
Abstract
The analytical tool for analysis of pesticide residues in honey must still be improved. Therefore, a sensitive, selective, rapid, and reliable analytical method for the analysis of pesticide residue belonging to 29 different classes at very low concentration levels was developed. Over 130 insecticides were extracted from the honey matrix using different amounts of sample, concentrations of the extraction solvent, and cleanup sorbents. Acceptable extraction recoveries accounting for matrix effect were obtained using the cleanup sorbent combination chitosan/aluminum oxide/C8 (200/200/200 mg) and 1% FA in ACN. Determinations were made by liquid chromatography-tandem mass spectrometry (LC–MS/MS). For most compounds, the recoveries range from 70 to 120%, with relative standard deviations < 20%. Linear correlation coefficients (r2) were higher than 0.99 at concentration levels of 0.001–0.100 mg kg−1. A positive matrix effect was observed for up to 95% of organophosphates and 43% of pyrethroids and a negative effect for 37% of neonicotinoids. This work showed that the presented method is convenient and reliable for quick monitoring of insecticides in honey samples.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Honey is a highly consumed product due to its nutritional and antimicrobial properties. It is often consumed by children, the elderly, and sick persons. However, it may contain residues of plant protection chemicals, which are the consequence of chemical plant protection.
Insecticides representing a very wide group of compounds with a diverse chemical structures play the main role in plant protection. Pyrethroids (PYR), organochlorines (ORG), carbamates (CAR), neonicotinoids (NEO), and organophosphorus (OP) are commonly used on rice, maize and sunflower, rape, potato, sugar beet, vegetable, and fruit crops (Biever et al. 2003). Many of these compounds are dangerous to human health. For example, pirimicarb belonging to the carbamate group acts through inhibition of the AChE neurotransmitter and is suspected to be carcinogenic (Zheng et al. 2016). Cypermethrin, belonging to the synthetic pyrethroid group, is classified as moderately hazardous and interacts with the sodium channels in nerve cells, through which sodium enters the cell in order to transmit a nerve signal and interferes with other receptors in the nervous system (Sekhar et al. 2009).
When collecting pollen and nectar from flowers, bees transfer chemical contaminants to their beehives, with the result that they are detected in honey (Orso et al. 2016; Tette et al. 2016a; Barganska et al. 2013). These chemical pollutans are grouped into three toxicity classes (low, medium, and high) Highly toxic insecticides (constituting the largest group of the compounds) are dangerous to pollinating organisms, even in sublethal doses (Kaczyński et al. 2017).
To ensure the safety and quality control of honey, it is necessary to monitor chemical pollution in honey in order to be sure that the natural product does not contain any toxic residues at levels that would be harmful to consumers (Zacharis et al. 2012). However, the monitoring of pesticides in honey performed in recent years indicates an increase in the levels of chemical pollutants in this bee products (Rial-Otero et al. 2007). Therefore, sensitive, selective, rapid, and reliable analytical methods for insecticide residue analysis in honey must still be in the initial development.
The maximum residue levels (MRLs) of insecticides in honey are regulated by the European Union. Many compounds have MRLs in the range of 0.01–0.05 mg kg−1, but there are exceptions, for example, fipronil MRL = 0.005 mg kg−1. Therefore, the determination of pesticide residues in honey is a challenge, especially because of the low concentration of analytes and large amounts of interfering substances which can be coextracted with analytes, and in most cases, adversely affect analysis results (Chudzinska et al. 2012; Kujawski and Namiesnik 2008). Honey contains a large amount of sugars, proteins, amino acids, vitamins, antioxidants, and pigments (Zhou et al. 2014; Ferreira et al. 2009). The amount of these compounds largely depends upon the variety of the honey.
As already mentioned, the group of insecticides is very wide and chemically diverse, which makes sample preparation difficult. The melting point of insecticides ranges from − 49 to 222.6 °C, and the octanol–water partition coefficient (log P) ranges from − 0.85 to 8.1.
Over the last decade, different sample preparation techniques based on liquid-liquid extraction (Blasco et al. 2004), solid-phase extraction (Łozowicka 2013), QuEChERS method, and its combinations (Tette et al. 2016b) have been developed in order to determine insecticide residues in honey samples. To our best knowledge, no rapid method of analysis of up to 132 insecticides belonging to 29 chemical classes in honey samples, using QuEChERS/LC–MS/MS has been described in the literature. Works available in the literature describe methods that make it possible to determine individual groups of insecticides: benzoylurea (Wang et al. 2016), neonicotinoids (Gbylik-Sikorska et al. 2015; Jovanov et al. 2013, 2014), organochlorines (Zacharis et al. 2012), pyrethroids (Li et al. 2013), organophosphorus (Naggar et al. 2015; Blasco et al. 2004), or several groups of insecticides, but never so many. For instance, Paradis et al. (2014) reported the simultaneous determination of 22 insecticides of three chemical classes in honey. Shendy et al. (2016) developed a method for determination of 200 pesticides including 90 insecticides of ten chemical classes in honey. In another study, Kasiotis et al. (2014) developed a method to investigate the occurrence of 115 pesticides including 43 insecticides of seven chemical classes in honey, bee pollen and honeybees.
