
Abstract
Glioma is the most common and lethal intrinsic primary tumor of the brain. Its controversial origins may contribute to its heterogeneity, creating challenges and difficulties in the development of therapies. Among the components constituting tumors, glioma stem cells are highly plastic subpopulations that are thought to be the site of tumor initiation. Neural stem cells/progenitor cells and oligodendrocyte progenitor cells are possible lineage groups populating the bulk of the tumor, in which gene mutations related to cell-cycle or metabolic enzymes dramatically affect this transformation. Novel approaches have revealed the tumor-promoting properties of distinct tumor cell states, glial, neural, and immune cell populations in the tumor microenvironment. Communication between tumor cells and other normal cells manipulate tumor progression and influence sensitivity to therapy. Here, we discuss the heterogeneity and relevant functions of tumor cell state, microglia, monocyte-derived macrophages, and neurons in glioma, highlighting their bilateral effects on tumors. Finally, we describe potential therapeutic approaches and targets beyond standard treatments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gliomas, traditionally named due to their close resemblance to glial cells, are the most frequent intrinsic primary tumors of the brain [1,2,3]. Different from other oncological diseases that benefit from multimodal therapy, limited progress has been made in the management of gliomas [4, 5]. Therefore, ongoing efforts to understand their highly heterogeneous nature and complicated reciprocal microenvironmental communication have been undertaken [6, 7]. Among their forms, diffuse gliomas, which have an unfavorable prognosis and high morbidity in adult patients, have been historically diagnosed as one of three categories outlined in the 2016 WHO central nervous system (CNS) classification [8, 9]: oligodendroglioma, astrocytoma, or glioblastoma (GBM). These subtypes share several molecular features and functional characteristics with their normal counterparts. Recent profiling efforts have identified subclassifications of diffuse gliomas by integrating histopathological analysis and genetic events [10]. Importantly, isocitrate dehydrogenase (IDH) status and chromosome 1p/19q co-deletion [11], have been identified as predictive genetic landmarks of favorable outcomes and have had a profound impact on treatment strategies and the design of clinical trials [12]. In particular, robust biomarkers have also been described by Eckel-Passow et al., who classify gliomas into five principal groups with prognostic significance. Of these, the triple-negative gliomas (no mutations in IDH and TERT plus a 1p/19q non-codeletion) were the most prevalent in a Chinese cohort [13, 14]. Mutations of TP53 and H3.3-K27M in triple-negative gliomas implicated an unfavorable prognosis [14]. Notably, the fifth edition of the WHO CNS (2021 WHO CNS5) grouped gliomas according to these genetic changes to enable a complete diagnosis [15]. Other molecular signatures, such as cell-cycle regulatory elements (CDKN2A/B) and epidermal growth factor receptor (EGFR), have also contributed to the illustration of oncogenic pathways. Progress in genomics has validated diverse genetic alterations harbored in diffuse gliomas, rendering glioma cells distinct from one another. The expression patterns of genetic mutations suggest that astrocytomas and oligodendrogliomas originate from abnormal glial progenitors or stem cells. These findings have led to the hypothesis that the cellular heterogeneity of gliomas is affected by the glial developmental process, intercellular signaling, and microenvironment stress. This review discusses new advances in oncogenic glial lineage, and reciprocal interactions in gliomas (i.e. with neurons and microglia), offering new insights into the potential development of effective treatments.
Glioma Origin: From Neurogenesis to Oncogenesis
Glioma Stem Cells
Among the components constituting tumors, glioma stem cells (GSCs) are highly plastic subpopulations bearing stemness properties and are thought of as the site of tumor initiation. Similar to neural stem cells (NSCs), GSCs have the ability to self-renew, differentiate and resist DNA damage [16,17,18]. A series of biomarkers have been identified in GSC populations: CD133 (PROM1), SOX2 (a transcription factor widely expressed in potent stem cells), OCT-4 (a transcription factor that plays an essential role in stem cell pluripotency), and Nestin (an intermediate filament protein). Several studies have shown that the expression of these molecular markers is closely associated with pluripotency and stemness in gliomas. By intracranial grafting as few as 100 CD133+ cells, tumors have been effectively produced and resembled the phenotype of the original tumor type, whereas no transplanted tumor was observed after injection of 105 CD133– cells [19]. Ablation of Nestin+ stem-like cells was not able to halt tumor progression, indicating the involvement of other factors [20]. CD133+ Notch1+ GSCs have also been reported to be located at the frontier of invasive tissues, exhibiting white-matter-tract tropism. The positive-feedback loop involving Notch-SOX2 controls the invasive phenotype of GSCs along white matter tracts [21]. The stem-cell activity of CD133+ cells has also been found in medulloblastomas, pilocytic astrocytomas, and gangliogliomas. Higher tumor grade is correlated with an increased fraction of CD133+ cells in tumor cultures [20]. In addition, the non-GSC population is induced to a newly converted GSC-like state after treatment with chemotherapeutic agents (e.g., temozolomide), and has a more invasive phenotype with higher implantation efficacy [22]. These findings focus attention on the cellular state of GSCs in gliomas. Lin et al. described a single axis of gene signatures in proliferating GBM cells, ranging from proneural GSCs to mesenchymal GSCs. Lineage tracing in silico supports the idea that mGSCs, which correlate with poor predicted survival, are the progenitors of pGSCs in IDH wild-type GBM [23]. Via enriching GSCs from primary GBM specimens, Richards et al. found that GSCs exist in two cellular states from the perspective of transcriptional programs: developmental and injury-response programs [24]. The astrocyte maturation gradient in tumor cells has also been implicated in the transformation of GSCs, which comprise the bulk of the tumor. Thus, understanding the evolution and differentiation of GSCs is essential for developing effective targeting therapies and identifying the source of heterogeneity in gliomas.
Neural Stem/Progenitor Cells
Different from abnormal glioma stem cells that populate GBM, neural stem cells/progenitor cells (NSCs/NPCs) are the natural starting point for neuron/glial lineage development, and are highly regulated in the brain. It is essential to understand the tumorigenesis process and decipher the mechanisms through which glial developmental programs are used by tumor cells to populate the tumor. The largest NSC niches are located along the remote region of the lateral ventricles, named the subventricular zone (SVZ). These NSCs are relatively quiescent, maintain their stemness properties, and generate NPCs independent of the specific microenvironment around the perivascular niches. This complex microenvironment is composed of NPCs, oligodendrocyte progenitor cells (OPCs), astrocytes, microglia, macrophages, neurons, associated vasculature, and extracellular matrix. Interestingly, some typical markers of NSCs have been identified in GSCs such as Nestin, Sox2, CD44, and CD133 (Fig. 1) [25,26,27]. The striking similarities between NSCs and GSCs support the hypothesis that SVZ NPSCs play the role of apex cells in the hierarchy of gliomas. Chen et al. used a fluorescent reporter to label quiescent NSCs in the adult SVZ, and revealed the presence of neural stem-like cells in glioma tissue [28]. Deep genomic sequencing of a GBM patient cohort provided direct evidence for the hypothesis that astrocyte-like NSCs in the SVZ are the origin of GBM. More than 80% of patients diagnosed with GBM have tumor-free SVZ tissue that shares low-level driver mutations or cancer-driving genes with tumor samples [29]. Migration of astrocyte-like NSCs contributes to the generation of malignant gliomas in distinct regions of the brain [29]. Accordingly, genetically-engineered mouse models (GEMMs) are powerful tools for use in deciphering the lineage complexity of glial cells, and may reveal associations between progenitor cells and the broad spectrum of neoplasms throughout the brain. Parada et al. induced resultant malignant astrocytoma via early inactivation of Tp53 and Nf1 in mice [30], and demonstrated that manipulation of tumor suppressors (Nf1, Tp53, and Pten) in NSCs and NPCs in vivo is both necessary and sufficient for the formation of astrocytomas (Fig. 1). A recent study also showed that the histological and transcriptional heterogeneity of GBM is similar to genome-edited NSC-like cells such as sgTP53/NF1/PTEN or sgTP53/NF1 in human pluripotent stem cells [31]. High-grade gliomas exhibit inactivation of p16INK4a/p19ARF and activation of epidermal growth factor receptor (EGFR). Previous findings reported that co-deletion of p16INK4a/p19ARF in NSCs with constitutive EGFR activation induce the phenotype of high-grade glioma [32]. However, no evidence of tumor formation was reported after targeting these GBM-relevant tumor suppressors in neuroblasts, late-stage neuronal progenitors, and differentiated neurons [33]. Jacques et al. reported that deletion of tumor suppressors genes (TP53 and PTEN) in adult SVZ stem cells, but not astrocytes, gives rise to tumors. These studies imply that an increase in lineage restriction decreases the tumorigenic capacity of neuronal lineage cells [34]. Driving neuronal lineage differentiation is a potent antitumorigenic treatment strategy for GBM.

