
Abstract
Chronic myeloid leukemia (CML) is a clonal myeloproliferative neoplasm driven by BCR-ABL1 oncoprotein, which plays a pivotal role in CML pathology, diagnosis, and treatment as confirmed by the success of tyrosine kinase inhibitor (TKI) therapy. Despite advances in the development of more potent tyrosine kinase inhibitors, some mechanisms particularly in terms of CML leukemic stem cell (CML LSC) lead to intrinsic or acquired therapy resistance, relapse, and disease progression. In fact, the maintenance CML LSCs in patients who are resistance to TKI therapy indicates the role of CML LSCs in resistance to therapy through survival mechanisms that are not completely dependent on BCR-ABL activity. Targeting therapeutic approaches aim to eradicate CML LSCs through characterization and targeting genetic alteration and molecular pathways involving in CML LSC survival in a favorable leukemic microenvironment and resistance to apoptosis, with the hope of providing a functional cure. In other words, it is possible to develop the combination therapy of TKs with drugs targeting genes or molecules more specifically, which is required for survival mechanisms of CML LSCs, while sparing normal HSCs for clinical benefits along with TKIs.
Similar content being viewed by others


Introduction
Chronic myeloid leukemia (CML) is a clonal myeloproliferative neoplasm featured with uncontrolled proliferation of myeloid cells at every stage of differentiation [1]. CML is the first discovery of a link between cancer and chromosomal abnormality, which also called the Philadelphia (Ph) chromosome, due to the reciprocal translocation that happens between the Abelson murine leukemia virus (ABL) gene on chromosomes 9 and the breakpoint cluster region (BCR) on chromosomes 22 (t9;22) (q34;q11) [2]. The presence of fusion oncogene BCR-ABL1 and generating a 210 KD chimeric oncoprotein (P210) with the constitutive activity of tyrosine kinase leads to clonal expansion of hematopoietic stem cell (HSC) and the pathogenesis of CML [3]. BCR-ABL1 contributes to CML leukemia stem cells (LSCs), identified by survival promotion, the capacity of self-renewal, and differentiation to aberrant hematopoietic subsets and resistance to apoptosis through the activation of various signaling molecules and pathways in downstream of the BCR-ABL protein, to regulate leukemogenesis [4, 5]. The LSCs can be also a persistent problem of CML patients since regardless of BCR-ABL kinase activation, they alter the relationship with bone marrow niche and leave the quiescence state and reach to proliferation state via intracellular signaling changes and suppressive cytokines secretion that can disrupt metabolic processes, prepare an anti-apoptotic microenvironment, and dysregulate immunological activities [6].
Allogeneic transplantation (bone marrow (BM)/HSC transplantation) is considered as the curative treatment of CML, although finding a suitable donor is the problematic factor for the treatment. In recent years, the discovery of imatinib mesylate (IM), as the first designed BCR-ABL1 tyrosine kinase inhibitor (TKI) and additional second- (dasatinib, nilotinib (NIL), and bosutinib) and third-generation (ponatinib) TKIs have dramatically enhanced the chance of survival in CML patients [7]. However, the major barrier against treatment of CML via the family of TKIs is primary and acquired resistance resulted from the accumulation of LSCs with a high heterogeneity in the blood and BM [8]. In other words, CML therapies including cytotoxic chemotherapy and TKIs cannot kill the population of quiescent LSCs, which are in charge of the subsequent relapse and CML stage progression. CML is a triphasic stage disease that begins with a chronic phase (CP), which is characterized by a significant increase in myeloid precursors and lasts about 3–5 years; however, without therapeutic intervention or in cases of refractory disease after 3-18 months, the disease progresses spontaneously toward accelerated phase (AP), and eventually to highly aggressive blast crisis phase (BCP), with a rapid expansion of primary bone marrow cells that spread to the circulatory system [9, 10].
In brief, CML stem cells can be either expressive or independent of BCR-ABL1 and represent TKI-resistant [11]. Therefore, the discovery of uncertain CML LSCs resistance mechanisms can be a great target to facilitate the eradication of these cells. This review underlines the current known characteristics of CML LSCs besides possible mechanisms and pathways that lead to survival and resistance to targeted multimodal therapies that might help developing new drugs and treatment options, since current therapeutic strategies often represent limitations and failures.
Origins and evolution of CML LSCs
In 1960, an abnormal “minute chromosome” which later became known as the “Philadelphia (Ph) chromosome” was identified in patients with CML by Peter Nowell and David Hungerford, representing the first discovery of a link between chromosomes and cancer [12]. Thirteen years later, Janet Rowley observed that the Ph chromosome was the result of the reciprocal translocation between chromosomes 9 and 22 which usually takes place in HSCs as the top of the hematopoietic hierarchy [13]. Over ten years later, it became evident that the human homolog of the Abelson murine leukemia viral oncogene (ABL1) in the region of chromosome 9 translocates to breakpoint cluster region (BCR) on chromosome 22 [14, 15]. In the early 1990s, the first convincing evidence of CML stem cells came from early observations in which transfusion of unirradiated white blood cells obtained by leukapheresis from CML patients with high granulocyte counts into severely neutropenic recipients, resulting in the transient repopulated Ph+/BCR-ABL+ cells [16]. In the following years, it is believed that the cell of origin in CML is characterized through specific genomic alteration of the CD34+CD38− subset of HSCs leading to Ph chromosome and the expression of fusion tyrosine kinase BCR-ABL1 and leukemogenesis [9]. However, the BCR-ABL1 translocation can be formed to three types of chimeric proteins with different mass and biological features as a result of breakage in one of the three regions of the BCR gene, named major (M-bcr), minor (m-bcr), and micro-bcr (µ-bcr) [17]. Fusion of BCR exon 13 or 14 (e13/e14) with ABL1 exon 2 (a2) forms a fusion gene referred to as e13a2 (b2a2) or e14a2 (b3a2), leading to the P210BCR-ABL1 protein (M-bcr) seen in the majority of CML patients [17, 18]. In rare cases of CML and two-thirds of Ph-positive acute lymphoblastic leukemia (Ph+ ALL), the breakpoint occurs in the first intron on the BCR gene (m-BCR), creating the P190BCR-ABL1 from the e1a2 transcript. Approximately one-third of Ph+ ALL patients will express one of the longer BCR-ABL1 isoforms [17,18,19]. In 2009, both Mullighan et al., and Den Boer et al. described a new subtype of ALL, named Philadelphia chromosome-like (Ph-like) or BCR-ABL1-like ALL, respectively [20, 21]. In 2016, the World Health Organization (WHO) classifies BCR-ABL1-like ALL as a provisional entity of hematopoietic neoplasm with a lack of the BCR-ABL1 translocation (about 90%) but having a highly similar profile of gene expression to that seen in ALL with BCR-ABL1 (Ph+ ALL ) and also a high frequency of deletions of IKAROS family zinc finger 1 (IKZF1) [22]. Overall, BCR-ABL1 fusion is present in essentially all cases of CML and in about 3-5% of pediatric ALL and 25% of adult ALL (mostly B-ALL) [22], while BCR-ABL1-like ALL subset of patients goes for about 20% of B-ALL cases overall. Moreover, about 1% of all patients with acute myeloid leukemia (AML) have Philadelphia translocation, which is cytogenetically indistinguishable from Ph+ CML; meanwhile, the breakpoint on chromosome 22 in Ph+ AML is distinct from CML [23, 24].
In 1986, BCR-ABL1 molecular aberration defines as the start-up of a chronic phase (CP), which in the absence of effective therapy, leads toward acute phase by further genetic instability and acquisition of mutations as a result of the P210BCR-ABL1 activity [25, 26]. All CML patients in blast crisis phase co-express both co-express P210BCR-ABL1 and P190 BCR-ABL1 transcripts [27]. Mutations in ABL1, IKZF1, RUNX1, ASXL1, and TP53, including an overrepresentation of ABL1 and IKZF1 mutations in lymphoid blast crisis, are among the additional genetic aberrations that occur during progression to accelerated phase and aggressive blast crisis [28]. However, the detection of low levels of BCR-ABL1 transcripts in healthy individuals [29] implies that a translocation has happened in a cell that does not possess the ability to initiate leukemia. In other words, BCR-ABL is required but not sufficient for the production of CML LSCs, which may necessitate further BCR-ABL copy number multiplication, subsequent mutations, and genomic instability [9]. Over the total genetic alterations of HSCs into CML LSCs, activation of various signaling pathways leads to CML progression as an LSC-derived disease, along with dysfunction of underlying cellular processes.
Properties of CML LSCs
A main challenge in developing strategies to provide novel ways for therapeutic targeting of CML LSCs is how to isolate and characterize these cells to increase therapeutic effects and reduce toxicities. One of the ways to identify is extensive analyses of unique cell surface antigens on CML LSCs, which must be specific or have a distinct expression pattern, density, or dispersion. Besides, similar to normal HSCs, CML LSCs can self-renew, differentiate, or enter dormancy. Moreover, knowing signaling pathways and mechanisms involved in the differentiation and survival of CML LSCs is another step to develop different therapeutic strategies to eliminate them [11].
Cell surface markers
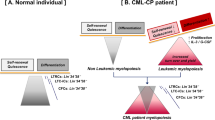
CML LSCs share many similarities with HSCs including cell surface phenotype such as Lin−CD34+CD38−. Many markers are reportedly expressed specifically on CML LSCs, including CD25, CD26, and interleukin-1 receptor accessory protein (IL-1RAP), which can be used as biomarkers for diagnostic, prognostic, or therapeutic purposes [30,31,32]. The increased expression of CD25 is related to the reduction in the proliferation capacity of CML LSCs and CML disease progression. Therefore, CD25 (IL2RA) is suggested as a marker of CML LSCs and a negative regulator of growth [33]. Furthermore, the IL-2-CD25 axis plays a critical role in the preservation of CML LSCs by a massive expansion of the LSCs during TKI resistance, which makes CD25 a novel marker and drug-sensitive suppressor in CML therapy [34, 35]. IL-1RAP, as an interleukin 1 receptor-1 (IL1R1) coreceptor, leads to increased proliferation of CML LSCs by binding of IL-1A or IL-1B and activation of the NF-κB, JNK, and P38-MAPK signaling pathways [33]. CD26 (dipeptidyl peptidase-4 (DPP4)) is involved in releasing CML LSCs into the bloodstream from the BM through cleaving stromal-derived factor (SDF-1: CXCL12)-CXCR4 (CD184) axis, which can lead to the disruption of interactions within the stem cell niche and spread of the disease regardless of specific niche regulations. Furthermore, as TKI resistance improved, the number of CD26+ CML LSCs increased [36]. Additionally, only CD26 appears to offer clinical application, although, in the preclinical era, other biomarkers of CML LSCs have been discovered [37]. CD33 (Siglec-3) expressed at greater levels in CML LSCs [38]. CD36 (scavenger receptor-B2 (SR-B2)) is a marker of immature CML cells that its upregulated expression on CD34+CD38low CML cells in chronic phase leads to less sensitivity to IM treatment [39]. In addition, in blast crisis CML, CD36 has indicated the mediation of fatty acid uptake with distinct metabolic properties [40]. There are other additional antigens, including CD44, CD47 (IAP), CD529 (Campath-1), CD56 (NCAM), CD90 (Thy-1), CD93, CD114 (granulocyte colony-stimulating factor (G-CSF) receptor), CD123 (IL-3RA), CD135 (FMS-like tyrosine kinase 3 (FLT3)), and CD295 (LEPR) that are also present on CML LSCs [34, 41,42,43]. CD44 is important for the engraftment of CML cells by its interaction with different bone marrow microenvironment (BMM)-associated proteins such as hyaluronan, osteopontin, or E-selectin [44]. The expression of CD93 is constant and selective in the subset of CML LSCs, which indicated self-renewal and proliferation capacity of this primitive cell subpopulation by the expression of selected genes such as ID2, MYB, PAK2, MEIS1, REL, CK1, CDK4, and CCND2. Furthermore, CD93, as a major integrin α5β1 mediator, along with CD44 can bind to fibronectin [45, 46] and might have a role in LSC adherence within the BM niche [37, 44]. Although CD93 is consistently and selectively expressed on CML LSCs, it can also be found on other cell types such as platelets and endothelial cells; therefore, CD93 is unlikely to serve as a therapeutic target, while it can be used as a prognostic biomarker to differentiate CML patients at high risk for molecular relapse after treatment discontinuation [47]. The possible profile of LSCs in CML could be Lin−CD34+CD38−CD45RA−/Low, CD117 (KIT)−, CD26+, and CD90+ that are identified as potential therapeutic targets at a single-cell level with a higher proliferative and colony-forming potential than normal HSCs [37, 48]. It has been reported that stem cell factor receptor (SCFR), also known as c-Kit receptor or CD117, was expressed on CD34+CD38low CML cells, although healthy cells have represented a greater level of SCFR expression [49]. However, the precise immunophenotype of these primitive CML cells is not completely determined, while the discovery of novel cell surface antigens on primitive CML cells might lead to novel treatment strategies.
Self-renewal and differentiation
LSCs have a self-renewal capacity to generate large numbers of leukemia progenitor cells (CD34+CD38+) through the activation of several signaling pathways including WNT/β-catenin, Hedgehog (Hh), Notch, transforming growth factor-β (TGF-β)/Forkhead box O (FOXO), and Musashi 2 (Msi2)-Numb signaling [50]. The P210BCR−ABL1 also regulates miRNA stability and induces an increase in self-renewal of CML LSCs by mediating the activation of Janus kinase (JAK)/STAT signaling pathway, increasing adenosine deaminases acting on double-strand RNA1 (ADAR1) enzyme levels, which hampers biogenesis of the miR precursor (miR-let7) and increased LIN28B pluripotency gene expression [51, 52]. Moreover, the expression of the krüppel-like factor 4 (KLF4) is essential for the regulation of LSC self-renewal and maintenance of CML. In this way, in the absence of KLF4 in CML LSCs, dual specificity tyrosine phosphorylation regulated kinase 2 (DYRK2) was remarkably upregulated, which in turn mediated the activation of P53 and c-MYC, and facilitated proteasomal degradation through phosphorylation, leading to apoptosis and decrease in the self-renewal in CML LSCs [53]. BMI1, a polycomb protein belonging to the polycomb repressive complex 1 (PRC1) family, works with BCR-ABL and is also involved in proliferation and self-renewal of LSCs [54]. MicroRNAs may be effective in self-renewal and maintenance of CML LSC, in particular miR-126, which regulates the dormancy of normal stem cells including CML LSCs [55]. CML stem cells could be able to maintain the energy output required for self-renewal capability by utilizing anaplerosis, which is a series of metabolic reactions in which critical TCA cycle intermediates, especially oxaloacetic acid, are produced [56]. BCR-ABL1 oncoprotein stimulates CML LSC self-renewal by increasing the expression of protein phosphatase 2A (PP2A) inhibitors, such as protein SET (SET), cancerous inhibitor of PP2A (CIP2A), and PP2A-Aα, resulting in inactivation of PP2A and MYC overexpression, LSC survival, and the establishment of a positive feedback loop for CML development to BP [57]. Besides, the high expression and activation of viral integration site 1 (EVI1) in CD34+CD38– stem cells leads to increase self-renewal capacity, leukemia development and the disease progression to BP [58]. LIGHT/lymphotoxin-β receptor (LTβR) signaling regulates quiescence and self-renewal of LSCs and HSCs by reducing cell proliferation and favoring symmetric cell division over asymmetric division. Since asymmetric division leads to differentiation and exhaustion of stem cells, targeting the LIGHT/LTβR pathway may offer a novel strategy to induce differentiation and eliminate LSCs [59].
