahman FZ, et al. Inflammatory bowel disease in CGD reproduces the clinicopathological features of Crohn’s disease. Am J Gastroenterol. 2009;104(1):117–24.
Hussain N, Fled JJ, Kleiner DE, et al. Hepatic abnormalities in patients with chronic granulomatous disease. Hepatology. 2007;45(3):675–83.
Feld JJ, Hussain N, Wright EC, et al. Hepatic involvement and portal hypertension predict mortality in chronic granulomatous disease. Gastroenterology. 2008;134(7):1917–26.
Walther MM, Malech H, Berman A, et al. The urological manifestations of chronic granulomatous disease. J Urol. 1992;147(5):1314–8.
Barese CN, Podestá M, Litvak E, et al. Recurrent eosinophilic cystitis in a child with chronic granulomatous disease. J Pediatr Hematol Oncol. 2004;26(3):209–12.
Claps A, Della Corte M, Gerocarni Nappo S, et al. How should eosinophilic cystitis be treated in patients with chronic granulomatous disease? Pediatr Nephrol. 2014;29(11):2229–33.
Akagi K, Kawai T, Watanabe N, et al. A case of macrophage activation syndrome developing in a patient with chronic granulomatous disease-associated colitis. J Pediatr Hematol Oncol. 2014;36(3):e169–72.
Bode SF, Ammann S, Al-Herz W, et al. The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: implications for differential diagnosis and pathogenesis. Haematologica. 2015;100(7):978–88.
Cale CM, Morton ML, Goldblatt D. Cutaneous and other lupus-like symptoms in carriers of X-linked chronic granulomatous disease: incidence and autoimmune serology. Clin Exp Immunol. 2007;148(1):79–84.
De Ravin SS, Naumann N, Cowen EW, et al. Chronic granulomatous disease as a risk factor for autoimmune disease. J Allergy Clin Immunol. 2008;122(6):1097–103.
Hauck F, Koletzko S, Walz C, et al. Diagnostic and treatment options for severe IBD in Female X-CGD carriers with non-random X-inactivation. J Crohns Colitis. 2016;10(1):112–5.
Battersby AC, Braggins H, Pearce MS et al. Inflammatory and autoimmune manifestations in X linked carriers of chronic granulomatous disease in the United Kingdom. J Allergy Clin Immunol 2017;140(2):628–630.
Marciano BE, Zerbe CS, Falcone EL et al. X-linked carriers of chronic granulomatous disease: Illness, lyonization and stability. J Allergy Clin Immunol 2017. http://doi.org/10.1016/j.jaci.2017.04.035.
Margolis DM, Melnick DA, Alling DW, Gallin JI. Trimethoprim-sulfamethoxazole prophylaxis in the management of chronic granulomatous disease. J Infect Dis. 1990;162(3):723–6.
Gallin JI, Alling DW, Malech HL, et al. Itraconazole to prevent fungal infections in chronic granulomatous disease. N Engl J Med. 2003;348(24):2416–22.
Segal BH, Barnhart LA, Anderson VL, et al. Posaconazole as salvage therapy in patients with chronic granulomatous disease and invasive filamentous fungal infection. Clin Infect Dis. 2005;40(11):1684–8.
The International Chronic Granulomatous Disease Cooperative Study Group. A controlled trial of interferon gamma to prevent infection in chronic granulomatous disease. N Engl J Med. 1991;324(8):509–16.
Marciano BE, Wesley R, De Carlo ES, et al. Long-term interferon-gamma therapy for patients with chronic granulomatous disease. Clin Infect Dis. 2004;39(5):692–9.
Lublin M, Bartlett DL, Danforth DN, et al. Hepatic abscess in patients with chronic granulomatous disease. Ann Surg. 2002;235(3):383–91.
Leiding JW, Freeman AF, Marciano BE, et al. Corticosteroid therapy for liver abscess in chronic granulomatous disease. Clin Infect Dis. 2012;54(5):694–700.
