
Abstract
Alopecia areata (AA), a prevalent inflammatory cause of hair loss, lacks FDA-approved therapeutics for extensive cases, which are associated with very poor rates of spontaneous hair regrowth and major psychological distress. Current treatments for severe cases include broad immune-suppressants, which are associated with significant adverse effects, precluding long-term use, with rapid hair loss following treatment termination. As a result of the extent of the disease in severe cases, topical contact sensitizers and intralesional treatments are of limited use. The pathogenesis of AA is not yet fully understood, but recent investigations of the immune activation in AA skin reveal Th1/IFN-γ, as well as Th2, PDE4, IL-23, and IL-9 upregulations. Tissue analyses of both animal models and human lesions following broad-acting and cytokine-specific therapeutics (such as JAK inhibitors and ustekinumab, respectively) provide another opportunity for important insights into the pathogenesis of AA. As reviewed in this paper, numerous novel therapeutics are undergoing clinical trials for AA, emphasizing the potential transformation of the clinical practice of AA, which is currently lacking. Dermatologists are already familiar with the revolution in disease management of psoriasis, stemming from better understanding of immune dysregulations, and atopic dermatitis will soon follow a similar path. In light of these recent developments, the therapeutic arena of AA treatments is finally getting more exciting. AA will join the lengthening list of dermatologic diseases with mechanism-targeted drugs, thus changing the face of AA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alopecia areata (AA) is a prevalent autoimmune disease causing loss of hair, with approximately 2% of the population suffering from the condition during their lifetime, most commonly starting before the age of 30 years [1, 2]. The typical lesion is a non-scarring, hairless, circular patch on the scalp, evolving to multiple patches, but extensive forms can progress to total loss of scalp hair—alopecia totalis (AT), or complete body hair loss—alopecia universalis (AU). Spontaneous hair growth is common when the disease is limited, but when extensive, spontaneous remission is rare. Relapses are common in AA patients, and only one-third of cases achieve long-lasting remission of 10–15 years [1]. Prognostic factors for AA include early onset, extensive involvement (especially AT or AU), and rapid disease progression [3, 4].
AA is associated with other dermatologic and autoimmune diseases, in particular atopic dermatitis (AD) [5], as well as vitiligo, lupus erythematosus, psoriasis, autoimmune thyroid disease, and allergic rhinitis [6,7,8]. Concomitant atopy is associated with higher risk for severe AA phenotype [6, 7, 9, 10].
Although AA is not life threatening, psychological comorbidities are common and result in major impact on patients’ lives [11].
Clinically, the severity of AA is commonly evaluated by the Severity of Alopecia Tool (SALT), a mathematical approach utilized to determine hair loss and hair regrowth. The percentages of scalp hair loss in four main areas are assessed independently (each of the sides, top, and top of the back, representing 18%, 18%, 40%, and 24% of the total scalp surface area, respectively). Relatively to their surface area, these subscores are summed for a final total percentage hair loss, designated as the SALT score [12]. Severe or extensive AA is usually defined as patients with at least 50% total scalp hair loss, including those with AT and AU. Lately, molecular tissue scores for treatment response were also proposed, including the baseline and post-treatment assessment of the expression level of hair keratins and inflammatory factors [13,14,15,16]. This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Current AA Treatment Paradigm
Extensive AA poses a huge therapeutic challenge due to the either lack of efficacy and/or side effects of the current available treatment options, with lack of systemic therapeutics that are FDA-approved for the disease. Topical treatments are largely not suitable for extensive cases of AA because of minimal efficacy [17, 18]. These include topical corticosteroids, tacrolimus, cryotherapy, minoxidil, anthralin, or ultraviolet light A combined with oral psoralens (PUVA), all of which have no robust evidence for their consistent efficacy in AA [17, 19,20,21]. Contact sensitizers, such as diphenylcyclopropenone (DPCP), considered by some as topical immunotherapy, have some efficacy and are also abundantly used, but may be associated with significant local adverse events, including vesicular reaction, pain, and scalp and facial edema [17, 20, 21]. Intralesional injections of corticosteroids are often effective but can only be considered for patients with limited involvement as well [22]. Since extensive AA, especially AT and AU, for which spontaneous regrowth is rare, is associated with devastating stigmatism and mental stress, patients seeking treatments are willing to risk significant adverse drug effects and attempt off-label use of various treatments. These include systemic corticosteroids, given as continuous or pulse therapy, cyclosporine A, mycophenolate mofetil, methotrexate as a monotherapy or in conjunction with corticosteroids, and azathioprine. All of these regimens are not FDA-approved for AA, and lack large, randomized placebo-controlled data for efficacy and safety in AA [22,23,24,25,26]. Although all are commonly used, these drugs have problematic safety profiles, with major systemic adverse effects and variable degree of immunosuppression, as well as rapid recurrence of hair loss following drug cessation, and are therefore unsuitable for the chronic nature of extensive AA [4]. In sum, despite being a relatively common condition, AA, including its extensive form, lacks good treatment options [18, 20]. Better clinical management with improved therapeutic modalities for AA, and particularly for widespread disease, is therefore highly desired.
AA in the Footsteps of Psoriasis
The understanding of immune activation, revolutionizing current dermatology by introducing new treatments targeting pathogenic immune pathways into daily clinical management of psoriasis [27, 28], has not been established yet for AA. Nevertheless, studies shed light on some immune markers upregulated in AA [13, 14]. In 1998, by induction of AA in immune-suppressed mice injected with autologous T cells isolated from involved AA human scalp, it was first clearly demonstrated that AA is mediated by T cells recognizing a follicular autoantigen [29]. About a decade later, NKG2D+ T cells were speculated to be the main drivers of the immune dysregulations in AA, associated with the collapse of hair follicle immune privilege [30]. The first cytokine to be identified as AA-related was IL-1 [31], followed by Th1/interferon (IFN)-γ [14]. Studies also showed that the levels of IFN-γ are downregulated in association with hair regrowth as a therapeutic response [14, 32]. Other immune pathways were also shown to be upregulated in AA, including Th2, IL-9, PDE4, and Th17/IL-23 [13]. The role of each immune axis in AA remained to be elucidated. Psoriasis, representing the best immune-characterized skin disease, was also studied through the investigation of targeted drugs, which revealed the role of different immune components in disease pathogenesis [27, 33]. AD, the most common inflammatory skin disease in adults, is undergoing a similar translational revolution to psoriasis, likely due to elucidation of pathogenic disease mechanisms [27, 34, 35]. The same approach, utilizing cytokine-specific drugs, as detailed in this review, will be able to clarify the contribution of the various upregulated markers and immune axes found in AA. Novel broad-acting therapeutics, such as apremilast and Janus kinase (JAK) inhibitors, appear to be promising strategies for severe AA, but will be less contributory in elucidating the effect of specific cytokines in producing the disease phenotype. We systemically review novel AA therapeutics, by searching PubMed, ClinicalTrials.gov, Google Scholar, and Science Direct using the words “alopecia areata”, “alopecia totalis”, “alopecia universalis”, and “extensive alopecia”, with “treatment”, “systemic treatment”, “drugs”, or with specific relevant therapeutics or immune markers such as “JAK inhibitors”, “anti-TNF”, “PDE4”, or “IL-23”. Acceleration in AA therapeutic research is well demonstrated by the fact that only ten clinical trials for AA were registered in ClinicalTrials.gov during the years 2010–2015, whereas in the following 2 years, 11 registered trials were found. We included ongoing trials as well as recently terminated trials of special interest. We also review treatment options that may be beneficial for future clinical trials in AA patients (Table 1; Fig. 1).