CML LSCs in chronic phase can simultaneously differentiate into a variety of malignant leukocytes that are morphologically mature and molecularly expressing BCR-ABL. As a result, the leukocyte population in CP are heterogeneous, with a limited number of LSCs and the bulk of leukemia non-stem cells [60]. Alternatively, leukemia non-stem cells including basophils and megakaryocytes (the platelet’s precursor) can play a role in CML pathogenesis and development [61]. Basophil-like CML cell-derived mediators such as hepatocyte growth factor (HGF) [62] and tryptase [63] might possibly trigger CML progression by altering the bone marrow niche, leading to increased fibrosis and a supportive growth environment for LSCs, as well as production of CCL3, which can facilitate CML LSCs proliferation [61]. BCR-ABL-expressing megakaryocytes in CML can induce senescence along with expression of TGF-β1 in a P16- and P21-dependent manner, supporting the maintenance of CML LSCs [64]. Furthermore, uncontrolled expression of c-MYC/miR-150 in LSC and Ph+ cells downregulates BCL-2 inhibitor (miR-153-3p), resulting in impairment of myeloid differentiation and increase TKI resistance [65]. It has been proposed that the enhancement of self-renewal and compromising differentiation can affect disease progression that appears to result from aberrant activation of self-renewal signaling pathways in a progenitor cell population or failure to shut down these pathways during differentiation processes. Therefore, they might be a promising therapeutic target for CML LSCs.
Survival and functional resistance of CML LSCs to therapies
Several factors are suggested to be important in the maintenance of CML LSCs through either the direct activation by kinase activity of BCR-ABL or independent of BCR-ABL kinase activity [6]. CML LSCs could avoid immune surveillance via a number of molecular pathways [66]. The other required factors in survival of CML LSCs are bone marrow resident cells and the support of LSCs in localization to the bone marrow niche [67]. It is thought that intrinsic regulatory systems and extrinsic microenvironmental signals are responsible for controlling quiescence of LSCs. In addition, the connection with the bone marrow niche and signaling molecules associated with self-renewal and survival are two key components in the long-term persistence of quiescent LSCs in CML, resulting in resistance to targeted therapy and increased risk of disease recurrence following therapy withdrawal [68, 69].
Bone marrow microenvironment (BMM)
Numerous types of cells in the bone marrow niche, including osteoblasts, endothelial cells, mesenchymal stromal/stem cells (MSCs), megakaryocytes, and neural cells, interact with normal HSCs via various mediators and signaling pathways [50]. BCR-ABL1T315I mutation leads to the maintenance of quiescent stem cells in the presence of TKIs through alternations in niche localization and increased level of integrin β3 and integrin-linked kinase (ILK) [70]. LSCs engraft in a different way than common HSCs, using distinct niche signals. There are controversial results regarding homing of CML LSCs, which displays impaired homing and persistence in the BM by inhibiting CXCL12 expression [71]. On the contrary, the elevated level of CXCR4 protein drives CML LSCs homing into the BMM leading to quiescence and TKI resistance. Hence, the decreased level of CXCR4 as a result of the inhibition of BCR-ABL1 tyrosine kinase activity can cause distribution of CML LSCs into the peripheral blood from the BMM [33]. Malfunction of β1-integrin (VLA4 or VLA5) causes redistribution of CML stem cells into the peripheral blood and different organs, which can potentially lead to uncontrolled extramedullary myeloproliferation and local LSC pools [72]. Multiple adhesion molecules expressed by LSCs, such as cadherins, CD44, and galectin-3, can also aid in the localization of LSCs to bone marrow niche to survive [61].
Furthermore, bone marrow resident cells support CML LSCs persistence by releasing various soluble substances such as JAGGED1 (a Notch ligand), TGF-β, bone morphogenic proteins (BMPs), chemokines, CXCL12, IL-1, and exosomes containing miR-126 [61]. Response of CML LSCs to the BMP/TGFβ superfamily is dysregulated to make the BMM more suitable for their survival by overexpression of the BMP type I receptors (BMBR1B) [73]. Furthermore, stimulation of the Notch signaling pathway in the BMM by the overexpression of Jagged2 reduces CML LSC cycling, indicating that Notch signaling fosters an antileukemic milieu, although the overexpression of Notch targets HES1 in CML patients in blast crisis, indicating that it may play a role in the development of CML from chronic phase to BP. As a result, the effects of Notch in CML LSCs need to be further investigated [74]. Altogether, the increased expression of BMPR1B, BMP2/4, and the embryonic transcription factor TWIST1, as well as the co-activation of SMAD1/5/8 and JAK2/STAT3 signaling are involved in LSC survival, treatment resistance and disease progression [75,76,77].
The CML-derived microvesicles (MVs) represent communication vehicles with the microenvironment which can shuttle miRNAs, amphiregulin, as well as BCR-ABL mRNAs to close stromal cells and cause reprogramming of niche cell functions [78]. CML-derived exosomes transfer miR-92a and activate SRC signaling, which induces phosphorylation of AKT and ERK1/2 and oncogenic signaling by BCR-ABL. However, this activation process can be reduced by TKI treatment [79, 80]. In addition, the secretion of exosomes containing amphiregulin along with CML LSCs can promote CML LSCs adhesion and their survival by activation the epidermal growth factor (EGFR) pathway in MSCs and increase secretion of IL-8, which stimulates human vascular endothelial cells to enhance the expression of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) [81, 82]. MSCs also play a role in maintaining CML stem cells in the BMM and TKI resistance through the upregulation of promyelocytic leukemia (PML), a tumor suppressor protein, and the cell quiescence regulator, to repair double-strand DNA breaks in MSCs that are used to upregulate inflammatory cytokines such as IL-6, IL-8 (CXCR2L) and CXCL1/CXCR2 as well as activation of various signaling pathways [83]. Therefore, targeting CXCR2 signaling might be a novel targeted therapy in TKI-resistant CML patients through the suppression of AKT/mTOR and c-MYC [84]. Moreover, MSCs can cause an increase in proliferation, self-renewal, and anti-apoptotic capability of CML LSCs and TKI resistance by the secretion of exogenous WNT and activation of WNT/β-catenin signaling during progression to the advanced phase of the disease [85,86,87]. The production of fibroblast growth factor 2 (FGF2) by BM-derived MSCs and the decreased level of reactive oxygen species (ROS) in CML LSCs also increases TKI resistance [88, 89].
Other way to enhance survival of CML progenitors is the reduced oxygen in the microenvironment formed a gradient of oxygen from the blood vessel to hypoxic areas [90]. In hypoxic BMM, there are increased levels of hypoxia-inducible factors (HIFs), being essential for regulation of proliferation, quiescence, TKI resistance, and survival of CML LSCs, as well as increased capacity of HSCs to transform into LSCs, mediated by increased glycolytic flux via glucose transporter 1 (GLUT1) and tumor M2-pyruvate kinase (PKM2), overexpression of P21, suppression of P53, increased transcription of antioxidant factors (FOXO and NRF2), and uptake of c-MYC [91,92,93]. In order to adjust to low oxygen concentrations in the BMM to facilitate dormancy and self-renewal of CML LSC, the STAT5/HIF-2α/CITED pathway is activated. However, peroxisome proliferator-activated γ (PPARγ), a negative regulator of this pathway, inhibits LSCs adhesion to the extracellular matrix and drives apoptosis [94, 95].
Autophagy
Autophagy is a vital pathway for survival and drug resistance of BCR-ABL1 expressing cells, resulting in reduced P53-mediated stress response and pro-apoptotic signals to avoid cell death, although the constitutive activation of BCR-ABL kinase can downregulate autophagy by activation of the phosphoinositide 3-kinase (PI3K)/AKT/FOXO4 pathway and the upregulation of the mTOR pathway [96, 97]. The increased expression of autophagy genes such as autophagy-related 4B cysteine peptidase (ATG4B) in CD34+ CML cells indicates that ATG4B may be a potential target for CML LSCs that its knockdown impairs proliferation and survival of CML stem cells [98]. Autophagy can also be linked to metabolism; in this way that knockdown of ATG7 caused a reduction in glycolysis, an elevation in oxidative phosphorylation, and an accumulation of mitochondrial ROS in CML cells [76].
Energy metabolism
In order to induce the survival and maintenance of stemness in LSCs, the metabolic alteration results in increased glucose flux, glycolysis, pyruvate transferring, anaplerosis, oxidative phosphorylation, and ROS accumulation [99]. BCR-ABL1 can stimulate aerobic glycolysis (the Warburg effect) in CML cells through activation of the PI3K/AKT pathway [100]. CML LSCs also represent a drastic increase in mitochondrial oxidative phosphorylation, which was controlled by activation of oxidative metabolism genes [56]. Interestingly, STAT3 has been connected to mitochondrial respiration; thus, it may assume that STAT3 involved in abnormal energy metabolism in CML LSCs and TKI-resistant. STAT3-mediated metabolic regulation might link to BMM-mediated quiescence and survival of LSCs [101, 102].
Fatty acid metabolism is also important for CML LSC survival [103]. CML LSCs show greater lipolysis and fatty acid oxidation capacity in comparison with differentiated CML cells [40, 56]. Fatty acid metabolism may be an additional factor in increasing the dependence of myeloid LSCs on oxidative phosphorylation. However, redundancy in fatty acid metabolism and signaling pathways, as well as a multiplicity of various fatty acid-binding proteins, may complicate targeting this process in LSCs [102]. Arachidonate lipoxygenase (ALOX5)-5 and ALOX15 are also involved in LSC self-renewal and fatty acid metabolism by changing arachidonic acid to leukotrienes such as leukotriene B4 (LTB4), which increases the expression of the leukotriene B4 receptor 2 (BLT2) in CML stem cells [104, 105]. Moreover, lysophospholipid metabolism plays a part in the maintenance of CML stem cells and TKI resistance by regulation of lysophospholipase D activity of GDPD3 in the BCR-ABL1-independent manner signaling pathway, and FOXO3A/β-catenin interaction [106].
In the hypoxia condition, metabolic reprogramming of CML LSCs is a pathway of BCR-ABL1-independent TKI resistance. The elevated amount of intracellular dipeptide amino acids in CML LSCs can cause CML LSC maintenance by the stimulation of the P38/MAPK pathway and phosphorylation of SMAD3/FOXO3A [107]. Besides, CML LSCs under hypoxic conditions induces upregulation of genes involved in carbohydrate metabolism to support energy production. Various enzymes associated with glucose metabolism are able to mediate CML cell survival despite TKI-mediated BCR-ABL1 inhibition, including the enzyme dihydrolipoamide S-acetyltransferase (DLAT), involved in the pyruvate dehydrogenase (PDH) complex [90]. The activation and upregulation of BCAA transaminase 1 (BCAT1), a cytosolic aminotransferase for branched-chain amino acids (BCAA), by Musashi2 is functionally needed for CML progression and metabolic reprogramming. The increased BCAA metabolism triggers activation of mTORC1 and increase BC-CML-initiating cells survival; hence, blocking BCAA metabolism and enzymatic activity of BCAT1 inhibits proliferation and causes cellular differentiation in blast crisis CML cells [103]. However, further research is needed to understand how CML LSCs become dependent on oxidative metabolism for survival.
Signaling pathways and potential molecules
The generation of BCR-ABL fusion gene in CML LSCs may have a role in a variety of biological processes, notably cell proliferation, differentiation, and apoptosis by encoding a constitutively activating tyrosine kinase as important mediator of signaling cascades such as RAS/mitogen-activated protein kinase (MAPK), PI3K/Akt/FOXO axis, WNT signaling, and JAK2/STAT3,5 signaling, which are implicated in the maintenance and survival of CML LSCs (Fig. 1) [61, 108]. As a result of the signaling pathways upregulation, the level of ROS and genomic instability increased that have a potential to create other mutations and chromosomal abnormalities, following the advancement of CML from the CP to the BP [109, 110].

Role of signaling pathways in survival of CML LSC
The high-affinity attachment of the growth factor receptor-bound protein 2 (GRB2) and the scaffolding protein Gab2 following the phosphorylation of tyrosine residue domain SH2 of ABL activates the Ras pathway [111]. Activation of RAF/mitogen-activated protein kinase (MAPK)/extracellular signal regulated kinase (ERK) signaling pathway in a RAS-dependent manner plays an important part in BCR-ABL-independent TKI resistance through an increase in the expression of protein kinase C (PKC) and protein kinase C eta (PRKCH), leading to additional proliferation along with the inhibition of CML stem cells apoptosis by the activation of transcription factors including c-JUN, c-MYC, and c-FOS, as well as the expression of genes involved in proliferation processes [17, 112]. Although for sustaining the proliferative properties of BCR-ABL+ cells, an active PI3K is required [17]. Moreover, the oncoprotein BCR-ABL can activate PI3K and AKT-mediated phosphorylation and cytosolic retention of FOXO transcription factors in different ways such as the association of the SRC homology 2 domain-containing (SHC) protein binding to the P85 regulatory subunit [113], besides binding to GRB2 [114] and the formation of complex c-CBL and adapter molecules (CRK/CRKL). The upregulation of FOXO target genes (e.g., the pro-survival factor BCL6, ATM, and cyclin-dependent kinase 1 (CDK1)) results in blocking the interaction of pro-apoptotic BAD proteins with anti-apoptotic proteins such as BCL-2 and B-cell lymphoma-extralarge (BCL-XL) and no release of pro-apoptotic factors besides the activation of caspase cascade. Therefore, BCR-ABL can directly activate and increase the expression of anti-apoptotic BCL-2 family members such as MCL-1 and BCL-XL, without the participation of JAK/STAT5 [96, 115, 116]. The pathway's third link is mTOR, which involves mTORC1, which is upregulated by AKT activation, and mTORC2, which mediates AKT activation [117]. In accordance with cross talk signaling, low level secretion of TGF-β by CML LSCs can support the survival of CML LSCs and leukemia progression through AKT activation, leading to FOXO3A cytosolic localization, mitochondrial dysfunction, and abnormal ROS production [118, 119]. Besides, the dysregulated PI3K/AKT/mTOR signaling in LSCs increases ROS production, which promotes the survival and drug resistance by the loss of negative the regulation of liver kinase B1 (LKB1) that controls the activity of AMP-activated kinase (AMPK) [117]. The activation of AMPK can induce caspase-3-mediated LSC apoptosis by upregulating P38 expression and c-JUN N-terminal kinase (JNK)-mediated phosphorylation of H2AX, in addition to induce downregulation of BCL-2 [120]. Overexpression of PTEN as a negative regulator of PI3K/AKT and downregulation of β-catenin expression by inhibiting AlOX15 expression can increase susceptibility of CML LSCs to TKI therapy and delayed disease progression [105].
The other signaling pathway in CML cells, which is mediated by binding of JAK2 to the SH2 domain of BCR-ABL1, leads to phosphorylation and activation of STAT3/5 [121]. The activated JAK/STAT signaling drives increased expression and activity of ADAR-1, which contributes to the malignant reprogramming of myeloid progenitors into LSC and CML development, as well as enhanced MDM2 expression and inhibition of the P53 tumor suppressor by creating misspliced isoforms of glycogen synthase kinase 3 beta (GSK3) [122]. The STAT3 activation confers survival and acquired TKI resistance of CML LSCs in BMM through BCR-ABL1-independent mechanisms, suggesting a different role for STAT3 in drug resistance of CML [123]. It has been observed that in both BCR-ABL-dependent STAT5-mediated pathway and a BCR-ABL-independent STAT4-mediated pathway, the expression of a pro-survival gene known as PIM2 is increased in CML LSCs leading to inhibition of the pro-apoptotic protein BAD [124]. Moreover, signal-transducing adapter protein-1 (STAP-1) is important for the preservation of CML LSCs because it suppresses apoptosis by regulating and reducing STAT5 phosphorylation, either directly or indirectly, via the PPAR signaling pathway [125]. The constitutive activation of STAT5 due to the presence of the BCR-ABL oncoprotein mediates the expression of anti-apoptotic proteins and maintenance of CML LSCs [126].
The WNT signaling pathway is composed to the canonical or “β-catenin dependent” pathway, and the non-canonical "WNT/calcium2+/nuclear factor of activated T-cells (NFAT)" pathway or "β-catenin-independent" pathway. BCR-ABL1 can mediate TKI resistance by the induction of constitutive release of WNT ligands, overexpression of frizzled-4 (FZD4) receptors for stabilization of β-catenin in the nucleus and its binding to DNA by interactions with other transcription factors (such as TCF/LEF family and FOXOs), leading to constitutive activation of target genes, such as survival genes (c-MYC and cyclinD1) [127,128,129]. In fact, the activation of WNT/β-catenin mediates the nuclear translocation of NFκB-P65, and its interaction with FOXM1/β-catenin that is essential for the survival of CML LSCs [130]. β-catenin physically interact with CREB-binding protein (CBP)/P300, identified as transcriptional coactivators in cell differentiation, indicating that CBP inhibition may be a new strategy for CML therapy [131]. Moreover, the increased accumulation of β-catenin nuclear accumulation by upregulation of prostaglandin E2 (PGE2) and their binding to E-prostanoid 1/2 (EP1/EP2) receptor leads to the improvement of the β-catenin/WNT signaling, conferring to LSC stemness, resistance to TKI and CML development [33, 132].