Freeman AF, Marciano BE, Anderson VL, et al. Corticosteroids in the treatment of severe Nocardia pneumonia in chronic granulomatous disease. Pediatr Infect Dis J. 2011;30(9):806–8.
Yamazaki-Nakashimada MA, Stiehm ER, Pietropaolo-Cienfuegos D, et al. Corticosteroid therapy for refractory infections in chronic granulomatous disease: case reports and review of the literature. Ann Allergy Asthma Immunol. 2006;97(2):257–61.
Uzel G, Orange JS, Poliak N, et al. Complications of tumor necrosis factor-α blockade in chronic granulomatous disease-related colitis. Clin Infect Dis. 2010;51(12):1429–34.
de Luca A, Smeekens SP, Casagrande A, et al. IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc Natl Acad Sci USA. 2014;111(9):3526–31.
Horwitz ME, Barrett AJ, Brown MR, et al. Treatment of chronic granulomatous disease with nonmyeloablative conditioning and a T-cell-depleted hematopoietic allograft. N Engl J Med. 2001;344(12):881–8.
Seger RA, Gungor T, Belohradsky BH, et al. Treatment of chronic granulomatous disease with myeloablative conditioning and an unmodified hemopoietic allograft: a survey of the European experience, 1985-2000. Blood. 2002;100(13):4344–50.
Soncini E, Slatter MA, Jones LB, et al. Unrelated donor and HLA-identical sibling haematopoietic stem cell transplantation cure chronic granulomatous disease with good long-term outcome and growth. Br J Haematol. 2009;145(1):73–83.
Schuetz C, Hoenig M, Gatz S, et al. Hematopoietic stem cell transplantation from matched unrelated donors in chronic granulomatous disease. Immunol Res. 2009;44(1–3):35–41.
Goździk J, Pituch-Noworolska A, Skoczeń S, et al. Allogeneic haematopoietic stem cell transplantation as therapy for chronic granulomatous disease—single centre experience. J Clin Immunol. 2011;31(3):332–7.
Martinez CA, Shah S, Shearer WT, et al. Excellent survival after sibling or unrelated donor stem cell transplantation for chronic granulomatous disease. J Allergy Clin Immunol. 2012;129(1):176–83.
Tewari P, Martin PL, Mendizabal A, et al. Myeloablative transplantation using either cord blood or bone marrow leads to immune recovery, high long-term donor chimerism and excellent survival in chronic granulomatous disease. Biol Blood Marrow Transplant. 2012;18(9):1368–77.
Hoenig M, Niehues T, Siepermann K, et al. Successful HLA haploidentical hematopoietic SCT in chronic granulomatous disease. Bone Marrow Transplant. 2014;49(10):1337–8.
Parta M, Hilligoss D, Kelly C, et al. Haploidentical hematopoietic cell transplantation with post-transplant cyclophosphamide in a patient with chronic granulomatous disease and active infection: a first report. J Clin Immunol. 2015;35(7):675–80.
Morillo-Gutierrez B, Beier R, Rao K, et al. Treosulfan-based conditioning for allogeneic HSCT in children with chronic granulomatous disease: a multicenter experience. Blood. 2016;128(3):440–8.
Zhou L, Dong LJ, Gao ZY et al. Haploidentical hematopoietic stem cell transplantation for a case with X-linked chronic granulomatous disease. Pediatr Transplant 2017;21(1). http://doi:org/10.1111/petr.12861.
Cole T, Pearce MS, Cant AJ, et al. Clinical outcome in children with chronic granulomatous disease managed conservatively or with hematopoietic stem cell transplantation. J Allergy Clin Immunol. 2013;132(5):1150–5.
Cole T, McKendrick F, Titman P, et al. Health related quality of life and emotional health in children with chronic granulomatous disease: a comparison of those managed conservatively with those that have undergone haematopoietic stem cell transplant. J Clin Immunol. 2013;33(1):8–13.
Ahlin A, Fugeläng J, de Boer M, et al. Chronic granulomatous disease—haematopoietic stem cell transplantation versus conventional treatment. Acta Pediatr. 2013;102(11):1087–94.
