
Abstract
Chronic myeloid leukemia (CML) is effectively treated with tyrosine kinase inhibitors (TKI) targeted against BCR-ABL. We previously reported the investigation of residual CML diseases during TKI treatment using FACS-sorting and quantitative RT-PCR of BCR-ABL among each population; total mononuclear cells, hematopoietic stem cells, and myeloid progenitors. The observations also implied that the second-generation of ABL-tyrosine kinase inhibitors (2nd TKIs), dasatinib or nilotinib therapy can be more promising approach for efficient reduction of the CML stem cells. Moreover, we need to develop the evaluation method of the residual CML diseases to establish rational therapy-cessation strategies in CML.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Properties of chronic myeloid leukemia stem cells
Chronic myeloid leukemia (CML) is a clonal myeloproliferative disorder that is characterized by the presence of a fusion oncogene, BCR-ABL, which encodes a protein with constitutive tyrosine kinase activity [1]. The mechanisms for TKI insensitivity of CML stem remains unclear; factors such as quiescence, high level of BCR-ABL expression, acquired mutations in the oncogene, and overexpression of membrane transporter proteins in these cells may play a role [2,3,4].
The normal hematopoiesis is sustained through the life by the regulated proliferative and differentiation activity of a large pool of hematopoietic stem cells (HSCs) (Fig. 1a). Cells within hematopoietic hierarchy can be distinguished by their proliferative and differentiation activity which they display under conditions designed to optimally elicit these, either in vivo (where the most primitive cells are called long-term repopulating cells, LTRCs) or in vitro (as long-term culture-initiating cells, LTC-ICs and CFCs) [5, 6]. Surface markers, such as CD34 and CD38 are differentially expresses upon differentiation, progenitors being mostly CD34+ CD38+ and HSCs exclusively CD34+ CD38− [7]. In patients with CML-chronic phase (CP), normal and leukemic cell population co-exist (Fig. 1b) [1, 4, 8, 9]. In the stem cell compartment, normal HSCs often outnumber the small numbers of their leukemic counterparts. However, current evidence suggests that the normal HSCs are outcompeted by the CML stem cells when these begin to proliferate and differentiate, which the CML stem cells also attempt more frequently due to their higher turnover and increased probability of differentiation. The autocrine secretion of IL-3 and Granulocyte colony stimulating factor (G-CSF) by primitive leukemic progenitors likely contributes to growth advantage of leukemic myeloid progenitors and mature cells in patients resulting in their dominance of peripheral blood and bone marrow of newly diagnosed CML patients with mature CML cells [6].

(adapted from Ref. [6])
CML-CP stem cells and leukemic myelopoiesis. a Schematic representation showing myelopoiesis in normal adults. Surface markers, such as CD34 and CD38 are differentially expressed upon differentiation. b Schematic representation showing how leukemic myelopoiesis is differently deregulated at different stages of hematopoiesis in patients with CML-CP
CD25
CML stem cell-specific surface molecules are potentially suitable markers for identification of LICs as well as targets for LIC eradication. Many markers are reportedly expressed specifically on CML stem cells, including IL1RAP, CD93, CD26, and CD25 [10,11,12,13]. Among these CD25 is well characterized in both human CML samples and murine CML models. CD25 is an α-chain of high affinity-IL-2 receptor encoded by IL2RA gene [14]. In mouse CML model, CD25 is expressed on the CML lineage(−)Sca-1(+)c-Kit(+) (LSK) fraction, including stem cells [13]. CD25+ LSK cells possess higher leukemia propagation capacity, whereas CD25- LSK cells can initiate leukemia to lesser extent. CD25+ CML LSK cells exhibit gene expression profile resembling mast cells with preferential differentiation toward FcεRI(+) mast cells. Of note, other CML stem cell markers IL1RAP [15] and CD26 [16] are also expressed on normal mast cells and CD25 is expressed on neoplastic mast cells [17], suggesting a common genetic mechanism regulated by BCR-ABL that underlies aberrant expression of CML stem cell-specific markers. Since basophils and mast cells are functionally and evolutionarily related cell types, it is interesting to ask whether this mechanism is also associated with increase in basophils in human CML.