Glioma origin hypothesis. Left, schematic of the normal neurogenesis process in the brain. Neural stem cells differentiate into several types of progenitor cell, which can transform into neurons, astrocytes, and oligodendrocytes. Right, schematic of the potential oncogenesis process in the brain. Glioma-stem cells, which populate adult-type diffuse gliomas, are labeled with several reported biomarkers. Gliomas produce oncometabolites in the tumor microenvironment, which correspondingly stimulate their progression. The dashed line between neurogenesis and oncogenesis represents the reprogrammed molecular mechanisms that have been previously reported. NSCs, Neural stem cells; GSCs, Glioma stem cells, NBs, Neuroblasts; APCs, Astrocyte progenitor cells; OPCs, Oligodendrocyte progenitor cells.
Other Glial Lineage States
Combining single-cell sequencing (scRNA-seq) with advanced computational algorithms allows researchers to comprehensively analyze cellular states across tumors [35, 36]. Four cellular states, three of which are anchored in neurodevelopment, are found in diverse malignant cells of glioblastoma: OPC-like, NPC-like, astrocyte (AC)-like, and mesenchymal (MES)-like. These cellular states have the potential for tumor plasticity and are influenced by genetic drivers, providing an understanding of the heterogeneity and therapeutic resistance of GBM. It is well-known that tumor phenotype transitions are derived from stage-specific fate-switches and transcriptional alterations in progenitor cells. Newly-generated cells including ACs and oligodendrocytes, which are considered to be the two broad categories of CNS glia, are continuously produced in the subgranular zone and ventricular SVZ [37]. Both oligodendrocytes and ACs perform a variety of functions for maintaining CNS homeostasis.
Although the precursors of ACs have not been clearly defined, some ACs can reenter the cell-cycle following traumatic brain injury [38]. A recent study reported that ASCL1+EGFR+ apical multipotent intermediate progenitor cells generated by cortical radial glial cells in the SVZ and VZ can differentiate into glial cells and olfactory bulb (OB) interneurons. Those progenitor cells transform into AC-lineage restricted progenitor cells in late embryogenesis in mice [39]. Injection of ACs carrying oncogenes leads to the genesis of malignant gliomas. Furthermore, all tumors expressed markers expected in astrocytomas, such as Gfap [32, 40,41,42]. Combinations of deletions of Pten, Tp53, and Rb1 in ACs in mature mice result in the progress of astrocytomas from grade III to grade IV [43]. Applying fluorescence-activated cell sorting–based strategy, a recent study reported five distinct AC subtypes across the adult brain and identified the specific subpopulations correlated with tumor invasion in gliomas [44]. OPCs, the most abundant cycling population in the adult CNS, is the last potential progenitor source of glioma origin. The OPC markers OLIG2 and NG2 are concurrently expressed in the major cycle-related cell population of the hippocampus [45]. Their correlates in mitotic characteristics give rise to the possibility that OPCs play a key role in tumorigenesis. Moreover, there is a large population of OLIG2+ cells (high Ki76 and CD133) in human gliomas, suggesting that proliferative OPCs may act as tumor-propagating cells [46, 47]. Several studies have found that oncogene mutations in OPCs are involved in the development of high-grade gliomas [48,49,50]. Overexpression of PDGF in OPCs, along with evidence of specific inactivation of Nf1 and tp53 in OPCs, are involved in the formation of malignant gliomas [51, 52]. Intriguingly, lineage tracing based on mosaic analysis with double markers (MADMs) has revealed that introducing p53/Nf1 mutations only in OPCs, but not NSCs, consistently leads to oncogenesis (Fig. 1). Phenotypic and transcriptomic analyses have identified the salient OPC features of these tumors [53]. By applying lineage-targeted scRNA-seq, Weng et al. elegantly identified a primitive OPC intermediate population in the neonatal cortex. Reprogrammed OPCs transformed into a stem-like state, resulting in the development of malignant tumors [54]. These studies revealed that OPCs can directly generate GBM via stepwise genetic and epigenetic reprogramming. We summarize reported gene-edited mouse models that mimic different types of gliomas in Table 1.
Oncometabolites
Mutation at Arg132 of IDH1 was thought to be an early initiating event driving the evolution of gliomas [55, 56]. Mutation of IDH enzymes results in the elevation of (R)-2-HG levels from 1 mmol/L to 3 mmol/L at the center of IDH mutant gliomas [57]. 2-HG, known to be an important oncometabolite, is a competitive substrate of α-ketoglutarate-dependent epigenetic enzymes [58,59,60]. A high concentration of 2-HG in vivo inhibits histone lysine demethylases and TET hydroxylases, leading to impairment of DNA demethylation and eventual hypermethylation in gliomas [61]. Intriguingly, accumulation of (R)-2-HG also causes impairment of collagen protein maturation, which is associated with the endoplasmic reticulum stress response and basement membrane aberrations, leading to a microenvironment favorable to gliomas [62]. Notably, expression of IDH1R132H cooperates with platelet-derived growth factor A expression and loss of Cdkn2a, Atrx, and Pten in glioma to mimic the proneural subtype of human GBM, which exhibits a stronger GBM formation ability in vivo [63]. By enhancing D-2-hydroxyglutarate-mediated DNA methylation, conditionally expressing IDH1R132H in the NPCs of the murine SVZ increases the number of NSCs and their progeny. Regulated stem cells exhibit invasive characteristics and uncontrolled expansion, which may explain the process of oncogenesis in the early phase [64]. Platten’s research group conducted a phase I clinical trial in which 33 patients received treatment with an IDH1-specific peptide vaccine. The convincing clinical data showed that the vaccine is safe, and in terms of therapeutic effect, the IDH1-vaccine significantly prolongs the survival time of patients [65, 66]. These findings support the hypothesis that mutations in oncogenic metabolic enzymes dramatically affect the cellular status of gliomas, leading to mutations in other genes that collectively affect tumor transformation and promote tumorigenesis [67, 68]. Moreover, other groups have reported that tumor-derived kynurenine, IDO1, tryptophan 2,3-dioxygenase, and IL-4I1 mediate immunosuppressive activities in GBM [69,70,71]. Therefore, inhibitor therapy against these targets might be an alternative approach.
Interactions Between Glioma and Microglia/Macrophages
Heterogeneity Between Resident Microglia and Monocyte-Derived Macrophages (MDMs)
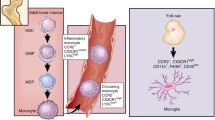
The microenvironment of gliomas consists of multiple interacting networks among cells, in which brain-resident microglia and infiltrating monocytes/MDMs contribute to a large fraction of the glioma immune landscape [72]. Microglia, derived from hematopoietic precursor cells of the yolk sac in the early developmental period, are crucial residential innate immune cells of the brain [73, 74]. They have an important influence in supporting neurogenesis, scavenging apoptotic cells, and refining synapses [73, 74]. Notably, different stages of glioma lead to differential compositions of the myeloid cell landscape. GBM can lead to partial disruption of the blood-brain barrier, enabling monocytes/MDMS to infiltrate the tumor. These distinct populations, termed tumor-associated macrophages (TAMs), have been widely reported as an important factor impinging on the intrinsic characteristics of tumor progression [75]. Using the head-protected irradiation and fluorescently tagged cell lineage tracing technique, microglia expressing high CD45 represent an inherent part of a glioma, while infiltrated tagged TAMs constitute up to 25% of the myeloid cell fraction after 21 days of tumor implantation [76]. The heterogeneity of time-lapse and spatial distribution in gliomas have been described through multiple timepoints and regional microdissection by scRNA-seq [77, 78]. Antunes et al. established the microglial fate-mapping system and revealed the similarities and differences in TAM distribution in newly-diagnosed GBM, recurrent GBM, and mouse GL261 models [79]. Moreover, microglia-derived TAMs or MDMs extracted from tumors are self-renewing populations that are unable to induce CD4+ T-cells or CD8+ T-cells, and compete for space in the tumor environment. Accordingly, the dominant myeloid population in glioma can progress from microglia-derived in the early phases to a mixture of microglia-derived TAMs and outnumbered MDMs in the later phase [89]. In addition, the aryl hydrocarbon receptor in monocytes boosts monocyte recruitment, and blocks antigen presentation expression in MDMs via the transcription factor KLF4 [69].