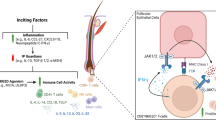
Adapted with permission from [118]
The immune pathways in lesional skin of alopecia areata (AA), with upregulated cytokines as therapeutic targets and corresponding antagonizing agents, as well as hair keratins decreased in different chronological stages of AA. The complex immune signature of AA is still poorly defined, with evidence supporting a pathogenic role of Th1/IFN-γ, Th2 (IL-4 and IL-13), IL-23/Th17, and Th9/IL-9 in the disease mechanism. Drugs highlighted in red represent treatment options that are currently tested in clinical trials or may be tested in future trials. Gray therapeutics that failed to show efficacy in AA. APC antigen-presenting cell, DC dendritic cell, JAK Janus kinase, PDE phosphodiesterase, dashed line indirect inhibition.
This review will encompass the current understanding of the complex immune activation of AA by reviewing AA pathogenesis by three main immune axes, with corresponding therapeutic approaches: broad T cell antagonism, Th-17/IL-23 inhibition, and Th2 antagonism.
Broad T Cell Antagonism
Since AA is associated with complex upregulation of various cytokines that are part of diverse immune pathways, broad-acting immune-modulating drugs, inhibiting common components shared between several immune axes, are being tested for the treatment of extensive AA cases. Such drugs include the JAK inhibitors, PDE4 inhibitors, and abatacept.
JAK Inhibitors
JAK inhibitors are group of small molecules that recently were shown to beneficially treat AA in mouse models and in small proof-of-concept clinical trials. These are antagonists of the various members of the JAK enzyme family, which consists of JAK1, JAK2, JAK3, and tyrosine kinase-2 (TYK2) [36]. JAKs enable the binding and activation of the transducer and activator of transcription (STAT), by phosphorylating the cytoplasmic domain of multiple cytokine receptors. This results in translocation of the STAT into the nucleus, which greatly affects transcription. JAK antagonism therefore blocks this signaling through STAT activation [37,38,39], targeting Th1/IFN-γ as well as common γc cytokines (shared between IL-2, IL-4, IL-9, IL-7, IL-15, and IL-21), and TYK2 also adds an IL-23 capability (Fig. 1) [14, 40, 41]. In AA, a subgroup of CD8+ T cells co-expressing receptor NKG2D+ was shown to be the predominant cellular infiltrate in the hair follicle in both mice and humans with AA, with potential to efficiently induce AA in mice [14]. Few cytokines were shown to support the autoreactive CD8+ T cells, including INF-γ, IL-2, and IL-15, and these cytokines are inhibited by JAK-STAT antagonism [14, 30, 42]. Both animal and in vitro models suggest that AA is characterized by a strong JAK3 expression, and JAK3 was found to be the only JAK that is overexpressed in human AA compared to controls [14, 43]. JAK3 is therefore of specific interest as a therapeutic target for AA. So far, three JAK inhibitors were shown to effectively treat AA, and these are currently being tested for extensive AA: ruxolitinib, tofacitinib, and baricitinib (NCT01950780, NCT02312882 and NCT02197455, and NCT02299297, respectively) [14, 32, 44,45,46,47]. Ruxolitinib and tofacitinib are blockers of multiple JAKs, and are FDA-approved for the treatment of hematological and reumatological diseases [37]. Baricitinib, a JAK1/2 inhibitor, is not yet approved by the FDA, but is being tested for numerous hemato-oncological, reumatological, and dermatological indications, including AD and AA (NCT02576938, NCT02299297, respectively; see Table 1). Ruxolitinib and tofacitinib cause significant immunosuppression, while baricitinib possibly has a better safety profile [39, 48]. The response to these JAK inhibitors in AA was well demonstrated in mouse models, as well as in human AA [14, 32, 44, 45]. JAK inhibition resulted in both clinical improvement with impressive hair growth, including regrowth in AT and AU patients [49], as well as immune response, characterized by downregulation of Th1/IFN-γ expression levels and upregulation of different hair keratins, likely representing tissue recovery [14, 32]. Tofacitinib was also able to reverse the dystrophic nail changes associated with AA [50], and showed general good safety and efficacy in a trial of 13 adolescents treated for AA [51]. Two new JAK inhibitors, PF-06700841 and PF-06651600, blocking JAK3 and TYK2/JAK1, respectively (the first representing a relatively specific inhibitor of the speculated main JAK member in AA), are also beginning clinical trials in AA patients, and include a placebo arm (NCT02974868). Recent studies show that within 3 months of discontinuation of oral JAKs, often there is loss of hair growth attributed to the drug [46, 47], necessitating long-term safety data to allow long-term use for severe patients. Topical JAK inhibitors, associated with minimal systemic immunosuppression, already showed efficacy in an allergic dermatitis animal model, as well as in psoriasis and AD clinical trials, and may prove beneficial for AA, especially for limited disease (NCT02553330) [52,53,54]. Local adverse reactions include application mild to moderate site pain, erythema, stinging/burning, and pruritus, with similar occurrence rates in the placebo groups [52, 54]. JAK inhibitors represent a promising treatment strategy, and the role and safety of various JAK antagonists in the treatment paradigm of extensive AA will be elucidated by large, placebo-controlled clinical trials.
PDE4 Inhibition
Another oral small molecule inhibiting broad T cell activation is apremilast—a PDE4 inhibitor. The levels of cyclic adenosine monophosphate (cAMP), an intracellular secondary messenger mediating inflammatory response, are monitored by PDE4 [55, 56]. Higher cAMP levels, as a result of PDE4 inhibition, decreases pro-inflammatory cytokines involved in the regulation of many biologic responses in humans, including inflammation, apoptosis, and lipid metabolism, such as IL-23 in psoriasis, resulting in clinical improvement of psoriasis and psoriatic arthritis [57, 58]. PDE4 antagonism was recently shown to effectively treat moderate to severe AD in a topical formulation (crisaborole) [59]. PDE4 is highly increased in human AA scalp lesions [13], representing a potential therapeutic target.