Targeting CD25+ cells using a monoclonal antibody delayed disease onset thus can be a good target for LIC eradication [13]. In humans, CD25 is highly expressed on the CD34+ CD38− stem cell fraction. The expression level of CD25 elevates along with disease progression from CP to AP and AP to BC [13]. CD25 is reportedly regulated by STAT5 activity, thus STAT5 inhibition with TKI treatment resulted in diminished CD25 expression [18]. Interestingly knockdown of CD25 by lentivirus system in human CML samples promoted proliferation of leukemic cells [18], which is apparently contrary to the efficacy of monoclonal antibody treatment against murine CML model. One explanation for this is targeting CD25 by monoclonal antibody not only blocks the signal from IL-2 but also activates antibody-dependent immune response to eliminate CML stem cells.
Given the specificity of CD25 expression in the stem cell fraction, it can also be a potential MRD marker, and now the clinical relevance of CD25 expression in the CD34+ CD38− fraction at diagnosis and during TKI treatment is being tested in a large cohort with 95 patients at Keio University [19].
Recent study examining single-cell gene expression profile of CML stem cells at diagnosis and during TKI treatment further revealed the stem cell-specific expression of CD25 along with CD26 and IL1RAP in CML patients while its expression diminishes after treatment as has been reported [20]. Combining CD25 with other surface markers would facilitate identification of CML stem cells and enable more precise prediction of the outcome through detection of MRD (Fig. 2).

CML-specific surface markers expressed on stem and progenitor cells. CD25, CD26, and CD93 are expressed specifically on the CD34+ CD38− stem cell fraction of CML, while IL1RAP is expressed on both of the CD34+ CD38− stem cell fraction and the CD34+ CD38+ progenitor fraction. The expression of CD25 is reportedly regulated by STAT5 activation
Residual CML stem cells during treatment with ABL-tyrosine kinase inhibitors
The use of tyrosine kinase inhibitors (TKI) such as imatinib mesylate (IM) targeted against BCR-ABL has proven successful in CML and long-term survival has become a reality [21, 22]. However, several mathematical models and ex-vivo examinations suggested that imatinib (IM) therapy does not eradicate CML stem cells [3, 8, 23,24,25]. We previously reported a method for investigation of CML-CP cases during TKI treatment using FACS-sorting and quantitative RT-PCR of BCR-ABL among each population; total mononuclear cells, HSC, and myeloid progenitors [9, 26, 27]. From each population, we collected at least 5000 cells (most samples were over 20,000 cells), and the limited number of sorting cells was one critical reason for the methodological limitation regarding subtle quantitative evaluation. As internal control, we compared ABL, GAPDH, and BCR. For this kind of evaluation using limited number of cells, ABL was not the best from the aspect of expression, and BCR was more suitable. In the HSC population by this method, more than 30% cells are supposed to have stem cell potential, likely as LTC-ICs (Fig. 1b). In optimal responders to IM therapy, BCR-ABL transcripts in the HSC populations tended to be more retentive than other populations. Treatment with the second-generation of ABL-tyrosine kinase inhibitors (2nd TKIs), dasatinib or nilotinib induced more rapid reduction of BCR-ABL transcripts even in the HSC population, which implied that 2nd TKI therapy can be a more promising approach than IM treatment for early reduction of CML stem cells [27]. However, these observations also implied that there was a methodological limitation for subtle quantitative evaluation around complete molecular remission (CMR) during 2nd-TKI treatments.
Mathematical models
The mathematical models based on the previous TKIs clinical studies, mathematicians made a standardized formula about the manner of the BCR-ABL decline, which consists of bi-exponential phases; α-slope with initial rapid decline and β-slope correspondent to kinetics of more residual cells [24]. Our observations were similar with biphasic decreasing in the CD34+38− population. Combined with the results, we developed a hypothesis that the β-slope corresponds mainly to the partial (quiescent, imatinib-insensitive stem cells) CD34+38− population, not to the entire one. Our results showed treatment with 2nd-TKI induced a steeper α-slope in comparison with IM treatment.