Additionally, the activation and upregulation of the smoothened (SMO) transmembrane protein in hedgehog (Hh) pathway can play a potential role in the maintenance of stem cells in myeloid leukemia. In fact, the Hh pathway in CML LSCs is triggered by attaching a ligand, sonic hedgehog (SHH), to the PTCH receptor, which removes PTCH-mediated suppression of SMO, and a conformational change of SMO, which stimulates the glioma-linked oncogene (GLI1 and GLI2) transcription factor, leading to reduced apoptosis [133, 134]. The persistence of Hh signaling unlike TKI therapy implies that this pathway is BCR-ABL1-independent [133, 134], and CML LSCs can use this pathways along with ALOX5 for their survival and TKI resistance [104]. Msi2-Numb signaling as a molecular pathway is correlated with HH and Notch and is involved in regulating the self-renewal properties of LSCs [135].
Epigenetic reprogramming
In addition to transcription factors, epigenetic regulation is other critical element for stem cell maintenance by modulating chromatin accessibility via DNA methylation and histone acetylation at gene expression, responsible for signaling pathways of survival, proliferation, and cell differentiation [136]. The dysregulation of leukemia-specific cellular pathways, such as an increase in the levels of cytidine deaminase and ROS lead to preleukemic lesions in epigenetic regulators (e.g., (cytosine-5)-methyltransferase 3A (DNMT3A), TET1/2, TP53) and acquisition of a hypermethylated phenotype, contributing to clonal hematopoiesis of intermediate potential (CHIP) as a significant element for hematological malignancies [4, 137]. The abnormal hypermethylation of the ABL promoter by methyltransferases appears to be a disease hallmark, suggesting genetic instability in CML progenitors [138]. The high level of DNA methylation at promoter site of tumor suppressor gene, MTSS1, in LSC leads to its downregulation and an increase in CML LSC proliferation [139]. CML LSCs indicate the increased expression of EZH2, a histone methyltransferase and the catalytic subunit of PRC2, which is associated with reprogramming of tri-methylation of histone H3 (H3K27me3) targets [140]). Aberrant activity of histone deacetylase (HDAC) in CML LSC blocks myeloid differentiation to save LSC [141]. Furthermore, overexpression of Sirtuin 1 deacetylase (SIRT1), part of the HDAC family, regulates the acetylation of numerous transcription factors, including P53, Ku70, and FOXO1, contributing to drug resistance and survival of CML LSCs by increasing the acquisition of genetic mutations [142]. The aberrant expressed protein arginine methyltransferase (PRMT5), which mediates histone methylation for RNA metabolism, can attach to BCR-ABL1 and form a positive feedback loop in the evolution of CML [145]. Indeed, both the inhibition of histone methylation and the modulation of DNA methylation can be a possible target for CML treatment in combination with TKIs.
MicroRNAs (miRNAs)
There are some evidences of miRNAs concerning their important roles in leukemogenesis and resistance of CML LSCs to treatment with TKI. The quiescent CML LSCs in BM can cause TKI resistance induced by the phosphorylation of SPRED1, increased levels of miR-126, and suppression of a miR-126 modulator [55]. The increased expression of CD70 through blocking miR-29, which activates CD27 and induces WNT signaling [143], as well as the further activation of NFAT leading to increase pro-survival cytokines and IM resistance [144]. When CML LSCs are exposed to IM, their levels of miR-21 rise, leading to TKI resistance [145]. Increases in miR-29a cause downregulation of tumor suppressor TET2 and antioxidant-coding EPAS1, as well as overexpression of anti-apoptotic genes BCL-2 and MCL-1, resulting in greater TKI resistance in CML LSC [146, 147]. In addition, low levels of tumor suppressor miR-142 are related to high levels of oncoproteins such as MCL-1 and c-KIT, which leads to anti-apoptotic, pro-survival, and therapy-resistant effects via reactivating PI3K/AKT, JAK/STAT, and RAS/RAF/MEK/ERK signaling in TKI-resistant LSCs [148, 149]. The expression of miR-30a is reduced following IM therapy, favoring LSC resistance to TKIs via a mechanism that involves Beclin1 and ATG5 [150]. The downregulation of miR‐494‐3p might contribute to the TKI resistance of CML LSCs by reduction in the TKI‐induced apoptosis, therefore be a novel target to effectively eradicate LSCs [147]. The dose-dependent PP2A activation and anti-proliferative functions of miR-300 may be upregulated by BMM signals, which induces growth arrest and expansion of the G0–G1 quiescent CML LSCs. The miR300-induced apoptosis is associated with downregulation of CCND2/CDK6, SET, and other PP2A-regulated CML survival-promoting factors such as JAK2, CTNNB1, Twist1, and MYC; therefore, the inactivation of PP2A is vital for survival of quiescent LSCs. Conversely, BCR-ABL1 downregulates miR300 in CML progenitors to prevent PP2A-mediated apoptosis via the upregulation of TUG1 long non-coding RNAs (lncRNAs). The modulation of miR300 and/or PP2A-activating treatment can trigger LSC apoptosis; thereby, making this TUG1/miR-300/PP2A signaling pathway important for both CML development and treatment [151]. Therefore, analyzing the mechanisms of miRNA activity in TKI-resistant cases may be beneficial for developing more efficient treatments.
Therapeutic implications of CML LSCs
In the early 1990s, CML treatment included splenic irradiation and use of cytostatic drugs such as busulfan and hydroxyurea. Later, HSC/BM transplant became the curative treatment, which has certain limitation such as challenge of availability of the compatible donor. In following for long-term survival of BMT cases that showed a continuous relapse over time, interferon alpha (IFN-α) in combination with hydroxyurea was used; however, over time response to them reduced due to resistance and adverse side effects [152]. In recent years, the treatment of CML has significant progression from radiotherapy and conventional chemotherapy to targeted therapies with TKIs and allogeneic hematopoietic stem cell transplantation (allo-HSCT) [153]. Inhibition of BCR-ABL tyrosine kinase activity with TKIs was found to be effective in nearly up to 50% of the vast majority of CML patients in chronic phase (mostly receiving IM), who may stop using TKIs and stay a therapy-free remission. However, patients in accelerated phase or blast crisis are frequently treated with later-generation TKIs and are also candidates for allo-HSCT as a last-resort salvage option [154, 155]. However, the CD34+CD38low CML LSCs are less responsive to TKI therapy and can stay quiescent and unaffected for extended periods of time [34], resulting in enhancement of TKI resistance and relapse at the level superior of a molecular response or disease progression [69]. Resistance to any or all TKIs may be provided by ABL1 hotspots mutations such as T315I and T315V, which are mostly susceptible to ponatinib [54]. Meanwhile, TKI application is associated with a number of adverse effects, including myelosuppression, gastrointestinal issues, hepatotoxicity, hyperglycemia, and cardiovascular events [156]. Therefore, utilizing the combination of TKIs and other drugs is of some interest concerning the inhibition of leukemia progression and elimination of CML LSCs.
Tyrosine kinase inhibitors (TKI) treatments
TKIs competitively bind to the ATP-binding site of the BCR-ABL1 to suppress downstream pathways and leukemogenesis by reducing aberrant phosphorylation of the dysregulated tyrosine kinase [157]. In the 2000s, imatinib mesylate (IM), a first-generation TKI, is quite efficient in apoptosis induction in CML stem cells by lowering programmed death receptor 1 (PD-1) expression on CD8+ T cells and monocytic myeloid-derived suppressor cells (MDSCs), resulting in increased cytotoxicity mediated by cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells; however, in another study it showed the enhanced expression of ATG4B and survival in CD34+ CML cells [158, 159]. Dasatinib, nilotinib (NIL), and bosutinib, as second-generation TKIs (2G-TKIs), are more effective and faster at cytogenetic and molecular response to treatment, but inactive toward T315I mutation. In the T315I mutation, the amino acid at position 315 of exon 6 of the ABL gene changed from threonine to isoleucine (Ile), hindering the high affinity binding of the inhibitor to the ATP-binding site of the kinase, thereby producing resistance. The metabolic risk of using second-generation TKI should be considered due to their major side effects [160,161,162]. Nilotinib is initially based on the IM scaffold and it binds to the same pocket as IM but with a much higher affinity that allows for effectiveness against certain IM-resistant point mutations [18]. Dasatinib, a dual SRC/ABL1 inhibitor, is successful in suppressing STAT5 and reducing mutant TP53, but not in eliminating LSCs [163]. Dasatinib increases expression of inhibitory KIR2DL1 receptors that can suppress NK cell toxicity against CML cells [164]. Similar to dasatinib, bosutinib inhibits BCR-ABL1 and SRC family kinases, while is minimally active against KIT and PDGFR [165]. Radotinib (RAD) is also a novel potent second-generation TKI with high inhibitory capacity for the BCR-ABL1 oncoprotein [166]. As an alternative 2G-TKIs in patients who are resistant to a 2G-TKI without specific mutations, a third-generation TKI, ponatinib is suggested due to its higher efficacy in eradicating CML stem cells and overcoming TKI resistance by replacing threonine with isoleucine at the ATP-binding site, as well as targeting the VEGFR, KIT, SRC, FGFR, PDGFR, FLT3, and KIT pathways [167]. However, some BCR-ABL1 or compound mutations (for example, T315M, T315V, and Y253H/T315I or E255V/T315I) can create ponatinib resistance [168]. Olverembatinib (HQP1351), a novel orally administered 3G-TKI, was found to be highly effective in CML patients who were resistant to existing TKI treatments, including some with T315I mutations in a phase II trial [169]. Vodobatinib (K0706) is another new orally bioavailable 3G-TKI with promising in vitro action against majority of BCR-ABL mutations, but not T315I. In a phase I trial of patients with CML who had failed to respond to ≥ 3 TKIs or lower, vodobatinib demonstrated an adequate safety profile [170]. Asciminib (ABL001), a FDA-approved fourth-generation TKI, is shown to be efficient against BCR-ABL1-dependent and independent mutations such as T315I with a moderate toxicity potential by blocking binding to the BCR-ABL1 myristoyl pocket (STAMP) and producing therapeutic synergism in lowering CRK-like protein phosphorylation for CML stem cells either as monotherapy or in combination with other TKIs [171, 172]. PF-114, a novel fourth-generation TKI, inhibits BCR-ABL1 and/or STAMP along with suppressing the constitutive activation of PI3K/AKT/ERK1/2 and JAK/STAT3/5 signaling and also elevating p27 levels to inhibit oncogenesis with less toxic complications [173,174,175]. Rebastinib (DCC-2036) is a newly discovered potent broad-spectrum inhibitor of BCR-ABL1 kinase and many other kinases including SRC kinases and FLT-3, but not KIT. It can inhibit many of the resistant mutants of BCR-ABL, including T315I [176].
Other drugs, targeting the survival mechanism of CML LSCs
It appears that CML LSCs are not completely reliant on BCR-ABL activity for survival so that these BCR-ABL-independent mechanisms may play a role in disease persistence [11]. Targeting BCR-ABL1-kinase independent pathways could regulate apoptosis, self-renewal, and fate of CML LSCs, also overcome survival pathways (Table 1). Knocking out resistance factors in combination with TKI treatment can sensitize drug-resistant CML stem cells to TKIs and decrease their proportion. Several clinical trials investigate the viability of licensed TKIs when used in combination with certain other agents (Table 2).
Anticancer drugs targeting autophagy
TKI therapy for CML causes autophagy as a drug resistance pathway employed for the survival of LSCs [177]; therefore, suppression of autophagy appears to be a therapeutic option for eliminating LSCs, especially combined with TKI therapy, despite its conflicting consequences in CML. As a result, knocking out either the ATG5 or ATG7 genes, which are involved in preautophagosome formation and activation, makes CML LSCs more susceptible to IM [177]. ATG7 silencing also decreases glucose levels in LSCs, inhibits formation of autophagosome by downregulating signaling pathways (e.g., WNT/catenin, PI3K/AKT/mTORC1, HIF, c-MYC), and activates cytochrome-c/caspase-9/caspase-3, resulting in mitochondrial translation, oxidative phosphorylation (OXPHOS), and oxygen consumption [56]. As mentioned in Table 1, chloroquine (CQ), Spautin-1, Lys05 (a dimeric analogue of chloroquine), and PIK-III, as autophagy inhibitors, can influence the maintenance and function of LSCs. Also, inhibition of BMI1, which is a key role in LSC self-renewal and advancement of CML to the acute level through blocking a defensive CCNG2-dependent autophagy pathway, combined with IM significantly reduces the clonogenic properties of CD34+ CML cells [178].
Immunotherapy strategies targeting cell surface markers
Although cell surface markers expressed on CML LSCs and normal HSCs are mostly similar, the expression levels of some markers are much greater in LSCs than in HSCs, providing an opportunity for molecules targeting cell surface receptors of CML LSCs using antibodies. Specific cell surface antigens can be used to develop CML-eradicating immunotherapies such as cell-targeting antibodies, antibody–toxin conjugates, and chimeric antigen receptor-engineered T (CAR-T) cell-based therapy. It seems that they are likely to have less toxic or off-target effects than inhibitors of factors essential for the survival of LSCs. However, unlike AML, CAR cell therapies have not been significantly developed in CML so far [42, 47]. Targeting surface markers particularly overexpressing on CML LSCs such as IL1RAP, CD26, CD44, CD70, and CD123 has demonstrated a powerful antileukemic effect in preclinical CML studies (Table 1), although the viability of some of them in the treatment of patients with CML needs to be further investigated, including anti-CD44 agents with regard to the expression of CD44 on HSCs [179].
Anticancer drugs targeting the interactions with BMM
Targeting the interaction of SDF1-CXCR4 by NOX-A12 and Plerixafor (AMD3100), CXCR4 antagonist prevents LSC homing and causes TKI sensitization. TKI-resistant mesenchymal cells enhanced BMP4 production, indicating a BMP autocrine loop may cause TKI resistance; hence, new anti-BMP pharmacological molecules could be used to target CML LSCs in their niche [76]. Furthermore, TGF-β stimulates the expression of plasminogen activator inhibitor-1 (PAI-1), a significant physiologic serine protease inhibitor (serpin) of the fibrinolytic network, leading to decrease membrane type-1 metalloprotease (MT1-MMP) activity and mobility of CML LSCs. Therefore, repression of PAI-1 can increase sensitivity of CML LSCs to TKIs through mobilization of CML LSCs from the niche as a measure to combat TKI resistance [180]. Moreover, in hypoxic microenvironment, a HIF-1 inhibitor (acriflavine) along with a TKI may possibly target CML LSCs and be responsible for the antileukemic response [181]. One of the recommended ways of eliminating CML LSCs is the inhibition of PPARγ by pioglitazone, rosiglitazone, and clofazimine. Despite various difficulties, such as adverse effects of PPARγ agonists, the discovery of PPARγ as a novel critical regulator for CML LSC survival offers fresh promises for targeting them [182]. Furthermore, overexpression of PPARα ligands (e.g., clofibrate) and increased expression of human organic cation transporter 1 (hOCT1) by WY-12643 promotes TKI-mediated apoptosis [182, 183]. To draw a conclusion upon these, understanding the interactions between LSCs and the bone marrow niche might lead to a new strategy for the elimination of CML stem cells.
Anticancer drugs targeting signaling pathways
One of the ways to prevent RAS/MEK/ERK/MAPK pathway is block isoprenoid group transfer as a posttranscriptional modification that induces membrane migration and activation of various proteins, including RAS and RAF by farnesyl transferase inhibitors (FT-Is). Although they show a few benefits, their combination with IM can be useful for CML patients who are not responding to IM monotherapy [184]. The combination of the inhibitors of RAS/MEK/ERK pathway with TKIs can reverse the resistance and cause drug synergistic effect [185, 186]. It appears that reactivating miR-185, which targets PAK6 transcripts, will make therapy-resistant cells more susceptible to TKIs by decreasing MAPK pathway activation, mitochondrial function, ROS production, and autophagy [187].