Apremilast, an oral, small molecule PDE antagonist is FDA-approved for psoriasis and psoriatic arthritis, and is currently tested in AD patients. It showed good clinical and safety data in an open-label pilot study in AD patients [55, 60,61,62,63,64], as well as in clinical trials for other inflammatory and dermatological conditions [61,62,63, 65], including AA (NCT02684123). Despite its broad activation, affecting many inflammatory cells (including neutrophils and peripheral blood mononuclear cells (PBMCs): monocytes, plasmacytoid DCs, T cells, and natural killer cells), the drug is well tolerated and requires minimal monitoring [57]. In AA, apremilast showed impressive clinical and molecular efficacy in a humanized mouse model, using human scalp grafts [65], resulting in preservation, but not regrowth (due to the used model), of hair follicles and significant downregulation of inflammatory markers (IFN-γ and TNF-α) [65]. In addition, in a three-patients trial of extensive AA of individuals successfully treated with IL-23 inhibitor, PDE genes were significantly upregulated at baseline and downregulated following hair regrowth in all patients [15]. These data support a possible role for apremilast in the treatment of extensive AA, as well as potentially for topical PDE4 inhibition for AA patients with limited disease. A clinical trial with apremilast in severe AA patients will elucidate the clinical potential of oral PDE inhibition in AA patients (NCT02684123).
Abatacept
Abatacept, a fusion protein, is a selective modulator of T cell co-stimulation, composed of cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) with a portion of IgG1 (CTLA-4Ig) [66]. Abatacept blocks the potential co-stimulatory interaction with antigen-presenting cells/APC, thus eliminating the process required for full T cell activation (Fig. 1) [66, 67]. In an animal model, abatacept’s robust anti-inflammatory effect resulted in reduction of T cell proliferation, as well as reduced production of key inflammatory cytokines, such as IFN-γ, TNF-α, and IL-2 [66]. Abatacept is FDA-approved for rheumatoid arthritis and polyarticular juvenile idiopathic arthritis [67, 68], and currently tested for type 1 diabetes and various other arthropathies, including psoriatic arthritis, with generally good efficacy and safety profile [69, 70]. Interestingly, a fusion molecule harnessing the inhibitory potential of CTLA-4 binding to partially activated T cells, similarly to abatacept, was investigated in AA mouse models several years before drug release to the market, in a setting of pre- and post-AA graft transplantations. Treatment results included prevention of the induced AA clinically, with hair follicles lacking the typical lymphocyte infiltrates [71]. Owing to the key role that activated T cells play in the complex immune activation of AA, both in vitro and in vivo, abatacept may prove beneficial for human AA as well [14, 29]. An ongoing open-label single arm clinical trial will further elucidate the therapeutic potential of abatacept for AA (NCT02018042).
A Few Broad T Cell Antagonists That Are Ineffective for AA
Few broad T cell antagonists failed to prove effectiveness for AA. These include alefacept, efalizumab, and anti-tumor necrosis factor (TNF) drugs (Table 2).
Alefacept is a soluble LFA-3-Ig fusion protein that interrupts the antigen-presenting process by preventing CD4+ and CD8+ T cell from binding to antigen-presenting cells (Fig. 1) [72, 73]. Alefacept was FDA-approved for psoriasis, and showed significant reduction of circulating CD4+ memory T cells, as well as downregulation of immune markers (e.g., IFN-γ, IL-8, and IL-23) in these patients, yet low efficacy resulted in drug withdrawal from the market in 2011 [72, 74]. A trial of alefacept for AD patients also failed to demonstrate significant clinical effectiveness [73, 75]. Although alefacept showed impressive hair regrowth in a case report of a patient with AU [76], a multicenter, double-blind, randomized, placebo-controlled trial of alefacept for AA patients showed disappointing results [77].
Efalizumab is a monoclonal antibody that targets the T cell adhesion molecule, leukocyte function-associated antigen-1/LFA-1, and thus blocks multiple T cell functions, including adhesion of T cells to keratinocytes, trafficking, and activation (Fig. 1) [78, 79]. It was FDA-approved for the treatment of psoriasis and showed a satisfactory efficacy for a specific resistant-to-therapy subtype, i.e., palmoplantar psoriasis [80]. Efalizumab was withdrawn from the market in 2009 because of four cases of progressive multifocal leukoencephalopathy (PML) in psoriasis patients [80]. In 2008, a randomized, double-blind, placebo-controlled trial of AA patients treated with efalizumab with intralesional scalp injection showed no clinical efficacy for the drug in AA [79]. Moreover, a psoriasis patient with history of chronic, mild AA experienced a rapid and severe worsening of his AA while treated with efalizumab for psoriasis [81].
TNF-α has a robust pro-inflammatory effect, due to the synergistic enhancement of both Th1 and Th17/IL-23 pathways, as shown in psoriasis [27, 33], and due to the induction of cell proliferation and differentiation [33, 82]. Three TNF inhibitors are FDA-approved for psoriasis and psoriatic arthritis: infliximab, adalimumb, and etanercept [27, 83], and anti-TNFs were shown to induce short- and long-term efficacy without major safety issues in multiple clinical trials of many autoimmune conditions, including various arthropothies and inflammatory bowel disease (IBD) [84]. Studies of AA blood and skin demonstrated elevated TNF-α levels, raising the possible role of anti-TNF for AA as well [13, 85]. An anecdotal report of a patient with AU receiving anti-TNF for other indications, resulting in impressive hair regrowth [86], suggested the possible benefit of anti-TNFs in AA. Nevertheless, an open-label clinical trial failed to show clinical response of AA to etanercept, with some participants even experiencing AA worsening during treatment [87]. Furthermore, there are reports on exacerbation of AA with all three anti-TNFs mentioned above [88]. Similar to AD [89, 90], these disappointing results eliminated a possible role for these biologics in AA, and we were not able to find ongoing clinical trials on anti-TNFs for AA.