Considering the distinct biology of the two cell populations, it is essential to identify stable biomarkers to distinguish these two groups. In humans, microglia and macrophages can be classified via fluorescence-activated cell sorting using CD45 and CD49D (known as α4 integrin and ITGA4) [75]. Accurate separation in mice can be obtained by Ly6C, CD11b, F4/80, CD45, and Cx3cr1 [79]. As previously reported, the classical signature markers for microglia (P2ry12 and Sall1) and MDMs (Ly6c and Ccr2) are reduced during glioma-induced activation or differentiation and are insufficient for use in classifying the two populations [6]. After infiltration into the CNS, MDMs has a higher microglia signature gene pattern (Cx3cr1 and Tmem119) and lower CD45 (Fig. 2). Nevertheless, Qian et al. found that Crybb1 and Ldhb are specific and stable markers across different tumor stages in mice, and the cluster of MDMs consistently featured with Iqgap1 corresponded to other clinical datasets [80]. A variety of cytokines (IL-6 and IL-10) and several genes encoding chemokines associated with wound healing (Ccl22, Ccl17, Cxcl2, and Cxcl3) are upregulated in TAMs [81]. Of interest, a pro-inflammatory subset of microglia-derived TAMs was found to exhibit increased expression of Il1b (encoding IL-1β), Ifnb1 (encoding IFNβ1), Ccl4 (encoding C-C Motif Chemokine Ligand 4), Il12 (encoding IL-12), and Tnf (encoding TNF) [82, 83]. In addition, time-of-flight mass cytometry was also combined to reveal the heterogeneity of TAMs in gliomas [79]. Multiple subsets were identified exhibiting downregulation of classical microglial signature genes and, to the contrary, with upregulation of pro-inflammatory cytokines, responses to type I interferons, and hypoxia-associated molecules [79]. Collectively, these results describe novel glioma-associated microglia phenotypes and their diverging functions, which may provide new potential avenues for therapeutic interventions.

Reported communications in the glioma microenvironment. The tumor microenvironment of gliomas is complicated. Left, the relationship between brain resting microglia and microglia-derived TAMs. Wedges indicate differential biomarker expression between the two groups. The classical biomarkers of microglia (CX3CR1 and TMEM119) are reduced in microglia-derived TAMs, rather than other activated markers (IL-1β, CXCL10). Right, the relationship between circulating monocytes and monocyte-derived TAMs. Upper, schematic of neuron-to-brain tumor synapses. Lower, signaling pathways between TAMs and tumor cells. VEGF, vascular endothelial growth factor; EGF, epidermal growth factor; TAM, tumor-associated macrophage; ECM, extracellular matrix.
Functional Characteristics of Microglia/Macrophages in Glioma
The role of microglia in glioma is controversial. In a model of organotypic slice cultures and in vivo implantation, the ablation of microglia impairs tumor growth and prolongs the survival of tumor-bearing mice. Further studies have revealed that glioma cells activate microglia but impair phagocytic activity [84,85,86]. Moreover, endogenous microglia derived from non-glioma subjects have a strong inhibitory effect on the expression of genes relevant to the cell cycle in tumor-initiating cells [87]. Microglia-activating substances such as GM-CSF and LPS can stimulate glioma cell migration cooperatively with endothelial cells, revealing that microglia do not merely react to tumor invasion but play a more complicated role in gliomas [88].
Compelling evidence underpins the perspective that genetic and molecular subtypes of GBM reflect distinct tumor microenvironment (TME), while secreted molecules or subsequently activated signaling from TAMs reciprocally remodels the cellular state of tumors. The functional interactions between GBM cells and components in the microenvironment play an important role in the modulation and infiltration of the brain. Liu et al. used scRNA-seq to characterize cell populations from IDH-WT and IDH-mutant samples and showed that the percentage of microglia and macrophages was higher in IDH-WT GBM [89]. Further, ~500 genes were found to be differently expressed in microglia isolated from IDH-WT and IDH-mutant samples [90]; but this cannot be exclusively considered as microglial heterogeneity. Using longitudinal scRNA-seq, Friedrich et al. examined myeloid cellular states in gliomas and demonstrated that differentiation of myeloid cells in IDH-mutant tumors is blocked by re-orchestration of tryptophan metabolism, leading to an immature phenotype [91]. Alteration of tryptophan metabolism in IDH-mutant gliomas reverses immunosuppression. It has also been reported that the mesenchymal subtype of GBM reported in The Cancer Genome Atlas is associated with an inferior prognosis and contains a higher proportion of TAMs compared to proneural or classical subtypes [36]. In addition, copy number amplifications such as CDK4, EGFR, and PDGFRA loci or mutation of the NF1 locus are correlated with different cellular states in GBM [36]. Mutations of NF1 result in a reduction of NF1 expression, which is predominantly found in the mesenchymal state of GBM, and are possibly responsible for the increase of TAM infiltration by NF1-regulated microglial chemotaxis [36]. Nevertheless, Hara et al. leveraged single-cell RNA sequencing and a mouse model to recapitulate mesenchymal GBM cellular states in vivo, exploring the crosstalk between macrophages and GBM subtype states. Analysis of ligand-receptor pairs suggests that oncostatin M in macrophages activates STAT3 signaling to induce a mesenchymal GBM cellular state via interaction with its receptors (or leukemia inhibitory factor receptor, in complex with GP130) in glioblastoma cells [92]. Moreover, in mouse GBM models, TAMs increase the levels of antigens presented, such as major histocompatibility complex type II expression, suggesting that TAMs in GBM can process antigens to T cells but are unable to activate the subsequent reaction [79]. Matrix metalloproteinases (MMPs) have been reported to be another group of proteins that are crucial to cells infiltration. MMP2 has been reported to be a marker of poor prognosis, facilitating the invasive and angiogenic properties of gliomas [93]. MMP2 is released in the form of pro-MMP2 and subsequently cleaved by MMP14 and converted into an active state which regulates the degradation of the extracellular matrix. In TME, the cleaved substrate pro-MMP2 is secreted by GBM cells, while microglia are the major source of MMP14 in TME. This reciprocal cooperation has been found to be regulated by its downstream signaling receptor TLR2 [94] and extracellular vesicles derived from GBM.
Furthermore, abundant and aberrant neovascularization is one of the defining characteristics of GBM. The process of angiogenesis and angiogenic factors have been extensively described. Resident microglia and peripheral macrophages collectively constitute perivascular niches, while a variety of pro-angiogenic molecules have been found to be upregulated. The CXCL2-CXCR2 signaling pathway is significantly upregulated during angiogenesis and has been shown to have a stronger angiogenic effect than VEGF in vitro [95]. Inhibiting the CXCL2-CXCR2 signaling pathway or selectively reducing resident microglia after tumor implantation decreases the tumoral vasculature count and tumor volume. Nevertheless, insulin-like growth factor-binding protein 1 (IGFBP1), released by microglia, is a novel factor mediating macrophage colony-stimulating factor-induced angiogenesis in GBM via an SYK-PI3K-NFκB-dependent mechanism [96]. Of interest, STAT3 expression (known as IL6/JAK/STAT3 signaling) in the glioma cell affects a variety of targeting genes and can propagate tumorigenesis by facilitating proliferation and angiogenesis. STAT3 upregulation, closely associated with abundant microglia and macrophages, is preferentially enriched in MES-like GBM. Further, ablation of receptors for advanced glycation end products (RAGE) prolongs the overall survival in a GL261 mouse model, and reduces the angiogenic factors secreted by TAMs. It has also been recently reported that microglial neuropilin-1 regulates vascular morphogenesis and affects its receptor VEGFR2 [97]. Administration of the inhibitor EG00229, which impairs the binding of tuftsin (Thr-Lys-Pro-Arg) with Nrp1, reverses the anti-inflammatory state of microglia through transforming growth factor beta signaling [97, 98]. Thus, the interactions between TAMs and GBM are complicated and multifactorial, and understanding the subtypes of TAMs presented in different primary gliomas is important for screening and developing subtype-specific targets.