As evident in Table 1, there are compounds which can also sensitize CML LSCs to TKIs and eliminate them by disrupting the PI3K/AKT/mTOR pathway. Moreover, inhibition of the PI3K/AKT signaling pathway by antagomiR-21 along with IM amplifies programmed cell death 4 (PDCD4) and PTEN, while decreases AKT phosphorylation and MYC expression, which recovers sensitivity of CML LSCs to TKIs by miR-21 depletion [145, 188]. A study showed that combination of IM and upregulation of miR-155 successfully triggered cell death of CD34+, CD38− CML stem cells by blocking of PI3K/mTOR pathway [189].
The other potential therapeutic target in combination with TKIs can be the of JAK2/STAT5 pathway and induces cell cycle arrest and apoptosis in CML cells by inhibitors such as pimozide, ruxolitinib, and fedratinib [126]. Since JAK2 inactivates PP2A and reactivation of PP2A leads to depletion of the LSCs by inhibiting STAT5, a synergism of JAK2 inhibitors and PP2A activators might be effective in the treatment of CML [190]. Also, MYC inhibitor and a SET antagonist can reactivate PP2A and apoptotic pathways in a PP2A-dependent manner [191, 192]; therefore, the impairment of PP2A inhibitors with a TKI can inhibit STAT5, and deplete LSCs [193, 194]. Combining IM and interferon-γ (IFN-γ) reduces STAT5 phosphorylation, whilst increases STAT1 phosphorylation that increases BCL6 expression and LSC survival [195]. The upregulation of C/EBPb and activation of STAT1 and STAT5 by IFN-α can leads to differentiation and exhaustion of CML stem cells [196]. Moreover, the inhibition of STAT3 by OP449 has a strong potential to stops TKI-resistant CML LSCs survival [197].
The disruption of the interaction between CBP and β/γ-catenin by PRI-724 can cause a decrease in self-renewal capability in leukemia-initiating cells in CML and eradication of drug-resistant primary CML cells [198]. In addition, CBP inhibitor therapy increases β-catenin binding to P300, which mediates cell differentiation, and causes P53/P21-dependent senescence in BCR-ABL mutant CML cells [131]. Inhibition of PGE2 by blocking the activity of cyclooxygenase-2 leads to decrease in both β-catenin and resistance to TKIs [199]. However, PGE1 shows protective roles against LSCs by suppression AP-1 factors such as FOSB that make it a target for CML stem cell eradication [33, 132]. Modulation of WNT signaling and other signaling pathways such as mTORC1, STAT3, NF-kB, and Notch pathways, by niclosamide impairs the ability of CML LSCs to survive and self-renew by disrupting [200]. Furthermore, the increased level of tyrosine phosphatase FAS-associated phosphatase 1 (FAP-1), which targets a β-catenin inhibitor (GSK3β), mediates persistence of CML LSCs [201]. The combination of porcupine (PORCN) inhibitor (WNT974) with NIL in transgenic mice models showed the inhibition of WNT/β-catenin signaling and suppression of c-MYC, cyclin-D1, and Axin-2 expression, contributing to an increase in the inhibition of proliferation and eradication of CML stem cells [127, 128].
Targeting the Hh signaling pathway by using the SMO inhibitors, (e.g., cyclopamine and LDE225 (sonidegib)), alone or in combination with TKIs, can be an effective treatment strategy for eradicating Hh-mediated self-renewal capacity of CML LSC and suppress their growth [202, 203]. The overexpression of GLI2 enhances the generation and commensurate of dormant LSCs, and also increases the repression of cell cycle regulatory genes, therefore providing glasdegib (PF-04449913), and an antagonist of the GLI2 transcriptional activator (SMO) stimulates CML LSCs to cell cycle and become sensitive to TKIs [204]. The inhibition of Hh pathway by vismodegib can treat the upregulation of autophagy in CML cells; hence, the combination of vismodegib with silencing of ATG5 and ATG7 can cause significant increase in CML cell death [205]. Moreover, overexpression of miR-326 results in an increased rate of apoptosis in CML CD34+ cells by downregulation of SMO [206].
Anticancer drugs targeting energy metabolism
The inhibition of mitochondrial function has antileukemia effects along with TKIs [90]. The important findings suggest that SIRT1 and/or elements downstream in the OXPHOS pathway may be potential therapeutic targets, for instance, inhibiting PPARγ coactivator-1α (PGC-1α) by SR-18292 in combination with TKIs could avoid CML relapse by the increase in on PGC-1 acetylation levels affected of SIRT1 [106].
As a result of shifting to mitochondrial metabolism, TKI-tolerant cells showed vulnerability to mitochondrial-druggable targets with antileukemia effects such as inhibition of the first enzyme in glutamine-dependent mitochondrial metabolism combined with TKI [207]. CML LSCs have also represent susceptibility to TKIs and tigecycline, an antibiotic from glycylcycline class, impairing mitochondrial protein synthesis and the mitochondrial respiration [56]. Furthermore, BCL-2 inhibitors (subutoclax and venetoclax) can increase the eradication of CML LSCs by disrupting energy metabolic pathways [208, 209]. Recently, an innovative strategy based on a liposome loaded with the BCL2 inhibitor (venetoclax) using an anti-CD26 antibody (begelomab) to selectively target CML LSCs showed that the immunoliposome could reduce cell growth and also induce apoptosis in CD26+ LSCs, along with synergistic effects by coadministration with imatinib or nilotinib [210]. Interestingly, inhibition of ALOX15 pathway involved in fatty acid metabolism with the BLT2-specific inhibitor triggered apoptosis and inhibited self-renewal in TKI-resistant CML cells in blast crisis phase [211]. Although blocking ALOX5 with zileuton decreases the survival of CML LSCs in mice, targeting ALOX5 is unlikely to be successful due to the low expression of ALOX5 in humans [33]. In another study, it was indicated that targeting integrin-linked kinase (ILK), which is highly upregulated in CML LSCs, can induce metabolic vulnerabilities by reduction in the CD36 expression, as a fatty acid receptor; therefore, an ILK inhibitor combined with dasatinib can increase the chance of recipient's survival [212].
Anticancer drugs targeting the epigenetic modification
Inhibition of EZH2 and its downregulation by TKI therapy blocks survival of leukemia-initiating cells and promotes CML LSCs apoptosis without impairing normal HSCs by modulation PTEN expression. Hence, inhibiting EZH2 along with TKI therapy increases activation of H3K27me3 targets such as CDKN2A and upregulates pro-apoptotic targets of P53 (e.g., NOXA, P53 upregulated modulator of apoptosis (PUMA), BAX, CDKN2A, TNFRS10B), causing TKI sensitivity to be restored and CML LSCs to be eradicated [70, 213]. The reactivation of P53 by an HDM2 antagonist or an MDM2 inhibitor restores sensitivity of quiescent CD34+ cells to BCL2 inhibitor and TKI-induced apoptosis [99, 214, 215]. Moreover, MI-219 can induce apoptosis through the alteration of HDM2 function, reactivation of P53 mediated by downregulation of c-MYC and upregulation of P21, and diminished essential genes for LSC self-renewal [99, 214, 215].
HDAC inhibitors, panobinostat, combined with TKIs can disrupt LSC quiescence and increase TKI-mediated apoptosis by acetylating HSP90 and increasing proteasomal degradation of key signaling proteins in CML LSCs [141, 216]. Another HDAC inhibitor, chidamide, induces apoptosis by increasing acetylation of histone H3, activation of caspase 3/9, reduction in the β-catenin levels by inhibiting the WNT–CBP–β-catenin pathway with limited toxicity to normal HSCs [217, 218]. Novel pan-HDAC inhibitor, MAKV-8, also contributes to LSC eradication through the stimulation of caspase 3/9 and ER stress [219]. Targeting SIRT1 in CML LSCs by Tenovin-6 or TV39OH leads to enhance apoptosis by increase acetylation of P53 [220, 221]. A PRMT5 inhibitor can induce apoptosis of CD34+CD38− cells by inhibiting the WNT/β-catenin pathway and inducing negative control on LSC renewal [222, 223].
Gene therapy
Advances in molecular biology and genetics have expanded our knowledge of genes involved in disease development. In line with this, recently, the common techniques used to monitor CML patients, detect the Ph chromosome, and recognize the BCR-ABL1 transcript are conventional cytogenetics and fluorescence in situ hybridization (FISH), and reverse transcriptase-polymerase chain reaction (RT-PCR) [60, 224, 225]. RT-PCR is also used to assess the following molecular response to treatment, defined as the ratio of BCR-ABL1 to ABL1 transcripts (known as molecular response (MR)) and categories in different groups including complete cytogenetic remission (MR ≤ 1%), major molecular response (MMR) (MR ≤ 0.1%), deep molecular response (DMR) ( MR4 ≤ 0.01%), and molecularly undetectable leukemia MR4.5 ≤ 0.0032% [60, 164].
Advances in genomic techniques have also led to the development of highly translational murine models of human hematologic malignancies [226]. In fact, the ideal murine model should replicate the genetic and molecular heterogeneity of tumors in immune-competent mice, while also offering a mechanism of monitoring clinical behavior of the human disease, progression, and treatment efficacy [227]. Xenograft models are highly beneficial for determining the efficacy of therapeutics on human tumor cells by proofing the concept by in vitro studies within in vivo conditions. However, there are some limitations that should be considered such as the lack of tumor microenvironment, inability to determine tumor interaction with the immune system, and inability to test complex genomic interactions in a single-cell system [226]. The transgenic mouse model has been used to mimic human cancers with an etiology based on genetic aberration by injecting a gene of interest in the vector form into a fertilized egg, allowing investigation of both microenvironment and immunity on the response of tumor in either preventative or long-term therapy. However, the manipulation of embryonic stem cell could associate with the potential of off-target mutation and genetic alteration during development. They are also genetically not as complex as human tumors [228, 229]. For the first time, the induction of CML-like myeloproliferative syndrome was observed in irradiated recipient mice transplanted with a retroviral vector encoding the BCR-ABL1 fusion protein to identify regions in this oncoprotein that are important for CML transformation in mice and design of TKIs [230]. Mouse models of retroviruses may evaluate the function of individual genes for CML development and progression, including the expression of STAT5 that is necessary for BCR-ABL1-mediated leukemogenesis [231]. Although the CP-CML depends on BCR-ABL1, the progression of acute blast crisis is mediated by additional genetic changes and mouse models of CML are needed to develop therapies for unresponsive patients to TKIs [232]. At present, CML could be induced in most of the inbred mouse strain, including C57BL/6, BALB/c, and viable gene knockout mice strain with high efficiency, providing an excellent model for studying CML LSCs and evaluating therapeutic agents for CML treatment based on their short latency (about 3 weeks) [233]. Therefore, retroviral mouse models have not only been instrumental in identifying the mechanisms of leukemogenesis but have also contributed to advances in the understanding of disease progression in CML and the identification of new therapeutic targets.
In general, TKIs only inactivate the oncoprotein, but the oncogene continues unaffected and treatment discontinuation is only an option for a small subset of patients; therefore, the interruption/deletion of the oncogenic sequence might be an effective new therapeutic option [224]. The emergence of clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 technology can be a treatment based on its capacity to induce a specific DNA double-strand break in precise locations and providing complete and permanent oncogene knockout by generating an animal carrying highly targeted genetic modification [224, 234]. Additionally, this system can be used to deliver combinations of guide RNAs to modify multiple genes in a single mouse hematopoietic stem cell, to more closely model the complexity of hematopoietic malignancy [235]. Although the risk of off-target editing is seen in cell-based systems, the accuracy of the CRISPR-mediated editing system is suggested in the embryonic system [236]. In 2015, the first CRISPR/Cas9 system that could correct acquired mutations in a human myeloid leukemia cell line was demonstrated and then successfully used in animal [237]. Three years later, the first clinical trial focused on HSCs by using CRISPR-Cas9-modified HSCs was approved [238]. Furthermore, there were mouse models for human HSC engrafting and mimicking human CML to provide opportunities to evaluate these CRISPR/Cas9 therapeutic applications [239]. In 2017, the CRISPR/Cas9 system effectively abrogates the BCR/ABL1 oncogene and reversed the tumorigenicity in edited CRISPR cells in a CML xenograft animal model [240]. In the following, other genome-editing nucleases (e.g., zinc finger nucleases (ZFN) targeting the exon 1 of BCR) achieved the abrogation of the BCR/ABL1 oncogene in both sensitive and resistant forms of K562 to IM [241]. Recently, for the first time, a CRISPR/Cas9 short deletion system interrupts the BCR/ABL1 oncogene in primary leukemic stem cells Sca1+ from a CML mouse model and CD34+ from human CML patients. They showed that edited LSCs had impaired tumorigenic activity and capacity for multipotency and also confer a significant therapeutic benefit on CML mouse models [242]. In addition to BCR/ABL1 oncogene, other genes involved in CML LSCs survival such as EZH2 can make LSCs more susceptible to TKIs by using CRISPR/Cas9-mediated gene editing [140]. Although the CRISPR-Cas9 system induces double-strand breaks at target sites in genomic DNA, it can also generate undesirable cleavages at off-target sites leading to mutations and the disruption of normal genes [243].
Conclusion
Over the past years, the inadequacy of many CML therapies results from their failure to target LSCs represents CML LSCs as the most critical target in the treatment. However, they are also the most difficult population to be targeted due to both their heterogeneity and phenotypic similarities with HSCs [9]. CML LSCs can be resistant to TKI therapy and remain after therapy, serving as a reservoir for residual disease and relapse due to independent BCR-ABL activity including the various molecular mechanisms and signaling pathways, which are often activated by epigenetic mechanisms and the impact of the bone marrow niche, as mentioned earlier in this study. Further investigating these different aspects of CML LSCs maintenance might enable us to provide novel therapeutic targets for the development of a TKI-based combinatorial therapy to eradicate CML stem cells. Altogether, a promising therapeutic strategy would combine TKI with drugs targeting alternative survival pathways. However, some of scientific research provides questionable or contradictory values, remained to be clarified to draw a proper conclusion.
Availability of data and materials
Not applicable.