Th17/IL-23 Inhibition
IL-23 was recently established as an important driver of the immune activation in many autoimmune diseases, including psoriasis, rheumatoid arthritis, vitiligo, and IBD [91, 92]. IL-23 induces intracellular pro-inflammatory molecules (JAK2 and Tyk2) in human keratinocytes [93]. Human keratinocytes express IL-23 receptor, therefore raising the possibility of a direct IL-23 effect on the skin, as was demonstrated in mouse psoriasis models [94]. The role of IL-23 was further elucidated through clinical trials in psoriasis, where IL-23-induced Th17 activation and production promote key disease pathways, and IL-23 cytokine inhibition using ustekinumab (anti-IL-12/IL-23p40 blocker) or an anti-IL-23p19 antagonist results in impressive clinical and tissue response in psoriasis [27, 95, 96]. In AA scalp tissues both p19 and p40 subunits of IL-23 were shown to be significantly upregulated [13]. Ustekinumab, an FDA-approved human monoclonal IgG1 antibody, binds the p40 subunit shared by IL-12 and IL-23 and inhibits their function. In AA, ustekinumab showed significant hair regrowth in three AA patients with extensive hair loss, including a patient with AU [15]. Hair restoration was coupled with marked downregulation of many inflammatory markers that were upregulated at baseline in scalp biopsies, including Th1 (CXCL10)-related but particularly Th2-related markers (CCL13, CCL26), as well as PDE4. The most significant response to ustekinumab was associated with shortest duration of disease (2 years) and highest immune activation at baseline, in an AU patient who grew full scalp hair following treatment. In addition, hair keratins showed increases in post-treatment lesional scalp of all patients [15]. This report of hair regrowth with specific cytokine antagonism in severe AA, including AU, associated with suppression of inflammatory pathways and improvement in tissue keratins, raises the possible role of ustekinumab in AA, but more importantly, suggests the investigation of other, specific therapeutic modalities in AA, including Th2-specific antagonism [15]. This preliminary report possibly implies a more complicated immune fingerprint in AA, rather than the exclusive Th1/IFN-γ skewing that was traditionally attributed to the disease. Of note, some case reports speculated that ustekinumab might induce mild AA, but failed to demonstrate direct causation [97,98,99].
As detailed above, IL-23 is an important factor in Th17 cell induction and maintenance. In human psoriasis, Th17 cells are producers of various important immune mediators, including IL-6, IL-17A, IL-17F, IL-21, IL-22, and TNF-α, and are key players in disease pathogenesis, as demonstrated by the dramatic efficacy of IL-17 inhibitors [27, 100,101,102]. Recent data support the association of IL17 and IL17RA gene polymorphism with AA [103], and IL-17 serum levels were significantly elevated in AA patients [104, 105], correlating with disease severity [104]. Nevertheless, both IL-17 and IL-22 cytokines and associated markers did not show increases in AA versus normal scalp [13], questioning a pathogenic role for the Th17/Th22 immune axes in AA. Secukinumab is a recombinant, high-affinity, fully human IgG1κ that selectively inhibits IL-17A, FDA-approved for the treatment of psoriasis [101, 102, 106]. In psoriasis, inhibition of IL-17 was shown to suppress both downstream immune activation, as expected, but also some upstream inhibition of the IL-23 axis, as well as IFN-γ expression [106]. IL-17 antagonism (NCT02599129) may hold some promise for AA through its suppressing effect on both IL-17, as well as indirectly through inhibition of IL-23 activation. Future placebo-controlled clinical trials should determine whether anti-IL-23 and/or IL-17 antagonism has a role in the treatment paradigm of extensive AA.
Th2 Antagonism
AA is commonly associated with AD, which is strongly Th2-skewed, and the diseases share a similar genetic background [4, 7]. Two key cytokines of the Th2 axis, IL-13 and IL-4, were associated with AA, as IL-13 gene susceptibility and IL-4 polymorphism were both found in AA patients [107, 108]. Additionally, AA shares some phenotypic similarities with AD, such as pruritus, elevated IgE, filaggrin mutations, and molecular activation that includes Th2, IL-23, and Th1 activation [10, 13, 109, 110]. Personal or familial history of atopy, especially AD, is the highest risk factor of AA development, and concomitant AA and AD results in higher risk for severe AA, such as the AU and AT subtypes [6, 7, 9, 10]. An analysis of AA lesional skin showed that the AA transcriptome is composed of Th1/IFN-γ genes, but is also heavily Th2-polarized [13]. Moreover, a study investigating the tissue response in AA following intralesional steroid injection and regrowth of hair showed pretreatment upregulation of Th2-related markers (IL-13, CCL17, and CCL18) in lesional compared to non-lesional skin, and downregulation of Th2 products post treatment (CCL18) [16].
Dupilumab
The Th2 inhibitor dupilumab acts as a strong Th2 antagonist by blocking IL-4 receptor α (IL-4Rα), inhibiting the two pivotal Th2 cytokines, IL-4 and IL-13, that signal through the targeted receptor (Fig. 1) [111, 112]. Dupilumab showed impressive clinical and tissue responses in advanced clinical trials of AD [112, 113]. The strong association between AD and AA, the Th2-related susceptibility basis in AA [107, 108], and the dominant Th2 upregulation in AA skin scalp lesions (including IL-13, CCL18, CCL26, thymic stromal lymphopoietin, and periostin) [13] suggest the possible utility of dupilumab in AA, also enabling one to elucidate whether Th2 skewing has a pathogenic role in AA (Table 1).
Tralokinumab
Another specific blocker of the Th2 axis, which targets IL-13 alone by specifically binding and neutralizing the cytokine, is tralokinumab, an IgG4 humanized monoclonal antibody. A structural characterization showed that tralokinumab inhibits the cytokine by binding to its heterodimeric receptor, composed of IL-4Rα (targeted by dupilumab) and IL-13Rα1 [114], as well as to its decoy receptor, IL-13Rα2 (Fig. 1) [115]. Tralokinumab is in clinical trials for asthma, idiopathic pulmonary fibrosis (IPF), IBD, AD, and AA (NCT02684097, Table 1). In asthma, tralokimunab showed the highest efficacy in a subset of asthma patients characterized by the highest sputum IL-13 levels, with an overall good safety profile [116, 117]. The clinical trial of tralokinumab for AA might shed light on the role of Th2, and specifically IL-13 in the AA inflammatory process.