Neurobiology of Gliomas
Epileptic seizures, memory disorders, and cognitive impairment are common manifestations of patients with gliomas. These clinical characteristics have long been thought of as the result of mechanical pressure caused by the occupying lesion, while little is known about the interactions between tumor cells and surrounding neurons. However, a close relationship between tumors and the CNS has been validated in that gliomas exhibit electrical activity and are integrated into neural networks [99], which is thought to be a milestone event in the rapidly emerging field termed cancer neuroscience [100]. Compared to other pro-tumor factors derived from adjacent normal cells, the supportive influence of neurons includes direct (electrical, synapses, or synapse-like structures) and indirect (chemical) effects. Using electron microscopy, Venkataramani et al. described subtypes of distinct synapses formed by gliomas [101]. There are three morphological categories of neuron-glioma synapses that are consistently formed in incurable human gliomas but hardly exist in oligodendrogliomas. A parallel study found broad expression of glutamate receptor genes in high-grade gliomas, including IDH-mutant glioma, IDH-wild-type glioma, and diffuse intrinsic pontine glioma. Targeted patch-clamp recordings showed the existence of spontaneous excitatory postsynaptic currents that are mediated by glutamate receptors of the AMPA subtype. Synchronized Ca2+ transients are generated by neuronal firing, while genetic perturbations of AMPA receptors or the AMPA receptor antagonist perampanel reduce the invasiveness of gliomas [101]. These studies suggest direct, biologically relevant glutamatergic communication between neurons and glioma cells (Fig. 2). Excessive glutamate released by glioma cells may explain the recurrent seizures in patients. Intriguingly, it has been reported that the expression of glutamate transporters is increased in para-tumor cells, and performs a neuroprotective function in animal models [102]. In addition, excessive glutamate release may also lead to opening of the blood-brain barrier via the activation of N-methyl-D-aspartate receptors [103], and this is beneficial to the efficacy of drug delivery. In a recent impressive study, Chen et al. used an autochthonous mouse model to recapitulate adult OPC-originated gliomagenesis and found that olfaction can directly regulate gliomagenesis via insulin-like growth factor 1 (IGF1) signaling [104]. The activity of olfactory receptor neurons (ORNs) has significant effects on the progress of gliomas, while specific knockout IGF1 receptors in mutant OPCs abolishes the influence derived from ORNs. According to these groundbreaking studies, gliomas have the ability to form electrical and functional synapses with surrounding neurons, which drive tumor growth and resistance [105, 106].
Notably, tumor cells from incurable gliomas share several features with developing neurons (in the process of axonal and dendritic outgrowth) and extend long and thin microtubes [105]. Several reports have found that neurotransmitters in TME drive tumor growth and invasion. Nevertheless, Venkatesh and colleagues [107, 108] revealed a novel mechanism behind this reciprocal influence, showing that neuron paracrine secretion of neuroligin-3 (NLGN3) facilitates tumor progression and in turn induces a synaptic gene signature in the tumor cell. Researchers applied the optogenetic approach in vivo and in vitro, demonstrating that the firing activity of neurons promotes the proliferation and growth of glioma cells. Moreover, NLGN3, secreted by cortical projection neurons and oligodendrocyte precursor cells, is the leading candidate mitogen regulating this process. NLGN3 is broadly expressed in excitatory synapses and affects glioma proliferation through the phosphoinositide 3-kinase–mammalian target of rapamycin pathway [107]. Remarkably, the growth of GBM xenografts is significantly impaired in Nlgn3-knockout mice. In addition, brain-derived neurotrophic factor (BDNF) has also been validated to play a central role in classical synaptic functions and has a stimulating effect in TME [109].
Of interest, the interactions between glioma and neurons might involve the intimate interplay of neurons with precursor cells (NPCs and OPCs). Neuron-to-non-neuron synapses were first described by Bergles et al. in 2000. They reported that neurons form bona fide synapses with OPCs and regulate their proliferation [110]. Electrophysiological analyses revealed that these neuron-glial synapses are similar to normal neuron-neuron synapses, sharing features such as rapid activation, quantal responses, facilitation, depression, and presynaptic inhibition. Previous evidence showed that gliomas mainly originate from NPCs and/or OPCs, which may explain these structural similarities. Moreover, Elizabeth et al. reported that NPCs in the SVZ stimulate invasion of glioma cells through the secretion of chemoattractant signals. Inhibition of Rho/ROCK signaling reduces invasion of glioma cells induced by factors secreted by SVZ NPCs [111]. This novel framework provides new insights into understanding the progression of cancer and sheds light on therapeutic opportunities that can disrupt these communications.
New Insights for Therapeutic Opportunities in High-Grade Glioma
Immune Checkpoint Therapy
The treatment of high-grade glioma is still mainly based on surgery with postoperative radiotherapy and chemotherapy [112]. It is promising that some novel treatment strategies have shown high promise. Tumor immunotherapy has attracted much attention, but owing to the lack of specificity of brain immunity, current immunotherapy strategies require further improvement before application in high-grade gliomas [113, 114]. A series of clinical trials that tested the safety and efficacy of targeting immune checkpoints showed no improved survival benefit in GBM patients [115,116,117]. In 2017, a phase III clinical trial comparing nivolumab (PD1 monoclonal antibody) with bevacizumab (VEGFA monoclonal antibody) showed that patients with recurrent glioblastoma did not benefit from nivolumab treatment (CheckMate-143) [117]. A phase III clinical trial comparing nivolumab plus radiotherapy with standard chemoradiotherapy further confirmed that patients with de novo O-6-methylguanine-DNA methyltransferase (MGMT) unmethylated glioblastoma did not benefit from nivolumab therapy (CheckMate-498) [118]. In addition, CheckMate-548 yielded similar negative results in a phase III trial which compared nivolumab plus standard chemoradiation versus standard chemoradiation in patients with MGMT-methylated glioblastoma [119]. Alternatively, a recent study demonstrated that changing the dosing strategy and administering PD1 antibodies using neoadjuvant therapy can prolong the median survival of patients with relapsed glioblastoma [120]. However, immune checkpoint inhibitors are unable to reverse immune exhaustion in GBM [121]. Mass cytometry time-of-flight analysis revealed that macrophages contributed to 72.6% of the leukocytes in the TME [122], most of which expressed multiple immunosuppressive markers. These data indicate that immune suppressive macrophages are an important confounder for attenuation of the T-cell response. Further understanding of the immune microenvironment within brain tumors is needed to improve the clinical efficacy of immune checkpoint therapy.
Cell-Based and Oncolytic Virus Therapy
Cell therapy based on chimeric antigen receptors (CARs), which involves grafting a specific designed receptor onto an effector cell, is also a research frontier in the treatment of high-grade gliomas [123]. Clinical trials targeting three antigens, EGFRvIII, HER2, and IL-13R alpha2, have confirmed that the application of CAR-T is safe, feasible, and potentially effective [124,125,126,127]. However, the application of CARs to brain tumors still faces challenges due to tumor heterogeneity and antigen loss. Antigen loss in recurrent tumors has been reported in both CAR-T therapy targeting EGFRvIII and IL-13R alpha2 [125, 127]. Interestingly, the major toxicity of CAR-T cells is cytokine release syndrome (CRS). Myeloid-derived macrophages have been found to contribute to the pathogenesis of CRS, mainly mediating the production of core cytokines including IL-6, IL-1, and interferon-γ [128]. In addition, Rodriguez-Garcia et al. demonstrated that CAR-T cell-mediated selective elimination of folate receptor β TAMs resulted in an increase in endogenous activated CD8+ T cells, decreased tumor burden, and prolonged survival [129]. Several reports have highlighted that engineering CAR macrophages is a valuable strategy in GBM. CAR-macrophages have been adapted and designed to produce pro-inflammatory cytokines, which convert subtypes of macrophages from M2 to M1. The polarization of macrophages increases T cell anti-tumor activity and further modulates the pro-inflammatory characteristics of TME [130, 131]. Thus, it is essential to develop new techniques to screen out suitable antigen sites of tumors or immunosuppressive cells, reduce antigen loss, and retard immune cell exhaustion.
Notably, therapeutic vaccination for brain tumors may be a promising treatment strategy. The EGFRvIII-based vaccine was successful in phase II clinical trials for glioblastoma, but failed to achieve positive results in phase III clinical trials. Tumor samples from relapsed patients showed immune escape, which is also a pressing problem during the vaccination treatment period and similar to the challenges of cell therapy [132,133,134,135]. Developing individualized vaccines based on specialized patient gene mutation patterns and expression profiles that collectively target the multiple glioma antigens is a potential future direction. Moreover, oncolytic virus therapy can activate antitumor immune responses, which are an important active immune therapy. A clinical trial using recombinant poliovirus in the treatment of recurrent glioblastoma suggested that this technique is effective and safe [136]. Therefore, improving the targeting of oncolytic viruses, slowing the clearance of oncolytic viruses by the immune system, and reducing the side-effects of oncolytic viruses are important methods for improving the clinical application value of oncolytic viruses.
Transdifferentiation Induction and Glioma Reprogramming
Owing to the similarity between GSCs and NSCs researchers have proposed that inducing GSCs to differentiate into terminally-differentiated cells, especially neurons, might be a supportive strategy to inhibit the progression of brain tumors. It has been reported that inhibition of the Notch pathway can significantly induce a subset of patient-derived GSCs with high ASCL1 expression to differentiate into neuron-like cells [137]. In addition, previous studies have shown that the pression of glioma cells can be retarded by inducing glial differentiation via activation of microRNA or BMP signaling [138, 139]. Overexpression of three neurogenic transcription factors (ASCL1, BRN2, and NGN2) reprogrammed 20%–40% of human glioma cells into TUBB3-positive neurons in vitro [140]. Cooperating with NGN2 and SOX11, intravenous injection of overexpressing viruses has been shown to improve the reprogramming efficiency of human glioma cells into terminally-differentiated neuron-like cells, thus delaying tumor progression and significantly prolonging the survival of tumor-bearing mice [141]. A similar result was found via overexpression of NGN2, ASCL1, and NeuroD1 in glioma cells. Unfortunately, gene regulation as a treatment for GBM are bound to face great challenges, and certain risks exist in the clinical application of transgenic technology and virus transfection such as off-target effects and neurotoxicity. However, these results suggest the potential for reprogramming of GBM cells into neurons [142,143,144].