Güngör T, Teira P, Slatter M, et al. Reduced-intensity conditioning and HLA-matched hematopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet. 2014;383(9915):436–48.
Mehta B, Mahadeo K, Kapoor N, et al. Low-dose total-body irradiation and alemtuzumab-based reduced-intensity conditioning regimen results in durable engraftment and correction of clinical disease among children with chronic granulomatous disease. Pediatr Transplant. 2015;19(4):408–12.
Khandelwal P, Bleesing JJ, Davies SM, Marsh RA. A Single-center experience comparing alemtuzumab, fludarabine, and melphalan reduced-intensity conditioning with myeloablative busulfan, cyclophosphamide, and antithymocyte globulin for chronic granulomatous disease. Biol Blood Marrow Transplant. 2016;22(11):2011–8.
Oshrine B, Morsheimer M, Heimall J, Bunin N. Reduced-intensity conditioning for hematopoietic cell transplantation of chronic granulomatous disease. Pediatr Blood Cancer. 2015;62(2):359–61.
Malech HL, Maples PB, Whiting-Theobald N, et al. Prolonged production of NADPH oxidase-corrected granulocytes after gene therapy of chronic granulomatous disease. Proc Natl Acad Sci USA. 1997;94(22):12133–8.
Grez M, Reichenbach J, Schwäble J, et al. Gene therapy of chronic granulomatous disease: the engraftment dilemma. Mol Ther. 2011;19(1):28–35.
Ott MG, Schmidt M, Schwarzwaelder K, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12(4):401–9.
Stein S, Ott MG, Schultze-Strasser S, et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16(2):198–204.
Bianchi M, Hakkim A, Brinkmann V, et al. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood. 2009;114(13):2619–22.
Siler U, Paruzynski A, Holtgreve-Grez H, et al. Successful combination of sequential gene therapy and rescue allo-HSCT in two children with X-CGD—importance of timing. Curr Gene Ther. 2015;15(4):416–27.
Kang HJ, Bartholomae CC, Paruzynski A, et al. Retroviral gene therapy for X-linked chronic granulomatous disease: results from phase I/II trial. Mol Ther. 2011;19(11):2092–101.
Xu X, Tailor CS, Grunebaum E. Gene therapy for primary immune deficiencies: a Canadian perspective. Allergy Asthma Clin Immunol. 2017;13:14.
Santilli G, Almarza E, Brendel C, et al. Biochemical correction of X-CGD by a novel chimeric promoter regulating high levels of transgene expression in myeloid cells. Mol Ther. 2011;19(1):122–32.
Chiriaco M, Farinelli G, Capo V, et al. Dual-regulated lentiviral vector for gene therapy of X-linked chronic granulomatosis. Mol Ther. 2014;22(8):1472–83.
Flynn R, Grundmann A, Renz P, et al. CRISPR-mediated genotypic and phenotypic correction of a chronic granulomatous disease mutation in human iPS cells. Exp Hematol. 2015;43(10):838–48.
De Ravin SS, Reik A, Liu PQ, et al. Targeted gene addition in human CD34 + hematopoietic cells for correction of X-linked chronic granulomatous disease. Nat Biotechnol. 2016;34(4):424–9.
De Ravin SS, Li L, Wu X et al. CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Sci Transl Med 2017;9(372). http://doi.org/10.1126/scitranslmed.aah3480.
Acknowledgements
No funding or sponsorship was received for this study or publication of this article. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published. During the peer review process, Horizon Pharma was offered an opportunity to comment on the article. Changes resulting from comments received were made by the author based on their scientific and editorial merit.
Disclosures
Danielle E. Arnold and Jennifer R. Heimall have nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Content
To view enhanced content for this article go to http://www.medengine.com/Redeem/DCCCF0601B323C17.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Arnold, D.E., Heimall, J.R. A Review of Chronic Granulomatous Disease. Adv Ther 34, 2543–2557 (2017). https://doi.org/10.1007/s12325-017-0636-2
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-017-0636-2