Conclusions
The clinical practice of dermatology was dramatically revolutionized in recent years, with a new era of pathogenesis-centered therapeutics, a result of the rapidly progressing translational research of inflammatory dermatologic diseases [27]. This paradigm shift was first implemented in psoriasis, with multiple targeted treatments introduced in the last decade, resulting in outstanding clinical responses [28]. Next to follow is AD, for which targeted agents are showing excellent efficacy, and will soon be commercially available [111, 112]. Many other clinical trials will further change the current treatment paradigm of moderate to severe AD [34]. Similar to psoriasis prior to the introduction of the biologics, the therapeutic armamentarium for AA is insufficient. A complex immune signature, with animal models and human AA showing Th1/IFN-γ, Th2, IL-9, PDE4, and Th17/IL-23 skewing, characterizes AA [13, 14, 65]. New, broad-acting drugs, such as JAK inhibitors [14, 32, 44,45,46,47], are holding promise for AA; yet surprisingly, cytokine-specific agents such as ustekinumab also showed clinical and tissue response in AA [14, 15, 32]. The latter will also be able to better dissect the contribution of single cytokines to disease pathogenesis. Clinical trials of broad-acting T cell antagonists (JAK inhibitors, PDE4 inhibitor), Th2 antagonists (tralokinumab), and IL-23/IL-17 antagonist (ustekinumab) are ongoing. Future clinical trials of IL-9 antagonists may also hold some promise [13].
Following psoriasis and AD, AA may represent the next inflammatory skin disease to undergo a dramatic alteration in its therapeutic paradigm, turning to disease-specific drugs with better efficacy and safety profile.
References
Tosti A, Bellavista S, Iorizzo M. Alopecia areata: a long term follow-up study of 191 patients. J Am Acad Dermatol. 2006;55(3):438–41
Safavi KH, Muller SA, Suman VJ, Moshell AN, Melton LJ 3rd. Incidence of alopecia areata in Olmsted County, Minnesota, 1975 through 1989. Mayo Clin Proc. 1995;70(7):628–33.
Vestey JP, Savin JA. Natural history of severe alopecia areata. Br J Dermatol. 1987;117(4):531.
Gilhar A, Etzioni A, Paus R. Alopecia areata. N Engl J Med. 2012;366(16):1515–25.
Huang KP, Mullangi S, Guo Y, Qureshi AA. Autoimmune, atopic, and mental health comorbid conditions associated with alopecia areata in the United States. JAMA Dermatol. 2013;149(7):789–94.
Lee NR, Kim BK, Yoon NY, Lee SY, Ahn SY, Lee WS. Differences in comorbidity profiles between early-onset and late-onset alopecia areata patients: a retrospective study of 871 Korean patients. Ann Dermatol. 2014;26(6):722–6.
Chu SY, Chen YJ, Tseng WC, et al. Comorbidity profiles among patients with alopecia areata: the importance of onset age, a nationwide population-based study. J Am Acad Dermatol. 2011;65(5):949–56.
Drucker AM, Thompson JM, Li WQ, et al. Incident alopecia areata and vitiligo in adult women with atopic dermatitis: Nurses’ Health Study 2. Allergy. 2017;72(5):831–4.
Barahmani N, Schabath MB, Duvic M, National Alopecia Areata Registry. History of atopy or autoimmunity increases risk of alopecia areata. J Am Acad Dermatol. 2009;61(4):581–91.
Betz RC, Pforr J, Flaquer A, et al. Loss-of-function mutations in the filaggrin gene and alopecia areata: strong risk factor for a severe course of disease in patients comorbid for atopic disease. J Invest Dermatol. 2007;127(11):2539–43.
Gulec AT, Tanriverdi N, Duru C, Saray Y, Akcali C. The role of psychological factors in alopecia areata and the impact of the disease on the quality of life. Int J Dermatol. 2004;43(5):352–6.
Olsen EA, Hordinsky MK, Price VH, et al. Alopecia areata investigational assessment guidelines–Part II. National Alopecia Areata Foundation. J Am Acad Dermatol. 2004;51(3):440–7.
Suarez-Farinas M, Ungar B, Noda S, et al. Alopecia areata profiling shows TH1, TH2, and IL-23 cytokine activation without parallel TH17/TH22 skewing. J Allergy Clin Immunol. 2015;136(5):1277–87.
Xing L, Dai Z, Jabbari A, et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat Med. 2014;20(9):1043–9.
Guttman-Yassky E, Ungar B, Noda S, et al. Extensive alopecia areata is reversed by IL-12/IL-23p40 cytokine antagonism. J Allergy Clin Immunol. 2016;137(1):301–4.
Fuentes-Duculan J, Gulati N, Bonifacio KM, et al. Biomarkers of alopecia areata disease activity and response to corticosteroid treatment. Exp Dermatol. 2016;25(4):282–6.
Shapiro J. Current treatment of alopecia areata. J Investig Dermatol Symp Proc. 2013;16(1):S42–4.
Delamere FM, Sladden MM, Dobbins HM, Leonardi-Bee J. Interventions for alopecia areata. Cochrane Database Syst Rev. 2008(2):CD004413.
Schmoeckel C, Weissmann I, Plewig G, Braun-Falco O. Treatment of alopecia areata by anthralin-induced dermatitis. Arch Dermatol. 1979;115(10):1254–5.
Shapiro J. Dermatologic therapy: alopecia areata update. Dermatol Ther. 2011;24(3):301.
Shapiro J. Alopecia areata. Update on therapy. Dermatol Clin. 1993;11(1):35–46.
MacDonald Hull SP, Wood ML, Hutchinson PE, Sladden M, Messenger AG, British Association of Dermatologists. Guidelines for the management of alopecia areata. Br J Dermatol. 2003;149(4):692–9.
Park KY, Jang WS, Son IP, et al. Combination therapy with cyclosporine and psoralen plus ultraviolet a in the patients with severe alopecia areata: a retrospective study with a self-controlled design. Ann Dermatol. 2013;25(1):12–6.
Olsen EA, Carson SC, Turney EA. Systemic steroids with or without 2% topical minoxidil in the treatment of alopecia areata. Arch Dermatol. 1992;128(11):1467–73.
Farshi S, Mansouri P, Safar F, Khiabanloo SR. Could azathioprine be considered as a therapeutic alternative in the treatment of alopecia areata? A pilot study. Int J Dermatol. 2010;49(10):1188–93.
Shreberk-Hassidim R, Ramot Y, Gilula Z, Zlotogorski A. A systematic review of pulse steroid therapy for alopecia areata. J Am Acad Dermatol. 2016;74(2):372–4 e1–5.
Noda S, Krueger JG, Guttman-Yassky E. The translational revolution and use of biologics in patients with inflammatory skin diseases. J Allergy Clin Immunol. 2015;135(2):324–36.
Nograles KE, Krueger JG. Anti-cytokine therapies for psoriasis. Exp Cell Res. 2011;317(9):1293–300.
Gilhar A, Ullmann Y, Berkutzki T, Assy B, Kalish RS. Autoimmune hair loss (alopecia areata) transferred by T lymphocytes to human scalp explants on SCID mice. J Clin Invest. 1998;101(1):62–7.