Strategy and issues based on the kinetics of CML stem cells
In the nonrandomized Stop Imatinib (STIM) study, IM treatment was discontinued in patients with CML who had achieved complete molecular remission (CMR) of more than 2-year duration [28]. Of the 69% of patients with complete follow-up, 61% relapsed from CMR states (nevertheless, all patients who relapsed responded safely to the reintroduction of IM). The remaining patients maintained CMR states, suggesting that TKI treatment may cure some proportion of patients with CML [29, 30]. Ross et al. proposed the sensitive measurement of minimal residual disease using genomic PCR method with patient-specific primers [31]. Moreover, we need to develop the clinically available biomarker and the evaluation method of the residual CML stem cells to establish rational TKI-cessation strategies in CML. At the same time, we should also continue to discuss about the classification and strategy based on the each clinical goal among CML patients.
ABL001 (Asciminib)
Both of first and second-generation TKIs have improved prognosis of patients with CML. However, CML cells acquire resistance during these TKI treatments. The resistance or intolerance mainly occurs because of BCR-ABL mutations following replacements of amino acids in BCR-ABL. Replacements of amino acids are observed in ABL-tyrosine kinase domains. The structure of BCR-ABL changed and these induce the lack of TKIs binding to ABL. Classic TKIs target the BCR-ABL ATP-binding catalystic site and reduce tyrosine kinase activities. Several mutations were reported [32], and second generation TKIs against most of mutations except for T315I mutation which located in the middle of ATP-binding site. Only ponatinib overcomes T315I mutation. However, new options are needed for patients with resistance or tolerance to ATP-binding TKIs or who do not achieve treatment goals with them.
Recently small molecules were reported to bind to the BCR-ABL myristoyl pocket and inhibit tyrosine kinase activity via an allosteric mechanism [33]. Unfortunately the first small molecule had limited potency and lacked activity against the BCR-ABL T315I mutation. However, second-generation allosteric inhibitors have similar cellular potencies but distinct patterns of resistance mutations [34, 35]. Asciminib (ABL001) is a potent and specific inhibitor of BCR-ABL with a mutation profile complementary to that of ATP-binding TKIs. Asciminib and nilotinib which is a second-generation ATP-binding TKI did not share the resistance with genetic barcoding studies [35]. In this study, rapid tumor regression was seen with single-agent asciminib or nilotinib in the mice xenograft model. However, the resistant emerged due to the A337V and P223S mutations with asciminib and T315I mutations with nilotinib. On the other hand, sustained tumor regression was observed with combination treatment of asciminib and nilotinib.
Based on this study, clinical trials performed to explore asciminib as both a single-agent and in combination with other TKIs in patients with CML-CP. In phase-I open-label study (NCT0281378) which investigates asciminib as a single-agent and in combination with imatinib, nilotinib, dasatinib, single-agent asciminib showed clinical activity and was generally well tolerated in patients with CML-CP who were resistant or intolerant to classic TKIs. In addition, phase-III study is underway to investigate the efficiency of asciminib versus bosutinib [36] in patients treated with classic TKIs (NCT03106779). Combination of ATP-binding TKIs and asciminib will have potential to achieve treatment-free remission in patients with CML.
AIC-47
Recently, anti-cancer drugs targeting energy metabolism is expected to be useful for overcoming drug resistance. One of the hallmarks of cancer glycolytic metabolism “Warburg effect” is achieved through regulated expression of pyruvate kinase isoforms, PKM1 and PKM2, by alternative splicers including PTBP1 [37, 38]. We found that knockdown of BCR-ABL led to perturbation of the Warburg effect through the PTBP1/PKM cascade [39]. Barger et al. also showed that BCR-ABL activates glycolysis and promotes glucose-dependent survival [40]. It is tempting to speculate that BCR-ABL functions one of the key molecules of glycolysis in CML cells.