Abbreviations
- ABL:
-
Abelson murine leukemia virus
- AP:
-
Accelerated phase
- ALL:
-
Acute lymphoblastic leukemia
- ADAR1:
-
Adenosine deaminases acting on double-strand RNA1
- AMPK:
-
AMP-activated kinase
- ALOX5:
-
Arachidonate lipoxygenase
- BCL-XL:
-
B-cell lymphoma-extralarge
- BCP:
-
Blast crisis phase
- BMM:
-
Bone marrow microenvironment
- BMPs:
-
Bone morphogenic proteins
- BCAA:
-
Branched-chain amino acids
- BCR:
-
Breakpoint cluster region
- CAR-T:
-
Chimeric antigen receptor-engineered T
- CML:
-
Chronic myeloid leukemia
- CP:
-
Chronic phase
- JNK:
-
c-JUN N-terminal kinase
- CBP:
-
CREB-binding protein
- CDK1:
-
Cyclin-dependent kinase 1
- DMR:
-
Deep molecular response
- DLAT:
-
Dihydrolipoamide S-acetyltransferase
- DPP4:
-
Dipeptidyl peptidase-4
- DYRK2:
-
Dual specificity tyrosine phosphorylation regulated kinase 2
- EGFR:
-
Epidermal growth factor
- ERK:
-
Extracellular signal regulated kinase
- FAP-1:
-
FAS-associated phosphatase 1
- FGF2:
-
Fibroblast growth factor 2
- FISH:
-
Fluorescence in situ hybridization
- FLT3:
-
FMS-like tyrosine kinase 3
- FOXO:
-
Forkhead box O
- FZD4:
-
Frizzled-4
- GLUT1:
-
Glucose transporter 1
- GSK3:
-
Glycogen synthase kinase 3 beta
- G-CSF:
-
Granulocyte colony-stimulating factor
- GRB2:
-
Growth factor receptor-bound protein 2
- Hh:
-
Hedgehog
- HSC:
-
Hematopoietic stem cell
- HGF:
-
Hepatocyte growth factor
- HHT:
-
Homoharringtonine
- hOCT1:
-
Human organic cation transporter 1
- HIFs:
-
Hypoxia-inducible factors
- IKZF1:
-
IKAROS family zinc finger 1
- IM:
-
Imatinib mesylate
- ILK:
-
Integrin-linked kinase
- ICAM-1:
-
Intercellular adhesion molecule-1
- IFN-α:
-
Interferon alpha
- IFN-γ:
-
Interferon-γ
- IL1R1:
-
Interleukin 1 receptor-1
- JAK:
-
Janus kinase
- KLF4:
-
Krüppel-like factor 4
- LSCs:
-
Leukemia stem cells
- LTB4:
-
Leukotriene B4
- LKB1:
-
Liver kinase B1
- lncRNAs:
-
Long non-coding RNAs
- LTβR:
-
Lymphotoxin-β receptor
- MMR:
-
Major molecular response
- MMP-9:
-
Matrix metalloproteinase-9
- MSCs:
-
Mesenchymal stromal/stem cells
- MVs:
-
Microvesicles
- miRNAs:
-
MicroRNAs
- MAPK:
-
Mitogen-activated protein kinase
- Msi2:
-
Musashi 2
- MDSCs:
-
Myeloid-derived suppressor cells
- NK cells:
-
Natural killer cells
- NIL:
-
Nilotinib
- NFAT:
-
Nuclear factor of activated T-cells
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- OMA:
-
Omacetaxine mepesuccinate
- OXPHOS:
-
Oxidative phosphorylation
- PUMA:
-
P53 upregulated modulator of apoptosis
- PPARγ:
-
Peroxisome proliferator-activated γ
- Ph:
-
Philadelphia (Ph) chromosome
- PTEN:
-
Phosphatase and tensin homolog
- PI3K:
-
Phosphoinositide 3-kinase
- PI3K:
-
Phosphatides inositol 3 kinase
- PAI-1:
-
Plasminogen activator inhibitor-1
- PGC-1α:
-
PPARγ coactivator-1α
- PD-1:
-
Programmed death receptor 1
- PML:
-
Promyelocytic leukemia
- PGE2:
-
Prostaglandin E2
- PKC:
-
Protein kinase C
- PP2A:
-
Protein phosphatase 2A
- PDH:
-
Pyruvate dehydrogenase
- RAD:
-
Radotinib
- ROS:
-
Reactive oxygen species
- RT-PCR:
-
Reverse transcriptase-polymerase chain reaction
- RUB:
-
Ruxolitinib
- SR-B2:
-
Scavenger receptor-B2
- SHC:
-
SRC homology 2 domain-containing
- SCFR:
-
Stem cell factor receptor
- TZD:
-
Thiazolidinediones
- TGF-β:
-
Transforming growth factor-β
- PKM2:
-
Tumor M2-pyruvate kinase
- TKI:
-
Tyrosine kinase inhibitor
- VPS34:
-
Vacuolar protein sorting 34
- VCAM-1:
-
Vascular cell adhesion molecule-1
- WHO:
-
World Health Organization
- ZFN:
-
Zinc finger nucleases
References
Arber D, Orazi A. The updated WHO classification of hematological malignancies: the 2016 revision to the WHO classification of myeloid neoplasms and acute leukemia. Blood J. 2016;127:2391–405.
Jain P, Kantarjian H, Patel KP, Gonzalez GN, Luthra R, Shamanna RK, et al. Impact of BCR-ABL transcript type on outcome in patients with chronic-phase CML treated with tyrosine kinase inhibitors. Blood. 2016;127(10):1269–75.
Goldman JM, Melo JV. Chronic myeloid leukemia—advances in biology and new approaches to treatment. N Engl J Med. 2003;349:1451–64.
Vetrie D, Helgason GV, Copland M. The leukaemia stem cell: similarities, differences and clinical prospects in CML and AML. Nat Rev Cancer. 2020;20:158–73.
Schepers K, Campbell TB, Passegué E. Normal and leukemic stem cell niches: Insights and therapeutic opportunities. Cell Stem Cell. 2015;16:254–67.
Loscocco F, Visani G, Galimberti S, Curti A, Isidori A. BCR-ABL independent mechanisms of resistance in chronic myeloid leukemia. Front Oncol. 2019;9(SEP):1–11.
Talati C, Pinilla-Ibarz J. Resistance in chronic myeloid leukemia: definitions and novel therapeutic agents. Curr Opin Hematol. 2018;25:154–61.
Chomel JC, Turhan AG. Chronic myeloid leukemia stem cells in the era of targeted therapies: Resistance, persistence and long-term dormancy. Oncotarget. 2011;2:713–27.
Holyoake TL, Vetrie D. The chronic myeloid leukemia stem cell: stemming the tide of persistence. Blood. 2017;129:1595–606.
Bocchia M, Sicuranza A, Abruzzese E, Iurlo A, Sirianni S, Gozzini A, et al. Residual peripheral blood CD26+ leukemic stem cells in chronic myeloid leukemia patients during TKI therapy and during treatment-free remission. Front Oncol. 2018;8:194.
Hamilton A, Helgason GV, Schemionek M, Zhang B, Myssina S, Allan EK, et al. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood. 2012;2119:1501–10.
Nowell C. The minute chromosome (Ph1) in chronic granulocytic leukemia. Blut Zeitschrift für die Gesamte Blutforsch [Internet]. 1962;8(2):65–6. https://doi.org/10.1007/BF01630378.
Rowley JD. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and giemsa staining. Nature [Internet]. 1973;243(5405):290–3.
Heisterkamp N, Stephenson JR, Groffen J, Hansen PF, de Klein A, Bartram CR, et al. Localization of the c-abl oncogene adjacent to a translocation break point in chronic myelocytic leukaemia. Nature [Internet]. 1983;306(5940):239–42.
Groffen J, Stephenson J, Heisterkamp N, Deklein A, Bartram C, Grosveld G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell [Internet]. 1984;36(1):93–9.
Sloma I, Jiang X, Eaves AC, Eaves CJ. Insights into the stem cells of chronic myeloid leukemia. Leukemia. 2010;24(11):1823–33.
Flis S, Chojnacki T. Chronic myelogenous leukemia, a still unsolved problem: Pitfalls and new therapeutic possibilities. Drug Des Dev Ther. 2019;37:530–42.
Braun TP, Eide CA, Druker BJ. Response and resistance to BCR-ABL1-targeted therapies. Cancer Cell. 2020;37:530–42.
Deininger MWN, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood [Internet]. 2000;96(10):3343–56.
Mullighan CG, Su X, Zhang J, Radtke I, Phillips LAA, Miller CB, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360:470–80.
Den Boer ML, van Slegtenhorst M, De Menezes RX, Cheok MH, Buijs-Gladdines JG, Peters ST, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009;10:125–34.
Tasian SK, Loh ML, Hunger SP. Philadelphia chromosome-like acute lymphoblastic leukemia. Blood. 2017;130:2064–72.
Piedimonte M, Ottone T, Alfonso V, Ferrari A, Conte E, Divona M, et al. A rare BCR-ABL1 transcript in Philadelphia-positive acute myeloid leukemia: case report and literature review. BMC Cancer. 2019;19:1–6.
Soupir CP, Vergilio JA, Dal Cin P, Muzikansky A, Kantarjian H, Jones D, et al. Philadelphia chromosome-positive acute myeloid leukemia: a rare aggressive leukemia with clinicopathologic features distinct from chronic myeloid leukemia in myeloid blast crisis. Am J Clin Pathol. 2007;127:642–50.
Goldman JM, Daley GQ. Chronic myeloid leukemia—a brief history. In: Melo JV, Goldman J, editors. Myeloproliferative disorders. Heidelberg; Springer, Berlin. 2007:p. 1–13.
Perrotti D, Jamieson C, Goldman J, Skorski T. Chronic myeloid leukemia: Mechanisms of blastic transformation. J Clin Investig. 2010;120:2254–64.
Lichty BD, Keating A, Callum J, Yee K, Croxford R, Corpus G, et al. Expression of p210 and p190 BCR-ABL due to alternative splicing in chronic myelogenous leukaemia. Br J Haematol. 1998;103:711–5.
Branford S, Wang P, Yeung DT, Thomson D, Purins A, Wadham C, et al. Integrative genomic analysis reveals cancer-associated mutations at diagnosis of CML in patients with high-risk disease. Blood. 2018;132:948–61.
Boquett JA, Alves JRP, de Oliveira CEC. Analysis of BCR/ABL transcripts in healthy individuals. Genet Mol Res. 2013;12:4967–71.
Taussig DC, Pearce DJ, Simpson C, Rohatiner AZ, Lister TA, Kelly G, et al. Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood. 2005;106:4086–92.
Inoue A, Kobayashi CI, Shinohara H, Miyamoto K, Yamauchi N, Yuda J, et al. Chronic myeloid leukemia stem cells and molecular target therapies for overcoming resistance and disease persistence. Int J Hematol. 2018;108(4):365–70.
Walcher L, Kistenmacher AK, Suo H, Kitte R, Dluczek S, Strauß A, et al. Cancer Stem Cells—Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front Immunol. 2020;11(August):1–33.
Houshmand M, Simonetti G, Circosta P, Gaidano V, Cignetti A, Martinelli G, et al. Chronic myeloid leukemia stem cells. Leukemia. 2019;33:1543–56.
Landberg, N. (2020). LUCC - Lund University Cancer Centre Translational Genomic and Functional Studies of Leukemia Division of Clinical Genetics Research output: Thesis › Doctoral Thesis (compilation).
Kobayashi CI, Takubo K, Kobayashi H, Nakamura-Ishizu A, Honda H, Kataoka K, et al. The IL-2/CD25 axis maintains distinct subsets of chronic myeloid leukemia-initiating cells. Blood. 2014;123:2540–9.
Herrmann H, Sadovnik I, Cerny-Reiterer S, Rülicke T, Stefanzl G, Willmann M, et al. Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood. 2014;123:3951–62.
Kinstrie R, Horne GA, Morrison H, Irvine D, Munje C, Castañeda EG, et al. CD93 is expressed on chronic myeloid leukemia stem cells and identifies a quiescent population which persists after tyrosine kinase inhibitor therapy. Leukemia. 2020;34:1613–25.
Herrmann H, Cerny-Reiterer S, Gleixner KV, Blatt K, Herndlhofer S, Rabitsch W, et al. CD34+/CD38− stem cells in chronic myeloid leukemia express Siglec-3 (CD33) and are responsive to the CD33-targeting drug gemtuzumab/ozogamicin. Haematologica. 2012;97:219–26.
Landberg N, von Palffy S, Askmyr M, Lilljebjörn H, Sandén C, Rissler M, et al. CD36 defines primitive chronic myeloid leukemia cells less responsive to imatinib but vulnerable to antibody-based therapeutic targeting. Haematologica. 2018;103:447–55.
Woolthuis CM, Stranahan AW, Park CY, Minhajuddin M, Gasparetto M, Stevens B, et al. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell. 2016;19:23–37.
Valent P, Sadovnik I, Ráčil Z, Herrmann H, Blatt K, Cerny-Reiterer S, et al. DPPIV (CD26) as a novel stem cell marker in Ph+ chronic myeloid leukaemia. Eur J Clin Investig. 2014;44:1239–45.
Valent P, Sadovnik I, Eisenwort G, Bauer K, Herrmann H, Gleixner KV, et al. Immunotherapy-based targeting and elimination of leukemic stem cells in AML and CML. Int J Mol Sci. 2019;20:4233.
Sadovnik I, Herrmann H, Eisenwort G, Blatt K, Hoermann G, Mueller N, et al. Expression of CD25 on leukemic stem cells in BCR-ABL1 + CML: potential diagnostic value and functional implications. Exp Hematol [Internet]. 2017;51:17–24.
Krause DS, Lazarides K, Von Andrian UH, Van Etten RA. Requirement for CD44 in homing and engraftment of BCR-ABL-expressing leukemic stem cells. Nat Med. 2006;12:1175–118.
Lugano R, Vemuri K, Yu D, Bergqvist M, Smits A, Essand M, et al. CD93 promotes β1 integrin activation and fibronectin fibrillogenesis during tumor angiogenesis. J Clin Invest. 2018;128:3280–97.
Riether C, Radpour R, Kallen NM, Bürgin DT, Bachmann C, Schürch CM, et al. Metoclopramide treatment blocks CD93-signaling-mediated self-renewal of chronic myeloid leukemia stem cells. Cell Rep. 2021;34:108663.
Soverini S, De Santis S, Monaldi C, Bruno S, Mancini M. Targeting leukemic stem cells in chronic myeloid leukemia: Is it worth the effort? Int J Mol Sci. 2021;22:7093.
Warfvinge R, Geironson L, Sommarin MNE, Lang S, Karlsson C, Roschupkina T, et al. Single-cell molecular analysis defines therapy response and immunophenotype of stem cell subpopulations in CML. Blood. 2017;129:2384–94.
Landberg N, Hansen N, Askmyr M, Ågerstam H, Lassen C, Rissler M, et al. IL1RAP expression as a measure of leukemic stem cell burden at diagnosis of chronic myeloid leukemia predicts therapy outcome. Leukemia. 2016;30:255–8.
Xie X, Feng M, Wang Q, Wang J, Yin R, Li Y, et al. Cellular and molecular state of myeloid leukemia stem cells. Adv Exp Med Biol. 2019;1143:41–57.
Chai SK, Nichols GL, Rothman P. Constitutive activation of JAKs and STATs in BCR-Abl-expressing cell lines and peripheral blood cells derived from leukemic patients. J Immunol. 1997;159:4720–8.
Zipeto MA, Court AC, Sadarangani A, Delos Santos NP, Balaian L, Chun HJ, et al. ADAR1 activation drives leukemia stem cell self-renewal by impairing Let-7 biogenesis. Cell Stem Cell. 2016;19:177–91.
Park CS, Lewis AH, Chen TJ, Bridges CS, Shen Y, Suppipat K, et al. A KLF4-DYRK2–mediated pathway regulating self-renewal in CML stem cells. Blood. 2019;134:1960–72.
Zabriskie MS, Eide CA, Tantravahi SK, Vellore NA, Estrada J, Nicolini FE, et al. BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell. 2014;26:428–42.
Zhang B, Nguyen LXT, Li L, Zhao D, Kumar B, Wu H, et al. Bone marrow niche trafficking of miR-126 controls the self-renewal of leukemia stem cells in chronic myelogenous leukemia. Nat Med. 2018;24:450–62.
Kuntz EM, Baquero P, Michie AM, Dunn K, Tardito S, Holyoake TL, et al. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat Med. 2017;23:1234–40.
Pippa R, Odero MD. The role of MYC and PP2A in the initiation and progression of myeloid leukemias. Cells. 2020;9:544.
Kataoka K, Kurokawa M. Ecotropic viral integration site 1, stem cell self-renewal and leukemogenesis. Cancer Sci. 2012;103:1371–7.
Höpner SS, Raykova A, Radpour R, Amrein MA, Koller D, Baerlocher GM, et al. LIGHT/LTβR signaling regulates self-renewal and differentiation of hematopoietic and leukemia stem cells. Nat Commun. 2021;12:1–16.
Hochhaus A, Baccarani M, Silver RT, Schiffer C, Apperley JF, Cervantes F, et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia. 2020;34:966–84.
Mukaida N, Tanabe Y, Baba T. Cancer non-stem cells as a potent regulator of tumor microenvironment: a lesson from chronic myeloid leukemia. Mol Biomed. 2021;2(1):1–16.
Cerny-Reiterer S, Ghanim V, Hoermann G, Aichberger KJ, Herrmann H, Muellauer L, et al. Identification of basophils as a major source of hepatocyte growth factor in chronic myeloid leukemia: A novel mechanism of BCR-ABL1-independent disease progression. Neoplasia (United States). 2012;14:572-IN10.
Samorapoompichit P, Kiener HP, Schernthaner GH, Jordan JH, Agis H, Wimazal F, et al. Detection of tryptase in cytoplasmic granules of basophils in patients with chronic myeloid leukemia and other myeloid neoplasms. Blood. 2001;98:2580–3.
Tanabe Y, Kawamoto S, Takaku T, Morishita S, Hirao A, Komatsu N, et al. Expansion of senescent megakaryocyte-lineage cells maintains CML cell leukemogenesis. Blood Adv. 2020;4:6175–88.
Li YL, Tang JM, Chen XY, Luo B, Liang GH, Qu Q, et al. MicroRNA-153–3p enhances the sensitivity of chronic myeloid leukemia cells to imatinib by inhibiting B-cell lymphoma-2-mediated autophagy. Hum Cell. 2020;33:610–8.