Ito T, Ito N, Saatoff M, Hashizume H, Fukamizu H, Nickoloff BJ, et al. Maintenance of hair follicle immune privilege is linked to prevention of NK cell attack. J Invest Dermatol. 2008;128(5):1196–206.
Hoffmann R, Happle R, Paus R. Elements of the interleukin-1 signaling system show hair cycle-dependent gene expression in murine skin. Eur J Dermatol. 1998;8(7):475–7.
Jabbari A, Dai Z, Xing L, Cerise JE, Ramot Y, Berkun Y, et al. Reversal of alopecia areata following treatment with the JAK1/2 inhibitor baricitinib. EBioMedicine. 2015;2(4):351–5.
Zaba LC, Suarez-Farinas M, Fuentes-Duculan J, et al. Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J Allergy Clin Immunol. 2009;124(5):1022–10 e1-395.
Oliva M, Renert-Yuval Y, Guttman-Yassky E. The ‘omics’ revolution: redefining the understanding and treatment of allergic skin diseases. Curr Opin Allergy Clin Immunol. 2016;16(5):469–76.
Malajian D, Guttman-Yassky E. New pathogenic and therapeutic paradigms in atopic dermatitis. Cytokine. 2015;73(2):311–8.
Williams NK, Bamert RS, Patel O, et al. Dissecting specificity in the Janus kinases: the structures of JAK-specific inhibitors complexed to the JAK1 and JAK2 protein tyrosine kinase domains. J Mol Biol. 2009;387(1):219–32.
Ghoreschi K, Gadina M. Jakpot! New small molecules in autoimmune and inflammatory diseases. Exp Dermatol. 2014;23(1):7–11.
Flanagan ME, Blumenkopf TA, Brissette WH, et al. Discovery of CP-690,550: a potent and selective Janus kinase (JAK) inhibitor for the treatment of autoimmune diseases and organ transplant rejection. J Med Chem. 2010;53(24):8468–84.
Papp K, Menter MA, Raman M, et al. A randomized phase 2b trial of baricitinib, an oral JAK1/JAK2 inhibitor, in patients with moderate-to-severe psoriasis. Br J Dermatol. 2016;174(6):1266–76.
Waldmann TA. The biology of IL-15: implications for cancer therapy and the treatment of autoimmune disorders. J Investig Dermatol Symp Proc. 2013;16(1):S28–30.
Ishizaki M, Akimoto T, Muromoto R, et al. Involvement of tyrosine kinase-2 in both the IL-12/Th1 and IL-23/Th17 axes in vivo. J Immunol. 2011;187(1):181–9.
Petukhova L, Duvic M, Hordinsky M, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature. 2010;466(7302):113–7.
Alves de Medeiros AK, Speeckaert R, Desmet E, Van Gele M, De Schepper S, Lambert J. JAK3 as an emerging target for topical treatment of inflammatory skin diseases. PLoS One. 2016;11(10):e0164080.
Harris JE, Rashighi M, Nguyen N, et al. Rapid skin repigmentation on oral ruxolitinib in a patient with coexistent vitiligo and alopecia areata (AA). J Am Acad Dermatol. 2016;74(2):370–1.
Craiglow BG, King BA. Killing two birds with one stone: oral tofacitinib reverses alopecia universalis in a patient with plaque psoriasis. J Invest Dermatol. 2014;134(12):2988–90.
Kennedy Crispin M, Ko JM, Craiglow BG, et al. Safety and efficacy of the JAK inhibitor tofacitinib citrate in patients with alopecia areata. JCI Insight. 2016;1(15):e89776.
Mackay-Wiggan J, Jabbari A, Nguyen N, et al. Oral ruxolitinib induces hair regrowth in patients with moderate-to-severe alopecia areata. JCI Insight. 2016;1(15):e89790.
Shi JG, Chen X, Lee F, et al. The pharmacokinetics, pharmacodynamics, and safety of baricitinib, an oral JAK 1/2 inhibitor, in healthy volunteers. J Clin Pharmacol. 2014;54(12):1354–61.
Liu LY, Craiglow BG, Dai F, King BA. Tofacitinib for the treatment of severe alopecia areata and variants: a study of 90 patients. J Am Acad Dermatol. 2017;76(1):22–8.
Ferreira SB, Scheinberg M, Steiner D, Steiner T, Bedin GL, Ferreira RB. Remarkable improvement of nail changes in alopecia areata universalis with 10 months of treatment with tofacitinib: a case report. Case Rep Dermatol. 2016;8(3):262–6.
Craiglow BG, Liu LY, King BA. Tofacitinib for the treatment of alopecia areata and variants in adolescents. J Am Acad Dermatol. 2017;76(1):29–32.
Bissonnette R, Papp KA, Poulin Y, et al. Topical tofacitinib for atopic dermatitis: a phase IIa randomized trial. Br J Dermatol. 2016;175(5):902–11.
Fukuyama T, Ehling S, Cook E, Baumer W. Topically administered Janus-kinase inhibitors tofacitinib and oclacitinib display impressive antipruritic and anti-inflammatory responses in a model of allergic dermatitis. J Pharmacol Exp Ther. 2015;354(3):394–405.
Ports WC, Khan S, Lan S, et al. A randomized phase 2a efficacy and safety trial of the topical Janus kinase inhibitor tofacitinib in the treatment of chronic plaque psoriasis. Br J Dermatol. 2013;169(1):137–45.
Samrao A, Berry TM, Goreshi R, Simpson EL. A pilot study of an oral phosphodiesterase inhibitor (apremilast) for atopic dermatitis in adults. Arch Dermatol. 2012;148(8):890–7.
Palfreeman AC, McNamee KE, McCann FE. New developments in the management of psoriasis and psoriatic arthritis: a focus on apremilast. Drug Des Devel Ther. 2013;7:201–10.
Schafer PH, Parton A, Capone L, et al. Apremilast is a selective PDE4 inhibitor with regulatory effects on innate immunity. Cell Signal. 2014;26(9):2016–29.
Schafer P. Apremilast mechanism of action and application to psoriasis and psoriatic arthritis. Biochem Pharmacol. 2012;83(12):1583–90.
Paller AS, Tom WL, Lebwohl MG, et al. Efficacy and safety of crisaborole ointment, a novel, nonsteroidal phosphodiesterase 4 (PDE4) inhibitor for the topical treatment of atopic dermatitis (AD) in children and adults. J Am Acad Dermatol. 2016;75(3):494–503 e4.
Edwards CJ, Blanco FJ, Crowley J, et al. Apremilast, an oral phosphodiesterase 4 inhibitor, in patients with psoriatic arthritis and current skin involvement: a phase III, randomised, controlled trial (PALACE 3). Ann Rheum Dis. 2016;75(6):1065–73.