Disruption of Neuron-Glioma Communication
Glutamatergic synaptic structures and gap junctions have been identified in diffuse gliomas and can evoke long-lasting depolarizing currents, Ca2+ flux, and subsequent electrical network reactions. This cascade of electrical responses in glioma subpopulations ultimately promotes cell invasion and mitosis [99, 101]. Noninvasive brain stimulation (NiBS) is a group of techniques applied to the scalp that are broadly used in clinical practice to modulate neural activity via transcranial electrical or magnetic fields (transcranial magnetic stimulation, TMS; tumor-treating fields, TTFields). Interestingly, NiBS can increase or decrease neural activity depending on different stimulation patterns, of which the mechanisms are considered to involve the regulation of synaptic plasticity [145]. Long-lasting effects across multiple regions of the brain have been reported after stimulation with magnetic or electrical fields [145, 146]. NiBS has also been reported to induce effects such as the modulation of glutamatergic transmission, BDNF-dependent plasticity, and the regulation of pathway activity [147, 148]. Thus, disruption of communication between glioma and neurons is a promising area of study. Considering the efficacy and safety of NiBS, both TMS and TTFields have been approved for the treatment of several psychiatric diseases [145]. As for neoplasms, the landmark EF-14 trial, showed that TTFields plus maintenance Temozolomide (TMZ) resulted in an increase in overall survival in patients with newly diagnosed GBM compared to TMZ alone (20.9 vs 16.0 months with TMZ alone). Furthermore, no systemic adverse events are associated with the addition of TTFields (48% vs 44% with TMZ alone) [149, 150]. Indeed, TTFields deliver a low intensity (1–3 V/cm) at medium frequency (100–300 kHz) to the tumor region, alternating extra physiological currents which do not affect neural activity but rather impede cancer cell mitosis. The formation of microtubules, which are essential structures for neuron-glioma communications [101, 151], is disrupted by TTFields. Recent reports also revealed that TTFields induce an increased release of micronuclei from tumor cells, leading to activation of cGAS/STING and AIM2/Caspase-1 [152]. After treatment with TTFields, T-cell activation and clonal expansion have been reported in samples and derived from the upregulation of adaptive immunity. Taylor et al. genetically or pharmacologically blocked BDNF-TrkB signaling in a xenograft model of pediatric glioblastoma, abrogating the tumor-promoting effects of BDNF on synapses and prolonging survival [109]. Thus, growing evidence suggests that the application of NiBS may be used to suppress glioma progression and tumor-promoting neuronal communication.
Discussion
Understanding of biology and immunology in gliomas has advanced at an impressive pace in recent years. The brain TME comprises heterogeneous populations of cells exhibiting differences in genetic characteristics and performing various modes of reciprocal interaction to mediate tumor initiation, progression, and therapeutic response. Combining advancing technologies in genetic engineering and sequencing enables promising capabilities in diagnosis and personalized treatment, and deciphering the origins of tumor-supporting cells at the single cell level as well. Studying developmental programs is a promising strategy for understanding the process of oncogenesis, and essential targets for disrupting disease progression or remission may be found. We here reviewed advances in gene-edited mouse models that mirror human disease and discussed potential glioma-initiated progenitor cells that may be used for further investigations. In addition, interactions between tumor subtypes, microglia, MDMs, and neurons in the brain TME play an important role in tumor progression. In light of many mechanisms of tumor/non-tumor cell crosstalk and their accompanying outgrowths, these implications in complex TME caused by these interactions are important components of the major driver in glioma biology. We here highlight a nascent but fast-growing field termed cancer neuroscience, which mainly focuses on the tumor-neuron network and its role in the progress of cancer growth. Interesting questions that remain to be answered include: (1) how to specifically target tumor cells in the tumor-neuron axis and integrate neural regulation methods into existing clinical strategy, (2) why and how tumors communicate with neurons in the brain, and (3) whether histopathological subtypes of glioma have a neuron communication preference. Lessons learned from TME suggest that disruption of the tumor/non-tumor cell dialogue could be helpful in inventing potential novel therapeutic approaches beyond standard treatment such as immune checkpoint inhibitors, cell-based biotechniques, and noninvasive brain stimulation. These therapies could potentially become the keystones of clinical practice in the future.
References
Zong H, Verhaak RG, Canoll P. The cellular origin for malignant glioma and prospects for clinical advancements. Expert Rev Mol Diagn 2012, 12: 383–394.
Shibahara I, Sonoda Y, Kanamori M, Saito R, Kumabe T, Tominaga T. New insights into glioma classification based on isocitrate dehydrogenase 1 and 2 gene status. Brain Tumor Pathol 2011, 28: 203–208.
Yan W, Zhang W, You G, Zhang J, Han L, Bao Z. Molecular classification of gliomas based on whole genome gene expression: A systematic report of 225 samples from the Chinese Glioma Cooperative Group. Neuro Oncol 2012, 14: 1432–1440.
Ashby LS, Ryken TC. Management of malignant glioma: Steady progress with multimodal approaches. Neurosurg Focus 2006, 20: E3.
Wen PY, Reardon DA. Progress in glioma diagnosis, classification and treatment. Nat Rev Neurol 2016, 12: 69–70.
Andersen BM, Faust Akl C, Wheeler MA, Chiocca EA, Reardon DA, Quintana FJ. Glial and myeloid heterogeneity in the brain tumour microenvironment. Nat Rev Cancer 2021, 21: 786–802.
Jung E, Osswald M, Ratliff M, Dogan H, Xie R, Weil S, et al. Tumor cell plasticity, heterogeneity, and resistance in crucial microenvironmental niches in glioma. Nat Commun 2021, 12: 1014.
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol 2016, 131: 803–820.
Wesseling P, Capper D. WHO 2016 classification of gliomas. Neuropathol Appl Neurobiol 2018, 44: 139–150.
Stupp R, Brada M, van den Bent MJ, Tonn JC, Pentheroudakis G, Group ESMOGW. High-grade glioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2014, 25: iii93–iii101.
Mizoguchi M, Yoshimoto K, Ma X, Guan Y, Hata N, Amano T, et al. Molecular characteristics of glioblastoma with 1p/19q co-deletion. Brain Tumor Pathol 2012, 29: 148–153.
Reifenberger J, Reifenberger G, Liu L, James CD, Wechsler W, Collins VP. Molecular genetic analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p. Am J Pathol 1994, 145: 1175–1190.
Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, et al. Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med 2015, 372: 2499–2508.
Chan AK, Mao Y, Ng HK. TP53 and histone H3.3 mutations in triple-negative lower-grade gliomas. N Engl J Med 2016, 375: 2206–2208.
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro Oncol 2021, 23: 1231–1251.
Li Y, Laterra J. Cancer stem cells: Distinct entities or dynamically regulated phenotypes? Cancer Res 2012, 72: 576–580.
Tang DG. Understanding cancer stem cell heterogeneity and plasticity. Cell Res 2012, 22: 457–472.
Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001, 414: 105–111.
Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature 2004, 432: 396–401.
Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res 2003, 63: 5821–5828.
Wang J, Xu SL, Duan JJ, Yi L, Guo YF, Shi Y, et al. Invasion of white matter tracts by glioma stem cells is regulated by a NOTCH1–SOX2 positive-feedback loop. Nat Neurosci 2019, 22: 91–105.
Auffinger B, Tobias A, Han Y, Lee G, Guo D, Dey M, et al. Sc-02conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Neuro Oncol 2014, 16: v197.
Wang L, Babikir H, Müller S, Yagnik G, Shamardani K, Catalan F, et al. The phenotypes of proliferating glioblastoma cells reside on a single axis of variation. Cancer Discov 2019, 9: 1708–1719.
Richards LM, Whitley OKN, MacLeod G, Cavalli FMG, Coutinho FJ, Jaramillo JE, et al. Gradient of Developmental and Injury Response transcriptional states defines functional vulnerabilities underpinning glioblastoma heterogeneity. Nat Cancer 2021, 2: 157–173.
Xu HS, Qin XL, Zong HL, He XG, Cao L. Cancer stem cell markers in glioblastoma - an update. Eur Rev Med Pharmacol Sci 2017, 21: 3207–3211.
Steed TC, Treiber JM, Taha B, Engin HB, Carter H, Patel KS, et al. Glioblastomas located in proximity to the subventricular zone (SVZ) exhibited enrichment of gene expression profiles associated with the cancer stem cell state. J Neurooncol 2020, 148: 455–462.