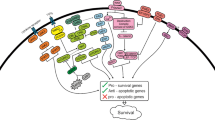
We recently reported medium-chain fatty-acid derivative AIC-47 as a novel anti-cancer agent for the CML therapy [39, 41]. AIC-47 induces transcriptional repression of BCR-ABL and disruption of glycolysis through the down-regulation of c-Myc (Fig. 3). The greatest difference between AIC-47 and TKIs is the effect on the expression of BCR-ABL. TKIs inhibit only the phosphorylation, whereas, AIC-47 suppresses the expression of BCR-ABL itself. It suggests that AIC-47 could affect BCR-ABL-mutant cells. As a consequence of the down-regulation of c-Myc and BCR-ABL, AIC-47 changes the ratio of PKM1/PKM2 through the down-regulation of PTBP1. We also showed that IM perturbs the Warburg effect through the PTBP1/PKM cascade.

AIC-47. Schematic diagram showing the mechanism of AIC-47 in CML cells. Cancer cells achieve the Warburg effect by dominant expression of PKM2, which is regulated by the c-Myc/BCR-ABL/PTBP1 signaling. AIC-47 decreases c-Myc and BCR-ABL, and as a consequence, AIC-47 perturbs the Warburg effect. AIC-47 also blockades the compensatory activation of fatty-acid oxidation through the inhibition of LCAD activity
Glucose metabolism is a central source of energy for cancer cells; however, impaired glycolysis can be compensated by other energy source. Earlier studies showed that stem cells utilize glucose-independent metabolic pathway, such as fatty-acid oxidation (FAO; also known as β-oxidation) or specific dipeptide species [42, 43]. Notably, FAO is essential for maintenance and chemoresistance of LSCs, and proposed to be as a potential target for CML therapy [44]. Our findings indicate that the perturbation of the Warburg effect by IM activates FAO as compensatory energy supplier; however, AIC-47 inhibits FAO [41]. AIC-47 exhibited cytotoxicity in cultures of CD34+ cells by modulation of both glycolysis and FAO. The docking experiment indicated that AIC-47 has highly binding affinity to PPARγ [39]. Prost et al. showed that PPARγ activation decreases the expression of STAT5 and HIF-2a/CITED2, which are key guardians of the quiescence and stemness of CML LSCs [45]. The interaction of AIC-47 and PPARγ may be one of the causes of effects of AIC-47 on CD34+ cells. Furthermore, pharmacological blockade of FAO improves the sensitivity of IM in CD34+ cells [41]. We speculate that the modulation of both glycolysis and FAO could be a useful strategy for radical cure of CML. Further investigation into the workings of metabolism and resistance will help us to refine rational approaches to CML therapies for overcoming resistance.
References
Melo JV, Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer. 2007;7:441–53.
Chen Y, Peng C, Sullivan C, Li D, Li S. Critical molecular pathways in cancer stem cells of chronic myeloid leukemia. Leukemia. 2010;24:1545–54.
Jiang X, Zhao Y, Smith C, Gasparetto M, Turhan A, Eaves A, et al. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia. 2007;21:926–35.
Jiang X, Forrest D, Nicolini F, Turhan A, Guilhot J, Yip C, et al. Properties of CD34+ CML stem/progenitor cells that correlate with different clinical responses to imatinib mesylate. Blood. 2010;116:2112–21.
Coulombel L, Kalousek DK, Eaves CJ, Gupta CM, Eaves AC. Long-term marrow culture reveals chromosomally normal hematopoietic progenitor cells in patients with Philadelphia chromosome-positive chronic myelogenous leukemia. N Engl J Med. 1983;308:1493–8.
Sloma I, Jiang X, Eaves AC, Eaves CJ. Insights into the stem cells of chronic myeloid leukemia. Leukemia. 2010;24:1823–33.
Seita J, Weissman IL. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip Rev Syst Biol Med. 2010;2:640–53.
Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2011;121:396–409.
Jamieson CHM, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351:657–67.
Järås M, Johnels P, Hansen N, Agerstam H, Tsapogas P, Rissler M, et al. Isolation and killing of candidate chronic myeloid leukemia stem cells by antibody targeting of IL-1 receptor accessory protein. Proc Natl Acad Sci USA. 2010;107:16280–5.