Hatemi G, Melikoglu M, Tunc R, et al. Apremilast for Behcet’s syndrome—a phase 2, placebo-controlled study. N Engl J Med. 2015;372(16):1510–8.
Pathan E, Abraham S, Van Rossen E, et al. Efficacy and safety of apremilast, an oral phosphodiesterase 4 inhibitor, in ankylosing spondylitis. Ann Rheum Dis. 2013;72(9):1475–80.
Paul J, Foss CE, Hirano SA, Cunningham TD, Pariser DM. An open-label pilot study of apremilast for the treatment of moderate to severe lichen planus: a case series. J Am Acad Dermatol. 2013;68(2):255–61.
Krase IZ, Cavanaugh K, Curiel-Lewandrowski C. Treatment of refractory pityriasis rubra pilaris with novel phosphodiesterase 4 (PDE4) inhibitor apremilast. JAMA Dermatol. 2016;152(3):348–50.
Keren A, Shemer A, Ullmann Y, Paus R, Gilhar A. The PDE4 inhibitor, apremilast, suppresses experimentally induced alopecia areata in human skin in vivo. J Dermatol Sci. 2015;77(1):74–6.
Moreland L, Bate G, Kirkpatrick P. Abatacept. Nat Rev Drug Discov. 2006;5(3):185–6.
Lovell DJ, Ruperto N, Mouy R, et al. Long-term safety, efficacy, and quality of life in patients with juvenile idiopathic arthritis treated with intravenous abatacept for up to seven years. Arthritis Rheumatol. 2015;67(10):2759–70.
Piantoni S, Colombo E, Tincani A, Airo P, Scarsi M. Predictive factors of abatacept therapy discontinuation in patients with rheumatoid arthritis. Clin Rheumatol. 2016;35(4):1065–9.
Caso F, Del Puente A, Peluso R, et al. Emerging drugs for psoriatic arthritis. Expert Opin Emerg Drugs. 2016;21(1):69–79.
Orban T, Bundy B, Becker DJ, et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet. 2011;378(9789):412–9.
Carroll JM, McElwee KJ, King LE, Byrne MC, Sundberg JP. Gene array profiling and immunomodulation studies define a cell-mediated immune response underlying the pathogenesis of alopecia areata in a mouse model and humans. J Invest Dermatol. 2002;119(2):392–402.
Chamian F, Lowes MA, Lin SL, et al. Alefacept reduces infiltrating T cells, activated dendritic cells, and inflammatory genes in psoriasis vulgaris. Proc Natl Acad Sci USA. 2005;102(6):2075–80.
Simon D, Wittwer J, Kostylina G, Buettiker U, Simon HU, Yawalkar N. Alefacept (lymphocyte function-associated molecule 3/IgG fusion protein) treatment for atopic eczema. J Allergy Clin Immunol. 2008;122(2):423–4.
Lin VW, Ringold S, Devine EB. Comparison of ustekinumab with other biological agents for the treatment of moderate to severe plaque psoriasis: a Bayesian network meta-analysis. Arch Dermatol. 2012;148(12):1403–10.
Moul DK, Routhouska SB, Robinson MR, Korman NJ. Alefacept for moderate to severe atopic dermatitis: a pilot study in adults. J Am Acad Dermatol. 2008;58(6):984–9.
Bui K, Polisetty S, Gilchrist H, Jackson SM, Frederic J. Successful treatment of alopecia universalis with alefacept: a case report and review of the literature. Cutis. 2008;81(5):431–4.
Strober BE, Menon K, McMichael A, et al. Alefacept for severe alopecia areata: a randomized, double-blind, placebo-controlled study. Arch Dermatol. 2009;145(11):1262–6.
Kothary N, Diak IL, Brinker A, Bezabeh S, Avigan M, Dal Pan G. Progressive multifocal leukoencephalopathy associated with efalizumab use in psoriasis patients. J Am Acad Dermatol. 2011;65(3):546–51.
Price VH, Hordinsky MK, Olsen EA, et al. Subcutaneous efalizumab is not effective in the treatment of alopecia areata. J Am Acad Dermatol. 2008;58(3):395–402.
Prater EF, Day A, Patel M, Menter A. A retrospective analysis of 72 patients on prior efalizumab subsequent to the time of voluntary market withdrawal in 2009. J Drugs Dermatol. 2014;13(6):712–8.
Tosti A, Pazzaglia M, Starace M, Bellavista S, Vincenzi C, Tonelli G. Alopecia areata during treatment with biologic agents. Arch Dermatol. 2006;142(12):1653–4.
Rabeony H, Petit-Paris I, Garnier J, et al. Inhibition of keratinocyte differentiation by the synergistic effect of IL-17A, IL-22, IL-1alpha, TNFalpha and oncostatin M. PLoS One. 2014;9(7):e101937.
Gottlieb AB, Chamian F, Masud S, et al. TNF inhibition rapidly down-regulates multiple proinflammatory pathways in psoriasis plaques. J Immunol. 2005;175(4):2721–9.
Lawson MM, Thomas AG, Akobeng AK. Tumour necrosis factor alpha blocking agents for induction of remission in ulcerative colitis. Cochrane Database Syst Rev. 2006;(3):CD005112.
Kasumagic-Halilovic E, Prohic A, Cavaljuga S. Tumor necrosis factor-alpha in patients with alopecia areata. Indian J Dermatol. 2011;56(5):494–6.
Gorcey L, Gordon Spratt EA, Leger MC. Alopecia universalis successfully treated with adalimumab. JAMA Dermatol. 2014;150(12):1341–4.
Strober BE, Siu K, Alexis AF, et al. Etanercept does not effectively treat moderate to severe alopecia areata: an open-label study. J Am Acad Dermatol. 2005;52(6):1082–4.
Tauber M, Buche S, Reygagne P, et al. Alopecia areata occurring during anti-TNF therapy: a national multicenter prospective study. J Am Acad Dermatol. 2014;70(6):1146–9.
Mangge H, Gindl S, Kenzian H, Schauenstein K. Atopic dermatitis as a side effect of anti-tumor necrosis factor-alpha therapy. J Rheumatol. 2003;30(11):2506–7.
Chan JL, Davis-Reed L, Kimball AB. Counter-regulatory balance: atopic dermatitis in patients undergoing infliximab infusion therapy. J Drugs Dermatol. 2004;3(3):315–8.
Vaccaro M, Cannavo SP, Imbesi S, et al. Increased serum levels of interleukin-23 circulating in patients with non-segmental generalized vitiligo. Int J Dermatol. 2015;54(6):672–4.