Stiles CD, Rowitch DH. Glioma stem cells: A midterm exam. Neuron 2008, 58: 832–846.
Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488: 522–526.
Lee JH, Lee JE, Kahng JY, Kim SH, Park JS, Yoon SJ, et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 2018, 560: 243–247.
Zhu Y, Guignard F, Zhao D, Liu L, Burns DK, Mason RP, et al. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell 2005, 8: 119–130.
Wang X, Zhou R, Xiong Y, Zhou L, Yan X, Wang M, et al. Sequential fate-switches in stem-like cells drive the tumorigenic trajectory from human neural stem cells to malignant glioma. Cell Res 2021, 31: 684–702.
Bachoo RM, Maher EA, Ligon KL, Sharpless NE, Chan SS, You MJ, et al. Epidermal growth factor receptor and Ink4a/Arf: Convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell 2002, 1: 269–277.
Alcantara Llaguno S, Sun D, Pedraza AM, Vera E, Wang Z, Burns DK, et al. Cell-of-origin susceptibility to glioblastoma formation declines with neural lineage restriction. Nat Neurosci 2019, 22: 545–555.
Jacques TS, Swales A, Brzozowski MJ, Henriquez NV, Linehan JM, Mirzadeh Z, et al. Combinations of genetic mutations in the adult neural stem cell compartment determine brain tumour phenotypes. EMBO J 2010, 29: 222–235.
Tanay A, Regev A. Scaling single-cell genomics from phenomenology to mechanism. Nature 2017, 541: 331–338.
Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 2019, 178: 835-849.e21.
Winner B, Winkler J. Adult neurogenesis in neurodegenerative diseases. Cold Spring Harb Perspect Biol 2015, 7: a021287.
Bardehle S, Krüger M, Buggenthin F, Schwausch J, Ninkovic J, Clevers H, et al. Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nat Neurosci 2013, 16: 580–586.
Li X, Liu G, Yang L, Li Z, Zhang Z, Xu Z, et al. Decoding cortical glial cell development. Neurosci Bull 2021, 37: 440–460.
Endersby R, Zhu X, Hay N, Ellison DW, Baker SJ. Nonredundant functions for Akt isoforms in astrocyte growth and gliomagenesis in an orthotopic transplantation model. Cancer Res 2011, 71: 4106–4116.
Paugh BS, Zhu X, Qu C, Endersby R, Diaz AK, Zhang J, et al. Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res 2013, 73: 6219–6229.
Radke J, Bortolussi G, Pagenstecher A. Akt and c-Myc induce stem-cell markers in mature primary p53−/− astrocytes and render these cells gliomagenic in the brain of immunocompetent mice. PLOS ONE 2013, 8: e56691.
Chow LML, Endersby R, Zhu X, Rankin S, Qu C, Zhang J, et al. Cooperativity within and among Pten, p53, and Rb pathways induces high-grade astrocytoma in adult brain. Cancer Cell 2011, 19: 305–316.
Lin CCJ, Yu K, Hatcher A, Huang TW, Lee HK, Carlson J, et al. Identification of diverse astrocyte populations and their malignant analogs. Nat Neurosci 2017, 20: 396–405.
Geha S, Pallud J, Junier MP, Devaux B, Leonard N, Chassoux F, et al. NG2+/Olig2+ cells are the major cycle-related cell population of the adult human normal brain. Brain Pathol 2010, 20: 399–411.
Ligon KL, Huillard E, Mehta S, Kesari S, Liu H, Alberta JA, et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron 2007, 53: 503–517.
Lu F, Liu Y, Inoue A, Suzuki T, Zhao K, Zhang Y. Establishing chromatin regulatory landscape during mouse preimplantation development. Cell 2016, 165: 1375–1388.
Assanah M, Lochhead R, Ogden A, Bruce J, Goldman J, Canoll P. Glial progenitors in adult white matter are driven to form malignant gliomas by platelet-derived growth factor-expressing retroviruses. J Neurosci 2006, 26: 6781–6790.
Alcantara Llaguno SR, Wang Z, Sun D, Chen J, Xu J, Kim E, et al. Adult lineage-restricted CNS progenitors specify distinct glioblastoma subtypes. Cancer Cell 2015, 28: 429–440.
Lindberg N, Kastemar M, Olofsson T, Smits A, Uhrbom L. Oligodendrocyte progenitor cells can act as cell of origin for experimental glioma. Oncogene 2009, 28: 2266–2275.
Uhrbom L, Hesselager G, Nistér M, Westermark B. Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res 1998, 58: 5275–5279.
Galvao RP, Kasina A, McNeill RS, Harbin JE, Foreman O, Verhaak RGW, et al. Transformation of quiescent adult oligodendrocyte precursor cells into malignant glioma through a multistep reactivation process. Proc Natl Acad Sci U S A 2014, 111: E4214–E4223.
Liu C, Sage JC, Miller MR, Verhaak RGW, Hippenmeyer S, Vogel H, et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 2011, 146: 209–221.
Weng Q, Wang J, Wang J, He D, Cheng Z, Zhang F, et al. Single-cell transcriptomics uncovers glial progenitor diversity and cell fate determinants during development and gliomagenesis. Cell Stem Cell 2019, 24: 707-723.e8.
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1andIDH2Mutations in gliomas. N Engl J Med 2009, 360: 765–773.
Cohen AL, Holmen SL, Colman H. IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep 2013, 13: 345.
Linninger A, Hartung GA, Liu BP, Mirkov S, Tangen K, Lukas RV, et al. Modeling the diffusion of D-2-hydroxyglutarate from IDH1 mutant gliomas in the central nervous system. Neuro Oncol 2018, 20: 1197–1206.
Li X, Egervari G, Wang Y, Berger SL, Lu Z. Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat Rev Mol Cell Biol 2018, 19: 563–578.
Feinberg AP, Koldobskiy MA, Göndör A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet 2016, 17: 284–299.
Ye D, Ma S, Xiong Y, Guan KL. R-2-hydroxyglutarate as the key effector of IDH mutations promoting oncogenesis. Cancer Cell 2013, 23: 274–276.
Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483: 479–483.
Sasaki M, Knobbe CB, Itsumi M, Elia AJ, Harris IS, Chio IIC, et al. D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev 2012, 26: 2038–2049.
Philip B, Yu DX, Silvis MR, Shin CH, Robinson JP, Robinson GL, et al. Mutant IDH1 promotes glioma formation in vivo. Cell Rep 2018, 23: 1553–1564.
Bardella C, Al-Dalahmah O, Krell D, Brazauskas P, Al-Qahtani K, Tomkova M, et al. Expression of Idh1R132H in the murine subventricular zone stem cell niche recapitulates features of early gliomagenesis. Cancer Cell 2016, 30: 578–594.
Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014, 512: 324–327.
Huang Y, Wang Y, Huang Z. A specific peptide vaccine against IDH1(R132H) glioma. Neurosci Bull 2022, 38: 223–225.
You JS, Jones PA. Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell 2012, 22: 9–20.
Choi JD, Lee JS. Interplay between epigenetics and genetics in cancer. Genomics Inform 2013, 11: 164–173.
Takenaka MC, Gabriely G, Rothhammer V, Mascanfroni ID, Wheeler MA, Chao CC, et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat Neurosci 2019, 22: 729–740.
Rothhammer V, Quintana FJ. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat Rev Immunol 2019, 19: 184–197.
Gabriely G, Quintana FJ. Role of AHR in the control of GBM-associated myeloid cells. Semin Cancer Biol 2020, 64: 13–18.
Broekman ML, Maas SLN, Abels ER, Mempel TR, Krichevsky AM, Breakefield XO. Multidimensional communication in the microenvirons of glioblastoma. Nat Rev Neurol 2018, 14: 482–495.
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330: 841–845.
Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FMV. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 2007, 10: 1538–1543.
Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, et al. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep 2016, 17: 2445–2459.
Müller A, Brandenburg S, Turkowski K, Müller S, Vajkoczy P. Resident microglia, and not peripheral macrophages, are the main source of brain tumor mononuclear cells. Int J Cancer 2015, 137: 278–288.
Darmanis S, Sloan SA, Croote D, Mignardi M, Chernikova S, Samghababi P, et al. Single-cell RNA-seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Rep 2017, 21: 1399–1410.
Müller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol 2017, 18: 234.
Pombo Antunes AR, Scheyltjens I, Lodi F, Messiaen J, Antoranz A, Duerinck J, et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat Neurosci 2021, 24: 595–610.
Qian J, Wang C, Wang B, Li C, Fu K, Wang Y, et al. Dynamics of glioma-associated microglia and macrophages reveals their divergent roles in the immune response of brain. bioRxiv 2021, https://doi.org/10.1101/2021.07.11.451874.