Kinstrie R, Horne GA, Morrison H, Moka HA, Cassels J, Dunn K, et al. CD93 is a novel biomarker of leukemia stem cells in chronic myeloid leukemia. Blood. 2015;126:49–9.
Herrmann H, Sadovnik I, Cerny-Reiterer S, Rülicke T, Stefanzl G, Willmann M, et al. Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood. 2014;123:3951–62.
Kobayashi CI, Takubo K, Kobayashi H, Nakamura-Ishizu A, Honda H, Kataoka K, et al. The IL-2/CD25 axis maintains distinct subsets of chronic myeloid leukemia-initiating cells. Blood. 2014;123:2540–9.
Minami Y, Kono T, Miyazaki T, Taniguchi T. The IL-2 receptor complex: its structure, function, and target genes. Annu Rev Immunol. 1993;11:245–68.
Ali S, Huber M, Kollewe C, Bischoff SC, Falk W, Martin MU. IL-1 receptor accessory protein is essential for IL-33-induced activation of T lymphocytes and mast cells. Proc Natl Acad Sci USA. 2007;104:18660–5.
Gschwandtner M, Paulitschke V, Mildner M, Brunner PM, Hacker S, Eisenwort G, et al. Proteome analysis identifies L1CAM/CD171 and DPP4/CD26 as novel markers of human skin mast cells. Allergy. 2017;72:85–97.
Sotlar K, Horny H-P, Simonitsch I, Krokowski M, Aichberger KJ, Mayerhofer M, et al. CD25 indicates the neoplastic phenotype of mast cells: a novel immunohistochemical marker for the diagnosis of systemic mastocytosis (SM) in routinely processed bone marrow biopsy specimens. Am J Surg Pathol. 2004;28:1319–25.
Sadovnik I, Hoelbl-Kovacic A, Herrmann H, Eisenwort G, Cerny-Reiterer S, Warsch W, et al. Identification of CD25 as STAT5-dependent growth regulator of leukemic stem cells in Ph+ CML. Clin Cancer Res. 2016;22:2051–61.
Karigane D, Kasahara H, Sakurai M, Matsuki E, Tozawa K, Koda Y, et al. Correlation of expression of CD25 in hematopoietic stem/progenitor cell fraction of bone marrow cells with response to tyrosine kinase inhibitors in chronic myelogenous leukemia patients. Blood. 2016;128:3061–1.
Warfvinge R, Geironson L, Sommarin MNE, Lang S, Karlsson C, Roschupkina T, et al. Single-cell molecular analysis defines therapy response and immunophenotype of stem cell subpopulations in CML. Blood. 2017;129:2384–94.
Druker BJ. Translation of the Philadelphia chromosome into therapy for CML. Blood. 2008;112:4808–17.
Shami PJ, Deininger M. Evolving treatment strategies for patients newly diagnosed with chronic myeloid leukemia: the role of second-generation BCR-ABL inhibitors as first-line therapy. Leukemia. 2012;26:214–24.
Michor F, Hughes TP, Iwasa Y, Branford S, Shah NP, Sawyers CL, et al. Dynamics of chronic myeloid leukaemia. Nature. 2005;435:1267–70.
Roeder I, Horn M, Glauche I, Hochhaus A, Mueller MC, Loeffler M. Dynamic modeling of imatinib-treated chronic myeloid leukemia: functional insights and clinical implications. Nat Med. 2006;12:1181–4.
Chu S, McDonald T, Lin A, Chakraborty S, Huang Q, Snyder DS, et al. Persistence of leukemia stem cells in chronic myelogenous leukemia patients in prolonged remission with imatinib treatment. Blood. 2011;118:5565–72.
Abe A, Minami Y, Hayakawa F, Kitamura K, Nomura Y, Murata M, et al. Retention but significant reduction of BCR-ABL transcript in hematopoietic stem cells in chronic myelogenous leukemia after imatinib therapy. Int J Hematol. 2008;88:471–5.
Minami Y, Abe A, Minami M, Kitamura K, Hiraga J, Mizuno S, et al. Retention of CD34 + CML stem/progenitor cells during imatinib treatment and rapid decline after treatment with second-generation BCR-ABL inhibitors. Leukemia. 2012;26:2142–3.