Tarafdar A, Hopcroft LEM, Gallipoli P, Pellicano F, Cassels J, Hair A, et al. CML cells actively evade host immune surveillance through cytokine-mediated downregulation of MHC-II expression. Blood. 2017;129:199–208.
Zhou HS, Carter BZ, Andreeff M. Bone marrow niche-mediated survival of leukemia stem cells in acute myeloid leukemia: Yin and Yang. Cancer Biol Med. 2016;13:248–59.
Li L, Bhatia R. Stem cell quiescence: figure 1. Clin Cancer Res. 2011;17:4936–41.
Shan Y, DeSouza N, Qiu Q, Li S. Leukemia Stem Cells in Chronic Myeloid Leukemia. In: Zhang H, Li S. editors. Leukemia Stem Cells in Hematologic Malignancies. Advances in Experimental Medicine and Biology, vol 1143. Singapore; Springer. (2019). https://doi.org/10.1007/978-981-13-7342-8_9.
Scott MT, Korfi K, Saffrey P, Hopcroft LEM, Kinstrie R, Pellicano F, et al. Epigenetic reprogramming sensitizes CML stem cells to combined EZH2 and tyrosine kinase inhibition. Cancer Discov. 2016;6:1248–57.
Hoggatt J, Kfoury Y, Scadden DT. Hematopoietic stem cell niche in health and disease. Annu Rev Pathol Mech Dis. 2016;11:555–81.
Mitchell R, Copland M. Defining niche interactions to target chronic myeloid leukemia stem cells. Haematologica. 2020;105:2–4.
Toofan P, Busch C, Morrison H, O’Brien S, Jørgensen H, Copland M, et al. Chronic myeloid leukaemia cells require the bone morphogenic protein pathway for cell cycle progression and self-renewal. Cell Death Dis. 2018;9:1–19.
Gurska L.M., Ames K., Gritsman K. (2019) Signaling Pathways in Leukemic Stem Cells. In: Zhang H., Li S. editors. Leukemia Stem Cells in Hematologic Malignancies. Advances in Experimental Medicine and Biology, vol 1143. Singapore; Springer. https://doi.org/10.1007/978-981-13-7342-8_1
Wang RN, Green J, Wang Z, Deng Y, Qiao M, Peabody M, et al. Bone morphogenetic protein (BMP) signaling in development and human diseases. Genes Dis. 2014;1:87–105.
Houshmand M, Circosta P, Saglio G. Immature CML cells implement a BMP autocrine loop to escape TKI treatment. Transl Cancer Res. 2018;7:S722–5.
Jeanpierre S, Arizkane K, Thongjuea S, Grockowiak E, Geistlich K, Barral L, et al. The quiescent fraction of chronic myeloid leukemic stem cells depends on BMPR1B, Stat3 and BMP4-niche signals to persist in patients in remission. Haematologica. 2020;106:111–22.
Jurj A, Pasca S, Teodorescu P, Tomuleasa C, Berindan-Neagoe I. Basic knowledge on bcr-Abl1-positive extracellular vesicles. Biomark Med. 2020;14:451–8.
Chiba M, Kubota S, Sato K, Monzen S. Exosomes released from pancreatic cancer cells enhance angiogenic activities via dynamin-dependent endocytosis in endothelial cells in vitro. Sci Rep. 2018;8:1–9.
Mineo M, Garfield SH, Taverna S, Flugy A, De Leo G, Alessandro R, et al. Exosomes released by K562 chronic myeloid leukemia cells promote angiogenesis in a src-dependent fashion. Angiogenesis. 2012;15:33–45.
Corrado C, Saieva L, Raimondo S, Santoro A, De Leo G, Alessandro R. Chronic myelogenous leukaemia exosomes modulate bone marrow microenvironment through activation of epidermal growth factor receptor. J Cell Mol Med. 2016;20:1829–39.
Taverna S, Flugy A, Saieva L, Kohn EC, Santoro A, Meraviglia S, et al. Role of exosomes released by chronic myelogenous leukemia cells in angiogenesis. Int J Cancer. 2012;130:2033–43.
Guarnerio J, Mendez LM, Asada N, Menon AV, Fung J, Berry K, et al. A non-cell-autonomous role for Pml in the maintenance of leukemia from the niche. Nat Commun. 2018;9:1–11.
Kim JH, Lee SJ, Kang KW, Lee BH, Park Y, Kim BS. CXCR2, a novel target to overcome tyrosine kinase inhibitor resistance in chronic myelogenous leukemia cells. Biochem Pharmacol. 2021;190:114658. https://doi.org/10.1016/j.bcp.2021.114658.
Coluccia AML, Vacca A, Dũach M, Mologni L, Redaelli S, Bustos VH, et al. Bcr-Abl stabilizes β-catenin in chronic myeloid leukemia through its tyrosine phosphorylation. EMBO J. 2007;26:1456–66.
Zhang B, McDonald T, Holyoake TL, Moon RT, Campana D, Shultz L, et al. Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt signaling. Blood. 2012;120:912.
Han Y, Wang Y, Xu Z, Li J, Yang J, Li Y, et al. Effect of bone marrow mesenchymal stem cells from blastic phase chronic myelogenous leukemia on the growth and apoptosis of leukemia cells. Oncol Rep. 2013;30:1007–13.
Traer E, Javidi-Sharifi N, Agarwal A, Dunlap J, English I, Martinez J, et al. Ponatinib overcomes FGF2-mediated resistance in CML patients without kinase domain mutations. Blood. 2014;123:1516–24.
Bourgeais J, Ishac N, Medrzycki M, Brachet-Botineau M, Desbourdes L, Gouilleux-Gruart V, et al. Oncogenic STAT5 signaling promotes oxidative stress in chronic myeloid leukemia cells by repressing antioxidant defenses. Oncotarget. 2017;8:41876–89.
Ng KP, Manjeri A, Lee KL, Huang W, Tan SY, Chuah CTH, et al. Physiologic hypoxia promotes maintenance of CML stem cells despite effective BCR-ABL1 inhibition. Blood. 2014;123:3316–26.
Zhang H, Li H, Xi HS, Li S. HIF1α is required for survival maintenance of chronic myeloid leukemia stem cells. Blood. 2012;119:2595–607.
Chen H, Shen Y, Gong F, Jiang Y, Zhang R. HIF-α promotes chronic myelogenous leukemia cell proliferation by upregulating p21 expression. Cell Biochem Biophys. 2015;72:179–83.
Saito S, Lin YC, Tsai MH, Lin CS, Murayama Y, Sato R, et al. Emerging roles of hypoxia-inducible factors and reactive oxygen species in cancer and pluripotent stem cells. Kaohsiung J Med Sci. 2015;31:279–86.
Westerweel PE, te Boekhorst PAW, Levin MD, Cornelissen JJ. New approaches and treatment combinations for the management of chronic myeloid leukemia. Front Oncol. 2019;9:665.
Yousefi B, Samadi N, Baradaran B, Shafiei-Irannejad V, Zarghami N. Peroxisome proliferator-activated receptor ligands and their role in chronic myeloid leukemia: therapeutic strategies. Chem Biol Drug Des. 2016;88:17–25.
Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia. 2004;18:189–218.
Sheng Z, Ma L, Sun JE, Zhu LJ, Green MR. BCR-ABL suppresses autophagy through ATF5-mediated regulation of mTOR transcription. Blood. 2011;118:2840–8.
Rothe K, Lin H, Lin KBL, Leung A, Wang HM, Malekesmaeili M, et al. The core autophagy protein ATG4B is a potential biomarker and therapeutic target in CML stem/progenitor cells. Blood. 2014;123:3622–34.
Grant S. Recruiting TP53 to target chronic myeloid leukemia stem cells. Haematologica. 2020;105:1172–4.
Alvarez-Calderon F, Gregory MA, Pham-Danis C, DeRyckere D, Stevens BM, Zaberezhnyy V, et al. Tyrosine kinase inhibition in leukemia induces an altered metabolic state sensitive to mitochondrial perturbations. Clin Cancer Res. 2015;21:1360–72.
Lee M, Hirpara JL, Eu JQ, Sethi G, Wang L, Goh BC, et al. Targeting STAT3 and oxidative phosphorylation in oncogene-addicted tumors. Redox Biol. 2019;25:101073.
Bencomo-Alvarez AE, Rubio AJ, Gonzalez MA, Eiring AM. Energy metabolism and drug response in myeloid leukaemic stem cells. Br J Haematol. 2019;186:524–37.
Hattori A, Tsunoda M, Konuma T, Kobayashi M, Nagy T, Glushka J, et al. Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature. 2017;545:500–4.
Chen Y, Hu Y, Zhang H, Peng C, Li S. Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat Genet. 2009;41:783–92.
Chen Y, Peng C, Abraham SA, Shan Y, Guo Z, Desouza N, et al. Arachidonate 15-lipoxygenase is required for chronic myeloid leukemia stem cell survival. J Clin Invest. 2014;124:3847–62.
Naka K. New routes to eradicating chronic myelogenous leukemia stem cells by targeting metabolism. Int J Hematol. 2021;113:648–55.
Naka K, Jomen Y, Ishihara K, Kim J, Ishimoto T, Bae EJ, et al. Dipeptide species regulate p38MAPK-Smad3 signalling to maintain chronic myelogenous leukaemia stem cells. Nat Commun. 2015;6:1–14.
Wang JYJ. The capable ABL: what is its biological function? Mol Cell Biol. 2014;34:1188–97.
Antoszewska-Smith J, Pawlowska E, Blasiak J. Reactive oxygen species in BCR-ABL1-expressing cells—relevance to chronic myeloid leukemia. Acta Biochim Pol. 2017;64:1–10.
Dueva R, Iliakis G. Alternative pathways of non-homologous end joining (NHEJ) in genomic instability and cancer. Transl Cancer Res. 2013;2:163–77.
Ma G, Lu D, Wu Y, Liu J, Arlinghaus RB. Bcr phosphorylated on tyrosine 177 binds Grb2. Oncogene. 1997;14:2367–72.
Ma L, Shan Y, Bai R, Xue L, Eide CA, Ou J, et al. A therapeutically targetable mechanism of BCR-ABL—independent imatinib resistance in chronic myeloid leukemia. Sci Transl Med. 2014;6:252ra121.
Ren SY, Xue F, Feng J, Skorski T. Intrinsic regulation of the interactions between the SH3 domain of p85 subunit of phosphatidylinositol-3 kinase and the protein network of BCR/ABL oncogenic tyrosine kinase. Exp Hematol. 2005;33:1222–8.
Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5:172–83.
Fu Z, Tindall DJ. FOXOs, cancer and regulation of apoptosis. Oncogene. 2008;27:2312–9.
Pellicano F, Scott MT, Helgason GV, Hopcroft LEM, Allan EK, Aspinall-O’Dea M, et al. The antiproliferative activity of kinase inhibitors in chronic myeloid leukemia cells is mediated by FOXO transcription factors. Stem Cells. 2014;32:2334–7.
Jacquel A, Luciano F, Robert G, Auberger P. Implication and regulation of AMPK during physiological and pathological myeloid differentiation. Int J Mol Sci. 2018;19:2991.
Naka K, Hoshii T, Muraguchi T, Tadokoro Y, Ooshio T, Kondo Y, et al. TGF-Β-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature. 2010;463:676–80.
Chen YF, Liu H, Luo XJ, Zhao Z, Zou ZY, Li J, et al. The roles of reactive oxygen species (ROS) and autophagy in the survival and death of leukemia cells. Crit Rev Oncol Hematol. 2017;112:21–30.
Frazzi R, Guardi M. Cellular and molecular targets of resveratrol on lymphoma and leukemia cells. Molecules. 2017;22:885.
Gallipoli P, Cook A, Rhodes S, Hopcroft L, Wheadon H, Whetton AD, et al. JAK2/STAT5 inhibition by nilotinib with ruxolitinib contributes to the elimination of CML CD34+ cells in vitro and in vivo. Blood. 2014;124:1492–501.
Bavaro L, Martelli M, Cavo M, Soverini S. Mechanisms of disease progression and resistance to tyrosine kinase inhibitor therapy in chronic myeloid leukemia: an update. Int J Mol Sci. 2019;20:6141.
Kuepper MK, Bütow M, Herrmann O, Ziemons J, Chatain N, Maurer A, et al. Stem cell persistence in CML is mediated by extrinsically activated JAK1-STAT3 signaling. Leukemia. 2019;33:1964–77.
Ma L, Pak ML, Ou J, Yu J, Louis PS, Shan Y, et al. Prosurvival kinase PIM2 is a therapeutic target for eradication of chronic myeloid leukemia stem cells. Proc Natl Acad Sci USA. 2019;116:10482.
Toda J, Ichii M, Oritani K, Shibayama H, Tanimura A, Saito H, et al. Signal-transducing adapter protein-1 is required for maintenance of leukemic stem cells in CML. Oncogene. 2020;39:5601–15.
Valent P. Targeting the JAK2-STAT5 pathway in CML. Blood. 2014;124:1386–8.
Agarwal P, Zhang B, Ho Y, Cook A, Li L, Wang Y, et al. Inhibition of CML stem cell renewal by the porcupine inhibitor WNT974. Blood. 2015;126:54.
Agarwal P, Zhang B, Ho Y, Cook A, Li L, Mikhail FM, et al. Enhanced targeting of CML stem and progenitor cells by inhibition of porcupine acyltransferase in combination with TKI. Blood. 2017;129:1008–20.
Grassi S, Palumbo S, Mariotti V, Liberati D, Guerrini F, Ciabatti E, et al. The WNT pathway is relevant for the BCR-ABL1-independent resistance in chronic myeloid leukemia. Front Oncol. 2019;9:532.
Karabay AZ, Koc A, Ozkan T, Hekmatshoar Y, Altinok Gunes B, Sunguroglu A, et al. Expression analysis of Akirin-2, NFκB-p65 and β-catenin proteins in imatinib resistance of chronic myeloid leukemia. Hematology. 2018;23:765–70.
Yang K, Wang F, Zhang H, Wang X, Chen L, Su X, Wu X, Han Q, Chen Z, Chen ZS, Fu L. Target Inhibition of CBP Induced Cell Senescence in BCR-ABL- T315I Mutant Chronic Myeloid Leukemia. Front Oncol. 2021;8(10): https://doi.org/10.3389/fonc.2020.588641.
Li F, He B, Ma X, Yu S, Bhave RR, Lentz SR, et al. Prostaglandin E1 and its analog misoprostol inhibit human CML stem cell self-renewal via ep4 receptor activation and repression of AP-1. Cell Stem Cell. 2017;21:359-373e5.
Su W, Meng F, Huang L, Zheng M, Liu W, Sun H. Sonic hedgehog maintains survival and growth of chronic myeloid leukemia progenitor cells through β-catenin signaling. Exp Hematol. 2012;40:418–27.
Dierks C, Beigi R, Guo GR, Zirlik K, Stegert MR, Manley P, et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on hedgehog pathway activation. Cancer Cell. 2008;14:238–49.
Moradi F, Babashah S, Sadeghizadeh M, Jalili A, Hajifathali A, Roshandel E. Signaling pathways involved in chronic myeloid leukemia pathogenesis: the importance of targeting musashi2-numb signaling to eradicate leukemia stem cells. Iran J Basic Med Sci. 2019;22:581.
Antoniani C, Romano O, Miccio A. Concise review: epigenetic regulation of hematopoiesis: biological insights and therapeutic applications. Stem Cells Transl Med. 2017;6:2106–14.
Valent P, Kern W, Hoermann G, Feenstra JDM, Sotlar K, Pfeilstöcker M, et al. Clonal hematopoiesis with oncogenic potential (CHOP): separation from CHIP and roads to AML. Int J Mol Sci. 2019;20:789.
Asimakopoulos FA, Shteper PJ, Krichevsky S, Fibach E, Polliack A, Rachmilewitz E, et al. ABL1 methylation is a distinct molecular event associated with clonal evolution of chronic myeloid leukemia. Blood. 1999;94:2452–60.
Arrigoni E, Del Re M, Galimberti S, Restante G, Rofi E, Crucitta S, et al. Concise review: chronic myeloid leukemia: stem cell niche and response to pharmacologic treatment. Stem Cells Transl Med. 2018;7:305–14.
Xie H, Peng C, Huang J, Li BE, Kim W, Smith EC, et al. Chronic myelogenous leukemia-initiating cells require polycomb group protein EZH2. Cancer Discov. 2016;6:1237–47.