Gorbachev AV, Fairchild RL. Regulation of chemokine expression in the tumor microenvironment. Crit Rev Immunol 2014, 34: 103–120.
Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res 2017, 77: 2266–2278.
Friebel E, Kapolou K, Unger S, Núñez NG, Utz S, Rushing EJ, et al. Single-cell mapping of human brain cancer reveals tumor-specific instruction of tissue-invading leukocytes. Cell 2020, 181: 1626-1642.e20.
Markovic DS, Glass R, Synowitz M, Rooijen NV, Kettenmann H. Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J Neuropathol Exp Neurol 2005, 64: 754–762.
Markovic DS, Vinnakota K, van Rooijen N, Kiwit J, Synowitz M, Glass R, et al. Minocycline reduces glioma expansion and invasion by attenuating microglial MT1-MMP expression. Brain Behav Immun 2011, 25: 624–628.
Zhai H, Heppner FL, Tsirka SE. Microglia/macrophages promote glioma progression. Glia 2011, 59: 472–485.
Sarkar S, Döring A, Zemp FJ, Silva C, Lun X, Wang X, et al. Therapeutic activation of macrophages and microglia to suppress brain tumor-initiating cells. Nat Neurosci 2014, 17: 46–55.
Bettinger I, Thanos S, Paulus W. Microglia promote glioma migration. Acta Neuropathol 2002, 103: 351–355.
Liu H, Sun Y, Zhang Q, Jin W, Gordon RE, Zhang Y, et al. Pro-inflammatory and proliferative microglia drive progression of glioblastoma. Cell Rep 2021, 36: 109718.
Klemm F, Maas RR, Bowman RL, Kornete M, Soukup K, Nassiri S, et al. Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell 2020, 181: 1643-1660.e17.
Friedrich M, Sankowski R, Bunse L, Kilian M, Green E, Guevara CR, et al. Tryptophan metabolism drives dynamic immunosuppressive myeloid states in IDH-mutant gliomas. Nat Cancer 2021, 2: 723–740.
Hara T, Chanoch-Myers R, Mathewson ND, Myskiw C, Atta L, Bussema L, et al. Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma. Cancer Cell 2021, 39: 779-792.e11.
Du R, Petritsch C, Lu K, Liu P, Haller A, Ganss R, et al. Matrix metalloproteinase-2 regulates vascular patterning and growth affecting tumor cell survival and invasion in GBM. Neuro Oncol 2008, 10: 254–264.
Vinnakota K, Hu F, Ku MC, Georgieva PB, Szulzewsky F, Pohlmann A, et al. Toll-like receptor 2 mediates microglia/brain macrophage MT1-MMP expression and glioma expansion. Neuro Oncol 2013, 15: 1457–1468.
Brandenburg S, Müller A, Turkowski K, Radev YT, Rot S, Schmidt C, et al. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta Neuropathol 2016, 131: 365–378.
Nijaguna MB, Patil V, Urbach S, Shwetha SD, Sravani K, Hegde AS, et al. Glioblastoma-derived macrophage colony-stimulating factor (MCSF) induces microglial release of insulin-like growth factor-binding protein 1 (IGFBP1) to promote angiogenesis. J Biol Chem 2015, 290: 23401–23415.
Gelfand MV, Hagan N, Tata A, Oh WJ, Lacoste B, Kang KT, et al. Neuropilin-1 functions as a VEGFR2 co-receptor to guide developmental angiogenesis independent of ligand binding. eLife 2014, 3: e03720.
Nissen JC, Selwood DL, Tsirka SE. Tuftsin signals through its receptor neuropilin-1 via the transforming growth factor beta pathway. J Neurochem 2013, 127: 394–402.
Venkatesh HS, Morishita W, Geraghty AC, Silverbush D, Gillespie SM, Arzt M, et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019, 573: 539–545.
Monje M, Borniger JC, D’Silva NJ, Deneen B, Dirks PB, Fattahi F, et al. Roadmap for the emerging field of cancer neuroscience. Cell 2020, 181: 219–222.
Venkataramani V, Tanev DI, Strahle C, Studier-Fischer A, Fankhauser L, Kessler T, et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573: 532–538.
Sattler R, Tyler B, Hoover B, Coddington LT, Recinos V, Hwang L, et al. Increased expression of glutamate transporter GLT-1 in peritumoral tissue associated with prolonged survival and decreases in tumor growth in a rat model of experimental malignant glioma. J Neurosurg 2013, 119: 878–886.
Vazana U, Veksler R, Pell GS, Prager O, Fassler M, Chassidim Y, et al. Glutamate-mediated blood-brain barrier opening: Implications for neuroprotection and drug delivery. J Neurosci 2016, 36: 7727–7739.
Chen P, Wang W, Liu R, Lyu J, Zhang L, Li B, et al. Olfactory sensory experience regulates gliomagenesis via neuronal IGF1. Nature 2022, 606: 550–556.
Osswald M, Jung E, Sahm F, Solecki G, Venkataramani V, Blaes J, et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528: 93–98.
Weil S, Osswald M, Solecki G, Grosch J, Jung E, Lemke D, et al. Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro Oncol 2017, 19: 1316–1326.
Venkatesh HS, Johung TB, Caretti V, Noll A, Tang Y, Nagaraja S, et al. Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell 2015, 161: 803–816.
Venkatesh HS, Tam LT, Woo PJ, Lennon J, Nagaraja S, Gillespie SM, et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 2017, 549: 533–537.
Taylor KR, Barron T, Zhang H, Hui A, Hartmann G, Ni L, et al. Glioma synapses recruit mechanisms of adaptive plasticity. bioRxiv 2021, https://doi.org/10.1101/2021.11.04.467325.
Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, et al. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 2014, 344: 1252304.
Qin EY, Cooper DD, Abbott KL, Lennon J, Nagaraja S, MacKay A, et al. Neural precursor-derived pleiotrophin mediates subventricular zone invasion by glioma. Cell 2017, 170: 845-859.e19.
Gupta T, Sarin R. Poor-prognosis high-grade gliomas: Evolving an evidence-based standard of care. Lancet Oncol 2002, 3: 557–564.
Xu S, Tang L, Li X, Fan F, Liu Z. Immunotherapy for glioma: Current management and future application. Cancer Lett 2020, 476: 1–12.
Calinescu AA, Kamran N, Baker G, Mineharu Y, Lowenstein PR, Castro MG. Overview of current immunotherapeutic strategies for glioma. Immunotherapy 2015, 7: 1073–1104.
Garg AD, Vandenberk L, van Woensel M, Belmans J, Schaaf M, Boon L, et al. Preclinical efficacy of immune-checkpoint monotherapy does not recapitulate corresponding biomarkers-based clinical predictions in glioblastoma. OncoImmunology 2017, 6: e1295903.
Reardon DA, Omuro A, Brandes AA, Rieger J, Wick A, Sepulveda J, et al. OS10.3 randomized phase 3 study evaluating the efficacy and safety of nivolumab vs bevacizumab in patients with recurrent glioblastoma: CheckMate 143. Neuro Oncol 2017, 19: iii21.
Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: The CheckMate 143 phase 3 randomized clinical trial. JAMA Oncol 2020, 6: 1003–1010.
Phase B. CheckMate-498 study did not meet primary endpoint of overall survival with opdivo (nivolumab) plus radiation in patients with newly diagnosed MGMT-Unmethylated glioblastoma multiforme| BMS newsroom. BMS Newsroom 2019, 5.
Lim M, Weller M, Idbaih A, Steinbach J, Finocchiaro G, Raval RR, et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro Oncol 2022, https://doi.org/10.1093/neuonc/noac116.
Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med 2019, 25: 477–486.
Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res 2018, 24: 4175–4186.
de Groot JF, Penas-Prado M, Mandel JJ, O’Brien BJ, Weathers SPS, Zhou S, et al. Window-of-opportunity clinical trial of a PD-1 inhibitor in patients with recurrent glioblastoma. J Clin Oncol 2008, 2018: 36.
Sadelain M, Brentjens R, Rivière I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol 2009, 21: 215–223.
Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: A phase 1 dose-escalation trial. JAMA Oncol 2017, 3: 1094–1101.
Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res 2015, 21: 4062–4072.
Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med 2016, 375: 2561–2569.
O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 2017, 9: eaaa0984.
Hao Z, Li R, Meng L, Han Z, Hong Z. Macrophage, the potential key mediator in CAR-T related CRS. Exp Hematol Oncol 2020, 9: 15.
Rodriguez-Garcia A, Lynn RC, Poussin M, Eiva MA, Shaw LC, O’Connor RS, et al. CAR-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat Commun 2021, 12: 877.