Duvallet E, Semerano L, Assier E, Falgarone G, Boissier MC. Interleukin-23: a key cytokine in inflammatory diseases. Ann Med. 2011;43(7):503–11.
Kanda N, Watanabe S. IL-12, IL-23, and IL-27 enhance human beta-defensin-2 production in human keratinocytes. Eur J Immunol. 2008;38(5):1287–96.
Chan JR, Blumenschein W, Murphy E, et al. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med. 2006;203(12):2577–87.
Papp KA, Langley RG, Lebwohl M, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet. 2008;371(9625):1675–84.
Krueger JG, Ferris LK, Menter A, et al. Anti-IL-23A mAb BI 655066 for treatment of moderate-to-severe psoriasis: safety, efficacy, pharmacokinetics, and biomarker results of a single-rising-dose, randomized, double-blind, placebo-controlled trial. J Allergy Clin Immunol. 2015;136(1):116–24 e7.
Verros C, Rallis E, Crowe M. Letter: alopecia areata during ustekinumab administration: co-existence or an adverse reaction? Dermatol Online J. 2012;18(7):14.
Tauber M, Beneton N, Reygagne P, Bachelez H, Viguier M. Alopecia areata developing during ustekinumab therapy: report of two cases. Eur J Dermatol. 2013;23(6):912–3.
Slowinska M, Kardynal A, Warszawik O, Czuwara J, Rudnicka L. Alopecia areata developing paralell to improvement of psoriasis during ustekinumab therapy. J Dermatol Case Rep. 2010;4(1):15–7.
Krueger JG, Fretzin S, Suarez-Farinas M, et al. IL-17A is essential for cell activation and inflammatory gene circuits in subjects with psoriasis. J Allergy Clin Immunol. 2012;130(1):145–54 e9.
Martin DA, Towne JE, Kricorian G, et al. The emerging role of IL-17 in the pathogenesis of psoriasis: preclinical and clinical findings. J Invest Dermatol. 2013;133(1):17–26.
Langley RG, Elewski BE, Lebwohl M, et al. Secukinumab in plaque psoriasis—results of two phase 3 trials. N Engl J Med. 2014;371(4):326–38.
Lew BL, Cho HR, Haw S, Kim HJ, Chung JH, Sim WY. Association between IL17A/IL17RA gene polymorphisms and susceptibility to alopecia areata in the Korean population. Ann Dermatol. 2012;24(1):61–5.
Atwa MA, Youssef N, Bayoumy NM. T-helper 17 cytokines (interleukins 17, 21, 22, and 6, and tumor necrosis factor-alpha) in patients with alopecia areata: association with clinical type and severity. Int J Dermatol. 2016;55(6):666–72.
Elela MA, Gawdat HI, Hegazy RA, et al. B cell activating factor and T-helper 17 cells: possible synergistic culprits in the pathogenesis of alopecia areata. Arch Dermatol Res. 2016;308(2):115–21.
Chiricozzi A, Krueger JG. IL-17 targeted therapies for psoriasis. Expert Opin Investig Drugs. 2013;22(8):993–1005.
Jagielska D, Redler S, Brockschmidt FF, et al. Follow-up study of the first genome-wide association scan in alopecia areata: IL13 and KIAA0350 as susceptibility loci supported with genome-wide significance. J Invest Dermatol. 2012;132(9):2192–7.
Kalkan G, Karakus N, Bas Y, Takci Z, Ozuguz P, Ates O, et al. The association between interleukin (IL)-4 gene intron 3 VNTR polymorphism and alopecia areata (AA) in Turkish population. Gene. 2013;527(2):565–9.
Bakry OA, El Shazly RM, Basha MA, Mostafa H. Total serum immunoglobulin E in patients with alopecia areata. Indian Dermatol Online J. 2014;5(2):122–7.
Finner AM. Alopecia areata: clinical presentation, diagnosis, and unusual cases. Dermatol Ther. 2011;24(3):348–54.
Hamilton JD, Suarez-Farinas M, Dhingra N, et al. Dupilumab improves the molecular signature in skin of patients with moderate-to-severe atopic dermatitis. J Allergy Clin Immunol. 2014;134(6):1293–300.
Hamilton JD, Ungar B, Guttman-Yassky E. Drug evaluation review: dupilumab in atopic dermatitis. Immunotherapy. 2015;7(10):1043–58.
Beck LA, Thaci D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371(2):130–9.
Hussein YM, Ahmad AS, Ibrahem MM, et al. Interleukin 13 receptors as biochemical markers in atopic patients. J Investig Allergol Clin Immunol. 2011;21(2):101–7.
Popovic B, Breed J, Rees DG, et al. Structural characterisation reveals mechanism of IL-13-neutralising monoclonal antibody tralokinumab as inhibition of binding to IL-13Ralpha1 and IL-13Ralpha2. J Mol Biol. 2017;429(2):208–19.
Danese S, Rudzinski J, Brandt W, et al. Tralokinumab for moderate-to-severe UC: a randomised, double-blind, placebo-controlled, phase IIa study. Gut. 2015;64(2):243–9.
Piper E, Brightling C, Niven R, et al. A phase II placebo-controlled study of tralokinumab in moderate-to-severe asthma. Eur Respir J. 2013;41(2):330–8.
Renert-Yuval Y, Guttman-Yassky E. A novel therapeutic paradigm for patients with extensive alopecia areata. Expert Opin Biol Ther. 2016;16(8):1005–14.
Acknowledgements
No funding or sponsorship was received for this study or publication of this article. The article processing charges were funded by the authors. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Disclosures
Emma Guttman-Yassky is a board member for Sanofi Aventis, Regeneron, Stiefel/GlaxoSmithKline, MedImmune, Celgene, Anacor, AnaptysBio, Celsus, Dermira, Galderma, Glenmark, Novartis, Pfizer, Vitae, and Leo Pharma; has received consultancy fees from Regeneron, Sanofi, MedImmune, Celgene, Stiefel/GlaxoSmithKline, Celsus, BMS, Amgen, Drais, AbbVie, Anacor, AnaptysBio, Dermira, Galderma, Glenmark, LEO Pharma, Novartis, Pfizer, Vitae, Mitsubishi Tanabe, and Eli Lilly; and has received research support from Janssen, Regeneron, Celgene, BMS, Novartis, Merck, LEO Pharma, and Dermira. Yael Renert-Yuval has nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to http://www.medengine.com/Redeem/9818F060128D216F.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Renert-Yuval, Y., Guttman-Yassky, E. The Changing Landscape of Alopecia Areata: The Therapeutic Paradigm. Adv Ther 34, 1594–1609 (2017). https://doi.org/10.1007/s12325-017-0542-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-017-0542-7