Matsuda Y, Yamauchi T, Hosono N, Uzui K, Negoro E, Morinaga K, et al. Combination of panobinostat with ponatinib synergistically overcomes imatinib-resistant CML cells. Cancer Sci. 2016;107:1029–38.
Wang Z, Yuan H, Roth M, Stark JM, Bhatia R, Chen WY. SIRT1 deacetylase promotes acquisition of genetic mutations for drug resistance in CML cells. Oncogene. 2013;32:589–98.
Riether C, Schürch CM, Flury C, Hinterbrandner M, Drück L, Huguenin AL, et al. Tyrosine kinase inhibitor-induced CD70 expression mediates drug resistance in leukemia stem cells by activating Wnt signaling. Sci Transl Med. 2015;7:298ra119.
Gregory MA, Phang TL, Neviani P, Alvarez-Calderon F, Eide CA, O’Hare T, et al. Wnt/Ca2+/NFAT signaling maintains survival of Ph+ leukemia cells upon inhibition of Bcr-Abl. Cancer Cell. 2010;18:74–87.
Wang WZ, Pu QH, Lin XH, Liu MY, Wu LR, Wu QQ, et al. Silencing of miR-21 sensitizes CML CD34+ stem/progenitor cells to imatinib-induced apoptosis by blocking PI3K/AKT pathway. Leuk Res. 2015;39:1117–24.
Di Stefano C, Mirone G, Perna S, Marfe G. The roles of microRNAs in the pathogenesis and drug resistance of chronic myelogenous leukemia (Review). Oncol Rep. 2016;35:614–24.
Salati S, Salvestrini V, Carretta C, Genovese E, Rontauroli S, Zini R, et al. Deregulated expression of miR-29a-3p, miR-494-3p and miR-660-5p affects sensitivity to tyrosine kinase inhibitors in CML leukemic stem cells. Oncotarget. 2017;8:49451–69.
Lv M, Zhang X, Jia H, Li D, Zhang B, Zhang H, et al. An oncogenic role of miR-142–3p in human T-cell acute lymphoblastic leukemia (T-ALL) by targeting glucocorticoid receptor-α and cAMP/PKA pathways. Leukemia. 2012;26:769–77.
Yung Y, Lee E, Chu HT, Yip PK, Gill H. Targeting abnormal hematopoietic stem cells in chronic myeloid leukemia and philadelphia chromosome-negative classical myeloproliferative neoplasms. Int J Mol Sci. 2021;22:659.
Zou Z, Wu L, Ding H, Wang Y, Zhang Y, Chen X, et al. MicroRNA-30a sensitizes tumor cells to cis-platinum via suppressing beclin 1-mediated autophagy. J Biol Chem. 2012;287:4148–56.
Silvestri G, Trotta R, Stramucci L, Ellis JJ, Harb JG, Neviani P, et al. Persistence of drug-resistant leukemic stem cells and impaired NK cell immunity in CML patients depend on MIR300 antiproliferative and PP2A-activating functions. Blood Cancer Discov. 2020;1:48–67.
Hehlmann R, Hochhaus A, Baccarani M. Chronic myeloid leukaemia. Lancet. 2007;370:342–50.
Bower H, Björkholm M, Dickman PW, Höglund M, Lambert PC, Andersson TML. Life expectancy of patients with chronic myeloid leukemia approaches the life expectancy of the general population. J Clin Oncol. 2016;34:2851–7.
Jiang Q, Liu ZC, Zhang SX, Gale RP. Young age and high cost are associated with future preference for stopping tyrosine kinase inhibitor therapy in Chinese with chronic myeloid leukemia. J Cancer Res Clin Oncol. 2016;142:1539–47.
Bonifacio M, Stagno F, Scaffidi L, Krampera M, Di Raimondo F. Management of chronic myeloid leukemia in advanced phase. Front Oncol. 2019;9:1132.
Steegmann JL, Baccarani M, Breccia M, Casado LF, García-Gutiérrez V, Hochhaus A, et al. European LeukemiaNet recommendations for the management and avoidance of adverse events of treatment in chronic myeloid leukaemia. Leukemia. 2016;30:1648–71.
Jiao Q, Bi L, Ren Y, Song S, Wang Q, Wang YS. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol Cancer. 2018;17:1–12.
Lee MY, Park CJ, Cho YU, You E, Jang S, Seol CA, et al. Differences in PD-1 expression on CD8+ T-cells in chronic myeloid leukemia patients according to disease phase and TKI medication. Cancer Immunol Immunother. 2020;69:2223–32.
Ciarcia R, Vitiello MT, Galdiero M, Pacilio C, Iovane V, D’Angelo D, et al. Imatinib treatment inhibit IL-6, IL-8, NF-KB and AP-1 production and modulate intracellular calcium in CML patients. J Cell Physiol. 2012;227:2798–803.
Ciarcia R, Damiano S, Puzio MV, Montagnaro S, Pagnini F, Pacilio C, et al. Comparison of dasatinib, nilotinib, and imatinib in the treatment of chronic myeloid leukemia. J Cell Physiol. 2016;231:680–7.
Sacha T, Szmit S, Zozulińska-Ziółkiewicz D, Prejzner W, Góra-Tybor J. Recommendations for assessment of co-morbidities and tyrosine kinase inhibitor choice in patients suffering from chronic myeloid leukemia. Acta Haematol Pol. 2016;47:184–96.
Liu J, Zhang Y, Huang H, Lei X, Tang G, Cao X, et al. Recent advances in Bcr-Abl tyrosine kinase inhibitors for overriding T315I mutation. Chem Biol Drug Des. 2021;97:649–64.
Abraham SA, Hopcroft LEM, Carrick E, Drotar ME, Dunn K, Williamson AJK, et al. Dual targeting of p53 and c-MYC selectively eliminates leukaemic stem cells. Nature. 2016;534:341–6.
Saussele S, Richter J, Guilhot J, Gruber FX, Hjorth-Hansen H, Almeida A, et al. Discontinuation of tyrosine kinase inhibitor therapy in chronic myeloid leukaemia (EURO-SKI): a prespecified interim analysis of a prospective, multicentre, non-randomised, trial. Lancet Oncol. 2018;19:747–57.
Doan V, Wang A, Prescott H. Bosutinib for the treatment of chronic myeloid leukemia. Am J Health Syst Pharm. 2015;72:439.
Eskazan AE, Keskin D. Radotinib and its clinical potential in chronic-phase chronic myeloid leukemia patients: an update. Ther Adv Hematol. 2017;8:237–43.
Molica M, Scalzulli E, Colafigli G, Foà R, Breccia M. Insights into the optimal use of ponatinib in patients with chronic phase chronic myeloid leukaemia. Ther Adv Hematol. 2019;10:204062071982644.
Cortes J, Lang F. Third-line therapy for chronic myeloid leukemia: current status and future directions. J Hematol Oncol. 2021;14:1–18.
Jiang Q, Huang X, Chen Z, Niu Q, Shi D, Li Z, et al. Novel BCR-ABL1 tyrosine kinase inhibitor (TKI) HQP1351 (olverembatinib) is efficacious and well tolerated in patients with T315I-mutated chronic myeloid leukemia (CML): results of pivotal (Phase II) trials. Blood [Internet]. 2020;136(Suppl 1):50–1.
Cortes JE, Saikia T, Kim D-W, Alvarado Y, Nicolini FE, Khattry N, et al. Phase 1 trial of vodobatinib, a novel oral BCR-ABL1 tyrosine kinase inhibitor (TKI): activity in CML chronic phase patients failing TKI therapies including ponatinib. Blood. 2020;136:51–2.
Eide CA, Zabriskie MS, Savage Stevens SL, Antelope O, Vellore NA, Than H, et al. Combining the allosteric inhibitor asciminib with ponatinib suppresses emergence of and restores efficacy against highly resistant BCR-ABL1 mutants. Cancer Cell. 2019;36(431):443.e5.
Özgür Yurttaş N, Eşkazan AE. Novel therapeutic approaches in chronic myeloid leukemia. Leuk Res. 2020;91:106337.
Ivanova ES, Tatarskiy VV, Yastrebova MA, Khamidullina AI, Shunaev AV, Kalinina AA, et al. PF-114, a novel selective inhibitor of BCR-ABL tyrosine kinase, is a potent inducer of apoptosis in chronic myelogenous leukemia cells. Int J Oncol. 2019;55:289–97.
Rossari F, Minutolo F, Orciuolo E. Past, present, and future of Bcr-Abl inhibitors: From chemical development to clinical efficacy. J Hematol Oncol. 2018;11:1–14.
Agarwal A, Mackenzie RJ, Besson A, Jeng S, Carey A, LaTocha DH, et al. BCR-ABL1 promotes leukemia by converting p27 into a cytoplasmic oncoprotein. Blood. 2014;124:3260–73.
Cortes J, Talpaz M, Smith HP, Snyder DS, Khoury J, Bhalla KN, et al. Phase 1 dose-finding study of rebastinib (DCC-2036) in patients with relapsed chronic myeloid leukemia and acute myeloid leukemia. Haematologica. 2017;102:519–28.
Sinclair A, Latif AL, Holyoake TL. Targeting survival pathways in chronic myeloid leukaemia stem cells. Br J Pharmacol. 2013;169:1693–707.
Mourgues L, Imbert V, Nebout M, Colosetti P, Neffati Z, Lagadec P, et al. The BMI1 polycomb protein represses cyclin G2-induced autophagy to support proliferation in chronic myeloid leukemia cells. Leukemia. 2015;29:1993–2002.
Raheem R, Alsayed R, Yousif E, Hairunisa N. Coronavirus new variants: the mutations cause and the effect on the treatment and vaccination. Baghdad J Biochem Appl Biol Sci. 2021;2:71–9.
Yahata T, Ibrahim AA, Hirano KI, Muguruma Y, Naka K, Hozumi K, et al. Targeting of plasminogen activator inhibitor-1 activity promotes elimination of chronic myeloid leukemia stem cells. Haematologica. 2020;106:483.
Cheloni G, Tanturli M, Tusa I, DeSouza NH, Shan Y, Gozzini A, et al. Targeting chronic myeloid leukemia stem cells with the hypoxia-inducible factor inhibitor acriflavine. Blood. 2017;130:655–65.
Prost S, Relouzat F, Spentchian M, Ouzegdouh Y, Saliba J, Massonnet G, et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARγ agonists. Nature. 2015;525:380–3.
Glodkowska-Mrowka E, Manda-Handzlik A, Stelmaszczyk-Emmel A, Seferynska I, Stoklosa T, Przybylski J, et al. PPARγ ligands increase antileukemic activity of second- and third-generation tyrosine kinase inhibitors in chronic myeloid leukemia cells. Blood Cancer J. 2016;6:e377.
Massimino M, Stella S, Tirrò E, Romano C, Pennisi MS, Puma A, et al. Non ABL-directed inhibitors as alternative treatment strategies for chronic myeloid leukemia. Mol Cancer. 2018;17:1–15.
Tari Ashizawa A, Ohanian M, Cortes JE. BP1001, a novel therapeutic for chronic myelogenous leukemia. Blood. 2016;128:4239.
Chorzalska A, Ahsan N, Rao RSP, Roder K, Yu X, Morgan J, et al. Overexpression of Tpl2 is linked to imatinib resistance and activation of MEK-ERK and NF-κB pathways in a model of chronic myeloid leukemia. Mol Oncol. 2018;12:630–47.
Lin H, Rothe K, Chen M, Wu A, Babaian A, Yen R, et al. The miR-185/PAK6 axis predicts therapy response and regulates survival of drug-resistant leukemic stem cells in CML. Blood. 2020;136:596–609.
Minciacchi VR, Kumar R, Krause DS. Chronic myeloid leukemia: a model disease of the past, present and future. Cells. 2021;10:117.
Tu YX, Wang SB, Fu LQ, Li SS, Guo QP, Wu Y, et al. Ovatodiolide targets chronic myeloid leukemia stem cells by epigenetically upregulating hsa-miR-155, suppressing the BCRABL fusion gene and dysregulating the PI3K/AKT/mTOR pathway. Oncotarget. 2018;9:3267–77.
Raivola J, Haikarainen T, Abraham BG, Silvennoinen O. Janus kinases in leukemia. Cancers. 2021;13:800.
Neviani P, Santhanam R, Oaks JJ, Eiring AM, Notari M, Blaser BW, et al. FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphocytic leukemia. J Clin Invest. 2007;117:2408–21.
Agarwal A, MacKenzie RJ, Pippa R, Eide CA, Oddo J, Tyner JW, et al. Antagonism of SET using OP449 enhances the efficacy of tyrosine kinase inhibitors and overcomes drug resistance in myeloid leukemia. Clin Cancer Res. 2014;20:2092–103.
Yokoyama N, Reich NC, Miller WT. Determinants for the interaction between Janus kinase 2 and protein phosphatase 2A. Arch Biochem Biophys. 2003;417:87–95.
Neviani P, Harb JG, Oaks JJ, Santhanam R, Walker CJ, Ellis JJ, et al. PP2A-activating drugs selectively eradicate tki-resistant chronic myeloid leukemic stem cells. J Clin Invest. 2013;123:4144–57.
Ujvari D, Nagy N, Madapura HS, Kallas T, Kröhnke MCL, Stenke L, et al. Interferon γ is a strong, STAT1-dependent direct inducer of BCL6 expression in multiple myeloma cells. Biochem Biophys Res Commun. 2018;498:502–8.
Yokota A, Hirai H, Sato R, Adachi H, Sato F, Hayashi Y, et al. C/EBPb is a critical mediator of IFN-a–induced exhaustion of chronic myeloid leukemia stem cells. Blood Adv. 2019;3:476–88.
Eiring AM, Page BDG, Kraft IL, Mason CC, Vellore NA, Resetca D, et al. Combined STAT3 and BCR-ABL1 inhibition induces synthetic lethality in therapy-resistant chronic myeloid leukemia. Leukemia. 2015;29:586–97.
Ruan Y, Kim HN, Ogana H, Kim YM. Wnt signaling in leukemia and its bone marrow microenvironment. Int J Mol Sci. 2020;21:6247.
Heidel FH, Bullinger L, Feng Z, Wang Z, Neff TA, Stein L, et al. Genetic and pharmacologic inhibition of β-catenin targets imatinib-resistant leukemia stem cells in CML. Cell Stem Cell. 2012;10:412–24.
Jin B, Wang C, Li J, Du X, Ding K, Pan J. Anthelmintic niclosamide disrupts the interplay of p65 and FOXM1/β-catenin and eradicates leukemia stem cells in chronic myelogenous leukemia. Clin Cancer Res. 2017;23:789–803.
Huang W, Luan CH, Hjort EE, Bei L, Mishra R, Sakamoto KM, et al. The role of Fas-associated phosphatase 1 in leukemia stem cell persistence during tyrosine kinase inhibitor treatment of chronic myeloid leukemia. Leukemia. 2016;30:1502–9.
Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458:776–9.
Irvine DA, Zhang B, Kinstrie R, Tarafdar A, Morrison H, Campbell VL, et al. Deregulated hedgehog pathway signaling is inhibited by the smoothened antagonist LDE225 (Sonidegib) in chronic phase chronic myeloid leukaemia. Sci Rep. 2016;6:1–13.
Sadarangani A, Pineda G, Lennon KM, Chun HJ, Shih A, Schairer AE, et al. GLI2 inhibition abrogates human leukemia stem cell dormancy. J Transl Med. 2015;13:1–14.
Wang YH, Israelsen WJ, Lee D, Yu VWC, Jeanson NT, Clish CB, et al. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell. 2014;158:1309–23.
Babashah S, Sadeghizadeh M, Hajifathali A, Tavirani MR, Zomorod MS, Ghadiani M, et al. Targeting of the signal transducer Smo links microRNA-326 to the oncogenic Hedgehog pathway in CD34+ CML stem/progenitor cells. Int J Cancer. 2013;133:579–89.
Gallipoli P, Giotopoulos G, Tzelepis K, Costa ASH, Vohra S, Medina-Perez P, et al. Glutaminolysis is a metabolic dependency in FLT3 ITD acute myeloid leukemia unmasked by FLT3 tyrosine kinase inhibition. Blood. 2018;131:1639–53.