Morrissey MA, Williamson AP, Steinbach AM, Roberts EW, Kern N, Headley MB, et al. Chimeric antigen receptors that trigger phagocytosis. eLife 2018, 7: e36688.
Murray PJ. Macrophage polarization. Ann Rev Physiol 2017, 79: 541–566.
Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol 2010, 28: 4722–4729.
Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol 2017, 18: 1373–1385.
Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565: 234–239.
Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanović S, Gouttefangeas C, et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565: 240–245.
Desjardins A, Gromeier M, Herndon JE 2nd, Beaubier N, Bolognesi DP, Friedman AH, et al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med 2018, 379: 150–161.
Park NI, Guilhamon P, Desai K, McAdam RF, Langille E, O’Connor M, et al. ASCL1 reorganizes chromatin to direct neuronal fate and suppress tumorigenicity of glioblastoma stem cells. Cell Stem Cell 2017, 21: 209-224.e7.
Piccirillo SGM, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 2006, 444: 761–765.
Mei J, Bachoo R, Zhang CL. microRNA-146a inhibits glioma development by targeting Notch1. Mol Cell Biol 2011, 31: 3584–3592.
Zhao J, He H, Zhou K, Ren Y, Shi Z, Wu Z, et al. Neuronal transcription factors induce conversion of human glioma cells to neurons and inhibit tumorigenesis. PLOS ONE 2012, 7: e41506.
Su Z, Zang T, Liu ML, Wang LL, Niu W, Zhang CL. Reprogramming the fate of human glioma cells to impede brain tumor development. Cell Death Dis 2014, 5: e1463.
Niu W, Zang T, Zou Y, Fang S, Smith DK, Bachoo R, et al. In vivo reprogramming of astrocytes to neuroblasts in the adult brain. Nat Cell Biol 2013, 15: 1164–1175.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126: 663–676.
Heins N, Malatesta P, Cecconi F, Nakafuku M, Tucker KL, Hack MA, et al. Glial cells generate neurons: The role of the transcription factor Pax6. Nat Neurosci 2002, 5: 308–315.
Polanía R, Nitsche MA, Ruff CC. Studying and modifying brain function with non-invasive brain stimulation. Nat Neurosci 2018, 21: 174–187.
Collingridge GL, Peineau S, Howland JG, Wang YT. Long-term depression in the CNS. Nat Rev Neurosci 2010, 11: 459–473.
Houdayer E, Degardin A, Cassim F, Bocquillon P, Derambure P, Devanne H. The effects of low- and high-frequency repetitive TMS on the input/output properties of the human corticospinal pathway. Exp Brain Res 2008, 187: 207–217.
Huang YZ, Edwards MJ, Rounis E, Bhatia KP, Rothwell JC. Theta burst stimulation of the human motor cortex. Neuron 2005, 45: 201–206.
Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA 2017, 318: 2306–2316.
Stupp R, Taillibert S, Kanner AA, Kesari S, Steinberg DM, Toms SA, et al. Maintenance therapy with tumor-treating fields plus temozolomide vs temozolomide alone for glioblastoma: A randomized clinical trial. JAMA 2015, 314: 2535–2543.
Rominiyi O, Vanderlinden A, Clenton SJ, Bridgewater C, Al-Tamimi Y, Collis SJ. Tumour treating fields therapy for glioblastoma: Current advances and future directions. Br J Cancer 2021, 124: 697–709.
Chen D, Le SB, Hutchinson TE, Calinescu AA, Sebastian M, Jin D, et al. Tumor Treating Fields dually activate STING and AIM2 inflammasomes to induce adjuvant immunity in glioblastoma. J Clin Invest 2022, 132: e149258.
Weiss WA, Burns MJ, Hackett C, Aldape K, Hill JR, Kuriyama H, et al. Genetic determinants of malignancy in a mouse model for oligodendroglioma. Cancer Res 2003, 63: 1589–1595.
Persson AI, Petritsch C, Swartling FJ, Itsara M, Sim FJ, Auvergne R, et al. Non-stem cell origin for oligodendroglioma. Cancer Cell 2010, 18: 669–682.
Ding H, Shannon P, Lau N, Wu X, Roncari L, Baldwin RL, et al. Oligodendrogliomas result from the expression of an activated mutant epidermal growth factor receptor in a RAS transgenic mouse astrocytoma model. Cancer Res 2003, 63: 1106–1113.
Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev 2001, 15: 1913–1925.
Tchougounova E, Kastemar M, Bråsäter D, Holland EC, Westermark B, Uhrbom L. Loss of Arf causes tumor progression of PDGFB-induced oligodendroglioma. Oncogene 2007, 26: 6289–6296.
Dai C, Lyustikman Y, Shih A, Hu X, Fuller GN, Rosenblum M, et al. The characteristics of astrocytomas and oligodendrogliomas are caused by two distinct and interchangeable signaling formats. Neoplasia 2005, 7: 397–406.
Calzolari F, Appolloni I, Tutucci E, Caviglia S, Terrile M, Corte G, et al. Tumor progression and oncogene addiction in a PDGF-B-induced model of gliomagenesis. Neoplasia 2008, 10: 1373–IN10.
Nazarenko I, Hedrén A, Sjödin H, Orrego A, Andrae J, Afink GB, et al. Brain abnormalities and glioma-like lesions in mice overexpressing the long isoform of PDGF-a in astrocytic cells. PLOS ONE 2011, 6: e18303.
Jensen NA, Pedersen KM, Lihme F, Rask L, Nielsen JV, Rasmussen TE, et al. Astroglial c-myc overexpression predisposes mice to primary malignant gliomas. J Biol Chem 2003, 278: 8300–8308.
Abel TW, Clark C, Bierie B, Chytil A, Aakre M, Gorska A, et al. GFAP-Cre-mediated activation of oncogenic K-ras results in expansion of the subventricular zone and infiltrating glioma. Mol Cancer Res 2009, 7: 645–653.
Shannon P, Sabha N, Lau N, Kamnasaran D, Gutmann DH, Guha A. Pathological and molecular progression of astrocytomas in a GFAP: 12V-ha-ras mouse astrocytoma model. Am J Pathol 2005, 167: 859–867.
Wei Q, Clarke L, Scheidenhelm DK, Qian B, Tong A, Sabha N, et al. High-grade glioma formation results from postnatal pten loss or mutant epidermal growth factor receptor expression in a transgenic mouse glioma model. Cancer Res 2006, 66: 7429–7437.
Holland EC, Hively WP, DePinho RA, Varmus HE. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev 1998, 12: 3675–3685.
Hede SM, Hansson I, Afink GB, Eriksson A, Nazarenko I, Andrae J, et al. GFAP promoter driven transgenic expression of PDGFB in the mouse brain leads to glioblastoma in a Trp53 null background. Glia 2009, 57: 1143–1153.
Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet 2000, 25: 55–57.
Uhrbom L, Dai C, Celestino JC, Rosenblum MK, Fuller GN, Holland EC. Ink4a-Arf loss cooperates with KRas activation in astrocytes and neural progenitors to generate glioblastomas of various morphologies depending on activated Akt. Cancer Res 2002, 62: 5551–5558.
Hu X, Pandolfi PP, Li Y, Koutcher JA, Rosenblum M, Holland EC. mTOR promotes survival and astrocytic characteristics induced by pten/Akt signaling in glioblastoma. Neoplasia 2005, 7: 356–368.
Kwon CH, Zhao D, Chen J, Alcantara S, Li Y, Burns DK, et al. Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer Res 2008, 68: 3286–3294.
Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 2008, 455: 1129–1133.
Koga T, Chaim IA, Benitez JA, Markmiller S, Parisian AD, Hevner RF, et al. Longitudinal assessment of tumor development using cancer avatars derived from genetically engineered pluripotent stem cells. Nat Commun 2020, 11: 550.
Shih AH, Dai C, Hu X, Rosenblum MK, Koutcher JA, Holland EC. Dose-dependent effects of platelet-derived growth factor-B on glial tumorigenesis. Cancer Res 2004, 64: 4783–4789.
Acknowledgments
This review was supported by the National Key Research and Development Program of China (2020YFA0804200), the National Natural Science Foundation of China (82073166, 82273203), and a Shanghai Municipal Science and Technology Major Project (2018SHZDZX01). H.Y. is supported by the Program for Professors of Special Appointment (Eastern Scholar) at the Shanghai Institutions of Higher Learning (SSF151005). Q.Y. is supported by the Student Scientific Research Program (Fuqing Scholar) of Shanghai Medical College, Fudan University (FQXZ202117C).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhuang, Q., Yang, H. & Mao, Y. The Oncogenesis of Glial Cells in Diffuse Gliomas and Clinical Opportunities. Neurosci. Bull. 39, 393–408 (2023). https://doi.org/10.1007/s12264-022-00953-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12264-022-00953-3