Mahon F-X, Réa D, Guilhot J, Guilhot F, Huguet F, Nicolini F, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11:1029–35.
Deininger M. Hematology: curing CML with imatinib—a dream come true? Nat Rev Clin Oncol. 2011;8:127–8.
Pellicano F, Sinclair A, Holyoake TL. In search of CML stem cells’ deadly weakness. Curr Hematol Malig Rep. 2011;6:82–7.
Ross DM, Hughes TP, Melo JV. Do we have to kill the last CML cell? Leukemia. 2011;25:193–200.
Soverini S, Hochhaus A, Nicolini FE, Gruber F, Lange T, Saglio G, et al. Bcr-Abl kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. Blood. 2011;118:1208–15.
Adrián FJ, Ding Q, Sim T, Velentza A, Sloan C, Liu Y, et al. Allosteric inhibitors of Bcr-abl-dependent cell proliferation. Nat Chem Biol. 2006;2:95–102.
Zhang J, Adrián FJ, Jahnke W, Cowan-Jacob SW, Li AG, Iacob RE, et al. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature. 2010;463:501–6.
Wylie AA, Schoepfer J, Jahnke W, Cowan-Jacob SW, Loo A, Furet P, et al. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature. 2017;543:733–7.
Cortes JE, Gambacorti Passerini C, Deininger MW, Mauro MJ, Chuah C, Kim D-W, et al. Bosutinib versus imatinib for newly diagnosed chronic myeloid leukemia: results from the randomized BFORE trial. JCO. 2018;36:231–7.
Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008;452:181–6.
Clower CV, Chatterjee D, Wang Z, Cantley LC, Vander Heiden MG, Krainer AR. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc Natl Acad Sci USA. 2010;107:1894–9.
Shinohara H, Taniguchi K, Kumazaki M, Yamada N, Ito Y, Otsuki Y, et al. Anti-cancer fatty-acid derivative induces autophagic cell death through modulation of PKM isoform expression profile mediated by bcr-abl in chronic myeloid leukemia. Cancer Lett. 2015;360:28–38.
Barger JF, Gallo CA, Tandon P, Liu H, Sullivan A, Grimes HL, et al. S6K1 determines the metabolic requirements for BCR-ABL survival. Oncogene. 2013;32:453–61.
Shinohara H, Kumazaki M, Minami Y, Ito Y, Sugito N, Kuranaga Y, et al. Perturbation of energy metabolism by fatty-acid derivative AIC-47 and imatinib in BCR-ABL-harboring leukemic cells. Cancer Lett. 2016;371:1–11.
Naka K, Jomen Y, Ishihara K, Kim J, Ishimoto T, Bae E-J, et al. Dipeptide species regulate p38MAPK-Smad3 signalling to maintain chronic myelogenous leukaemia stem cells. Nat Commun. 2015;6:8039.
Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, et al. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med. 2012;18:1350–8.
Velez J, Pan R, Lee JTC, Enciso L, Suarez M, Duque JE, et al. Biguanides sensitize leukemia cells to ABT-737-induced apoptosis by inhibiting mitochondrial electron transport. Oncotarget. 2016;7:51435–49.
Prost S, Relouzat F, Spentchian M, Ouzegdouh Y, Saliba J, Massonnet G, et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARγ agonists. Nature. 2015;525:380–3.
Acknowledgements
The preparation of this review was partially supported by Grants-in-Aid from the National Institute of Biomedical Innovation and from the Ministry of Education, Culture, Sports, Science and Technology on Scientific Research, Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Y. Minami received research grants from Ono, Bristol-Myers Squibb, Novartis, and Kyowa-Kirin. They did not in any way influence the content of the paper.
About this article

Cite this article
Inoue, A., Kobayashi, C.I., Shinohara, H. et al. Chronic myeloid leukemia stem cells and molecular target therapies for overcoming resistance and disease persistence. Int J Hematol 108, 365–370 (2018). https://doi.org/10.1007/s12185-018-2519-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-018-2519-y