Goff DJ, Recart AC, Sadarangani A, Chun HJ, Barrett CL, Krajewska M, et al. A Pan-BCL2 inhibitor renders bone-marrow-resident human leukemia stem cells sensitive to tyrosine kinase inhibition. Cell Stem Cell. 2013;12:316–28.
Carter BZ, Mak PY, Mu H, Zhou H, Mak DH, Schober W, et al. Combined targeting of BCL-2 and BCR-ABL tyrosine kinase eradicates chronic myeloid leukemia stem cells. Sci Transl Med. 2016;8:355ra117.
Houshmand M, Garello F, Stefania R, Gaidano V, Cignetti A, Spinelli M, et al. Targeting chronic myeloid leukemia stem/progenitor cells using venetoclax-loaded immunoliposome. Cancers (Basel). 2021;13:1311.
Xiao M, Ai H, Li T, Rajoria P, Shahu P, Li X. Targeting of the BLT2 in chronic myeloid leukemia inhibits leukemia stem/progenitor cell function. Biochem Biophys Res Commun. 2016;472:610–6.
Rothe K, Babaian A, Nakamichi N, Chen M, Chafe SC, Watanabe A, et al. Integrin-linked kinase mediates therapeutic resistance of quiescent CML stem cells to tyrosine kinase inhibitors. Cell Stem Cell. 2020;27:110-124.e9.
Zhou J, Nie D, Li J, Du X, Lu Y, Li Y, et al. PTEN is fundamental for elimination of leukemia stem cells mediated by GSK126 targeting EZH2 in chronic myelogenous leukemia. Clin Cancer Res. 2018;24:145–57.
Peterson LF, Lo MC, Liu Y, Giannola D, Mitrikeska E, Donato NJ, et al. Induction of p53 suppresses chronic myeloid leukemia. Leuk Lymphoma. 2017;58:2165–75.
Carter BZ, Mak PY, Mak DH, Ruvolo VR, Schober W, McQueen T, et al. Synergistic effects of p53 activation via MDM2 inhibition in combination with inhibition of Bcl-2 or Bcr-Abl in CD34+ proliferating and quiescent chronic myeloid leukemia blast crisis cells. Oncotarget. 2015;6:30487–99.
Bamodu OA, Kuo KT, Yuan LP, Cheng WH, Lee WH, Ho YS, et al. HDAC inhibitor suppresses proliferation and tumorigenicity of drug-resistant chronic myeloid leukemia stem cells through regulation of hsa-miR-196a targeting BCR/ABL1. Exp Cell Res. 2018;370:519–30.
Bernardo PS, Lemos LGT, de Moraes GN, Maia RC. Unraveling survivin expression in chronic myeloid leukemia: molecular interactions and clinical implications. Blood Rev. 2020;43:100671.
He B, Wang Q, Liu X, Lu Z, Han J, Pan C, et al. A novel HDAC inhibitor chidamide combined with imatinib synergistically targets tyrosine kinase inhibitor resistant chronic myeloid leukemia cells. Biomed Pharmacother [Internet]. 2020;129:110390.
Lernoux M, Schnekenburger M, Losson H, Vermeulen K, Hahn H, Gérard D, et al. Novel HDAC inhibitor MAKV-8 and imatinib synergistically kill chronic myeloid leukemia cells via inhibition of BCR-ABL/MYC-signaling: effect on imatinib resistance and stem cells. Clin Epigenetics. 2020;12:1–26.
Li L, Wang L, Li L, Wang Z, Ho Y, McDonald T, et al. Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell. 2012;21:266–81.
Abraham A, Qiu S, Chacko BK, Li H, Paterson A, He J, et al. SIRT1 regulates metabolism and leukemogenic potential in CML stem cells. J Clin Invest. 2019;129:2685–701.
Zhu F, Rui L. PRMT5 in gene regulation and hematologic malignancies. Genes Dis. 2019;6:247–57.
Jin Y, Zhou J, Xu F, Jin B, Cui L, Wang Y, et al. Targeting methyltransferase PRMT5 eliminates leukemia stem cells in chronic myelogenous leukemia. J Clin Invest. 2016;126:3961–80.
Vuelta E, García-Tuñón I, Hernández-Carabias P, Méndez L, Sánchez-Martín M. Future approaches for treating chronic myeloid leukemia: CRISPR therapy. Biology. 2021;10:118.
Cross NCP, White HE, Müller MC, Saglio G, Hochhaus A. Standardized definitions of molecular response in chronic myeloid leukemia. Leukemia. 2012;26:2172–5.
Kohnken R, Porcu P, Mishra A. Overview of the use of murine models in leukemia and lymphoma research. Front Oncol. 2017;7:22.
Heyer J, Kwong LN, Lowe SW, Chin L. Non-germline genetically engineered mouse models for translational cancer research. Nat Rev Cancer. 2010;10:470–80.
Rosenthal N, Brown S. The mouse ascending: perspectives for human-disease models. Nat Cell Biol. 2007;9:993.
Richmond A, Yingjun S. Mouse xenograft models vs GEM models for human cancer therapeutics. DMM Dis Models Mech. 2008;1:78–82.
Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210 bcr/abl gene of the Philadelphia chromosome. Science (80-). 1990;247:824–30.
Ye D, Wolff N, Li L, Zhang S, Ilaria RL. STAT5 signaling is required for the efficient induction and maintenance of CMLin mice. Blood. 2006;107:4917–25.
Giotopoulos G, van der Weyden L, Osaki H, Rust AG, Gallipoli P, Meduri E, et al. A novel mouse model identifies cooperating mutations and therapeutic targets critical for chronic myeloid leukemia progression. J Exp Med. 2015;212:1551–69.
Peng C, Li S. Chronic myeloid leukemia (CML) mouse model in translational research. Methods Mol Biol [Internet]. 2016. https://doi.org/10.1007/978-1-4939-3661-8_13.
Qin W, Kutny PM, Maser RS, Dion SL, Lamont JD, Zhang Y, et al. Generating mouse models using CRISPR-Cas9-MEDIATED GENOME EDIting. Curr Protoc Mouse Biol. 2016;6:39–66.
Heckl D, Kowalczyk MS, Yudovich D, Belizaire R, Puram RV, McConkey ME, et al. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat Biotechnol. 2014;32:941–6.
Singh P, Schimenti JC, Bolcun-Filas E. A mouse geneticist’s practical guide to CRISPR applications. Genetics. 2015;199:1–15.
Cyranoski D. CRISPR gene-editing tested in a person for the first time. Nature. 2016;539:479.
Frangoul H, Bobruff Y, Cappellini MD, Corbacioglu S, Fernandez CM, de la Fuente J, et al. Safety and efficacy of CTX001 in patients with transfusion-dependent β-thalassemia and sickle cell disease: early results from the climb THAL-111 and climb SCD-121 studies of autologous CRISPR-CAS9-modified CD34+ hematopoietic stem and progenitor cells. Blood. 2020;136:3–4.
Peng C, Li S. Chronic myeloid leukemia (CML) mouse model in translational research. Methods Mol Biol. 2016;1438:225–43.
García-Tuñón I, Hernández-Sánchez M, Ordoñez JL, Alonso-Pérez V, Álamo-Quijada M, Benito R, et al. The CRISPR/Cas9 system efficiently reverts the tumorigenic ability of BCR/ABL in vitro and in a xenograft model of chronic myeloid leukemia. Oncotarget. 2017;8:26027–40.
Huang N, Huang Z, Gao M, Luo Z, Zhou F, Liu L, et al. Induction of apoptosis in imatinib sensitive and resistant chronic myeloid leukemia cells by efficient disruption of bcr-abl oncogene with zinc finger nucleases. J Exp Clin Cancer Res. 2018;37:1–14.
Vuelta E, Luis Ordoñez J, Alonso-Pérez V, Méndez L, Hernández-Carabias P, Saldaña R, et al. CRISPR/Cas9 technology abolishes the BCR/ABL1 oncogene effect in chronic myeloid leukemia and restores normal hematopoiesis. bioRxiv. 2020.
Li J, Hong S, Chen W, Zuo E, Yang H. Advances in detecting and reducing off-target effects generated by CRISPR-mediated genome editing. J Genet Genomics. 2019;46:513–21.
Helgason G, Mukhopadhyay A, Karvela M, Salomoni P, Calabretta B, Holyoake T. Autophagy in chronic myeloid leukaemia: stem cell survival and implication in therapy. Curr Cancer Drug Targets. 2013;13:724–34.
Kalluri R. The biology and function of exosomes in cancer. J Clin Investig. 2016;126:1208–15.
Baquero P, Dawson A, Mukhopadhyay A, Kuntz EM, Mitchell R, Olivares O, et al. Targeting quiescent leukemic stem cells using second generation autophagy inhibitors. Leukemia. 2019;33:981–94.
Zhang B, Chu S, Agarwal P, Campbell VL, Hopcroft L, Jørgensen HG, et al. Inhibition of interleukin-1 signaling enhances elimination of tyrosine kinase inhibitor-treated CML stem cells. Blood. 2016;128:2671–82.
Järås M, Johnels P, Hansen N, Ågerstam H, Tsapogas P, Rissler M, et al. Isolation and killing of candidate chronic myeloid leukemia stem cells by antibody targeting of IL-1 receptor accessory protein. Proc Natl Acad Sci USA. 2010;107:16280.
Ågerstam H, Karlsson C, Hansen N, Sandén C, Askmyr M, Von Palffy S, et al. Antibodies targeting human IL1RAP (IL1R3) show therapeutic effects in xenograft models of acute myeloid leukemia. Proc Natl Acad Sci USA. 2015;112:10786–91.
Warda W, Larosa F, Da Rocha MN, Trad R, Deconinck E, Fajloun Z, et al. CML hematopoietic stem cells expressing IL1RAP can be targeted by chimeric antigen receptor-engineered T cells. Cancer Res. 2019;79:663–75.
Willmann M, Sadovnik I, Eisenwort G, Entner M, Bernthaler T, Stefanzl G, et al. Evaluation of cooperative antileukemic effects of nilotinib and vildagliptin in Ph+ chronic myeloid leukemia. Exp Hematol. 2018;57:50-59.e6.
Zhou S, Li W, Xiao Y, Zhu X, Zhong Z, Li Q, et al. A novel chimeric antigen receptor redirecting T-cell specificity towards CD26+ cancer cells. Leukemia. 2021;35:119–29.
Yu B, Liu D. Gemtuzumab ozogamicin and novel antibody-drug conjugates in clinical trials for acute myeloid leukemia. Biomark Res. 2019;7:1–13.
Holm F, Mason CN, Runza V, Weigand S, Sadarangani A, Jamieson CHM. CD44 monoclonal antibody-enhanced clearance of chronic myeloid leukemia stem cells from the malignant niche. Blood. 2013;122:858.
Frolova O, Benito J, Brooks C, Wang RY, Korchin B, Rowinsky EK, et al. SL-401 and SL-501, targeted therapeutics directed at the interleukin-3 receptor, inhibit the growth of leukaemic cells and stem cells in advanced phase chronic myeloid leukaemia. Br J Haematol. 2014;166:862–74.
Alkharabsheh O, Frankel AE. Clinical activity and tolerability of SL-401 (Tagraxofusp): recombinant diphtheria toxin and interleukin-3 in hematologic malignancies. Biomedicines. 2019;7:6.
CX-01. Combined with azacitidine in the treatment of relapsed or refractory myelodysplastic syndrome and acute myeloid leukemia—full text view—ClinicalTrials.gov [Internet]. Cited 22 Apr 2021. https://clinicaltrials.gov/ct2/show/NCT02995655
A Clinical Research of CD123-targeted CAR-T in myeloid malignancies—full text view—ClinicalTrials.gov [Internet]. Cited 22 Apr 2021. https://clinicaltrials.gov/ct2/show/NCT02937103
Kaur H, Bruno JG, Kumar A, Sharma TK. Aptamers in the therapeutics and diagnostics pipelines. Theranostics. 2018;8:4016–32.
Agarwal A, Fleischman AG, Petersen CL, MacKenzie R, Luty S, Loriaux M, et al. Effects of plerixafor in combination with BCR-ABL kinase inhibition in a murine model of CML. Blood. 2012;120:2658–68.
Hallal R, Nehme R, Brachet-Botineau M, Nehme A, Dakik H, Deynoux M, et al. Acriflavine targets oncogenic STAT5 signaling in myeloid leukemia cells. J Cell Mol Med. 2020;24:10052–62.
Zhang H, Li H, Ho N, Li D, Li S. Scd1 plays a tumor-suppressive role in survival of leukemia stem cells and the development of chronic myeloid leukemia. Mol Cell Biol. 2012;32:1776–87.
Kumar H, Chattopadhyay S, Das N, Shree S, Patel D, Mohapatra J, et al. Leprosy drug clofazimine activates peroxisome proliferator-activated receptor-γ and synergizes with imatinib to inhibit chronic myeloid leukemia cells. Haematologica. 2019;105:971.
Cortes J, Quintás-Cardama A, Garcia-Manero G, O’Brien S, Jones D, Faderl S, et al. Phase 1 study of tipifarnib in combination with imatinib for patients with chronic myelogenous leukemia in chronic phase after imatinib failure. Cancer. 2007;110:2000–6.
Cortes J, Jabbour E, Daley GQ, O’Brien S, Verstovsek S, Ferrajoli A, et al. Phase 1 study of Lonafarnib (SCH 66336) and imatinib mesylate in patients with chronic myeloid leukemia who have failed prior single-agent therapy with imatinib. Cancer. 2007;110:1295–302.
Lim S, Saw TY, Zhang M, Janes MR, Nacro K, Hill J, et al. Targeting of the MNK-eIF4E axis in blast crisis Chronic myeloid leukemia inhibits leukemia stem cell function. Proc Natl Acad Sci USA. 2013;110:E2298–307.
Xin P, Li C, Zheng Y, Peng Q, Xiao H, Huang Y, et al. Efficacy of the dual PI3K and mTOR inhibitor NVP-BEZ235 in combination with imatinib mesylate against chronic myelogenous leukemia cell lines. Drug Des Dev Ther. 2017;11:1115–26.
Schuster K, Zheng J, Arbini AA, Zhang CC, Scaglioni PP. Selective targeting of the mTORC1/2 protein kinase complexes leads to antileukemic effects in vitro and in vivo. Blood Cancer J. 2011;1:34.
Dengler J, von Bubnoff N, Decker T, Peschel C, Duyster J. Combination of imatinib with rapamycin or RAD001 acts synergistically only in Bcr-Abl-positive cells with moderate resistance to imatinib [1]. Leukemia. 2005;19:1835–8.
Mancini M, Petta S, Martinelli G, Barbieri E, Santucci MA. RAD 001 (everolimus) prevents mTOR and Akt late re-activation in response to imatinib in chronic myeloid leukemia. J Cell Biochem. 2010;109:320–8.
Samanta A, Perazzona B, Chakraborty S, Sun X, Modi H, Bhatia R, et al. Janus kinase 2 regulates Bcr-Abl signaling in chronic myeloid leukemia. Leukemia. 2011;25:463–72.
Okabe S, Tauchi T, Katagiri S, Tanaka Y, Ohyashiki K. Combination of the ABL kinase inhibitor imatinib with the Janus kinase 2 inhibitor TG101348 for targeting residual BCR-ABL-positive cells. J Hematol Oncol. 2014;7:1–11.
Pan XN, Chen JJ, Wang LX, Xiao RZ, Liu LL, Fang ZG, et al. Inhibition of c-Myc overcomes cytotoxic drug resistance in acute myeloid leukemia cells by promoting differentiation. PLoS ONE. 2014;9:e105381.
Zhou H, Mak PY, Mu H, Mak DH, Zeng Z, Cortes J, et al. Combined inhibition of β-catenin and Bcr-Abl synergistically targets tyrosine kinase inhibitor-resistant blast crisis chronic myeloid leukemia blasts and progenitors in vitro and in vivo. Leukemia. 2017;31:2065–74.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
HM conceptualized the title, prepared the initial draft, and designed the figure. NY helped in drafting the manuscript, revised the manuscript, and prepared the final draft. NR helped in preparing the final draft, critically revised the manuscript, and supervised the project. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mojtahedi, H., Yazdanpanah, N. & Rezaei, N. Chronic myeloid leukemia stem cells: targeting therapeutic implications. Stem Cell Res Ther 12, 603 (2021). https://doi.org/10.1186/s13287-021-02659-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-021-02659-1