The goal of this work was to modify the QuEChERS technique to develop a rapid, sensitive, and selective method for determination of 132 insecticide residues belonging to 29 different chemical classes in honey samples using liquid chromatography with MS/MS detection. The proposed and validated procedure was used to determine insecticide residues in real samples of honey.
Materials and Methods
Chemicals, Reagents, and Insecticide Standards
Formic acid, LC–MS grade acetonitrile and methanol were purchased from Merck (Darmstadt, Germany). The water (LC grade 18 MΩ cm) was obtained from a MilliQ water purification system (Millipore Ltd., Bedford, MA, USA). Chitosan was obtained from Sigma-Aldrich (Steinheim, Germany), silica gel and aluminum oxide from Merck (Darmstadt, Germany). Florisil, octadecylsilane (C18), and octasilane (C8) were purchased from J. T. Baker (Deventer, Holand). QuEChERS kits with buffering citrate salts were purchased from Agilent Technologies (Santa Clara, USA).
Insecticide standards were obtained from Dr. Ehrenstorfer Laboratory (Germany). All the standards were of high purity grade (> 93.0%). An overview of the physicochemical and toxicological properties of the studied insecticides is given in Table 1.
Stock standard solutions (at 1000 μg mL−1) of individual insecticides were prepared by dissolving an accurately weighed amount of reference standard in methanol. Working standard solutions for plotting calibration curves were prepared by serial dilution of the stock solution in methanol. The working standard solutions were used for preparation of honey matrix-matched standards within the concentration range of 0.001–1.0 μg mL−1. All stock and working standard solutions were protected from direct light and stored in dark glass bottles in a freezer at about − 4 °C until analysis.
Sampling and Sample Preparation
Multiflower honey samples were provided by ecological producers of honey from the Podlasie region of Poland. Honey was characterized by golden color; it was derived from nectar collected by bees from different flowers in the spring and summer period, such as raspberry or cornflower. Samples were tested in order to check whether they were free from pesticide residues. The samples were stored in a refrigerator (4 °C) until analysis.
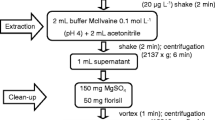
A honey sample (2, 5, or 10 g) was weighed into a 50-ml PTFE tube and dissolved in 10 mL deionized water by shaking for 1 min. Acetonitrile acidified with 0.1, 0.5, or 1% formic acid (10 mL), 4 g anhydrous magnesium sulfate, 1 g sodium chloride, 1 g trisodium citrate dehydrate, and 0.5 g disodium hydrogen citrate sesquihydrate were added and shaken vigorously for 1 min. Then, the samples were centrifuged at 4500 rpm for 5 min. For this purpose, 6 mL of the upper clear solution was transferred into a 15-mL polyethylene tube containing various sorbents or sorbent mixtures: aluminum oxide, chitosan, silica gel, C18, C8, Florisil, chitosan/aluminum oxide/C8, chitosan/aluminum oxide/C18, chitosan/Florisil, or PSA/C18/MgSO4. The extract was mixed, vigorously with the desiccant sorbent for 1 min and centrifuged at 4500 rpm for 10 min. Figure 1 shows the final parameters of the method.

Scheme of honey sample preparation
LC–MS/MS Conditions
Chromatographic analyses were performed using an Eksigent Ultra LC-100 liquid chromatography system (Eksigent Technologies, Dublin, CA, USA). A KINETEX XB analytical column (1.7 μm, 2.1 × 50 mm) from Phenomenex (Torrance, USA) was used for separation, and the column temperature was kept at 40 °C. The volume injected into the LC–MS/MS system was 10 μL. The mobile phase consisted of (A) water and (B) methanol, both containing 5 mmol L−1 ammonium formate and 0.2% formic acid. The elution gradient was as follows: 0–0.5 min (A: 95%, B: 5%), 5–7.5 min (A: 5%, B: 95%), 8–10 min (A: 95%, B: 5%).
Mass spectrometry analyses were conducted using a 6500 QTRAP system (AB Sciex Instruments, Foster City, CA) in multiple reaction monitoring mode. Instrumental parameters for MS/MS analysis were as follows: nitrogen (N2), employed as the nebulizer gas (GS1), auxiliary gas (GS2), and curtain gas (CUR), at a pressure of 60, 70, and 35 psi, respectively; ion spray voltage, 4500 V; temperature, 450 °C. The total chromatographic runtime was 25 min. The system’s operation, data acquisition, and analysis are controlled and processed by Analyst software version 1.6.2 (AB Sciex Instruments, Foster City, CA).
The optimal mass spectrometric parameters for each compound, including collision energy (CE), collision cell exit potential (CXP), declustering potential (DP), entrance potential (EP), and retention times are summarized in Table S1.
Validation Study
Validation datasets were gathered according to SANTE/11945/2015 Document (2016). The parameters, including matrix effect, selectivity, linearity and working range, limit of detection (LOD), recovery (accuracy) and repeatability (precision) were evaluated.
Results and Discussion
Sample preparation is one of the most important steps in trace pesticide analysis, with a direct and important influence on both the quantification and detection limits achieved. The extraction efficiency strongly depends on the organic solvents used, the nature of the sample, and the chemical properties of the pesticide residues (Cunha and Fernandes 2011). In order to obtain the optimal QuEChERS conditions, various parameters affecting the extraction performance of the method, such as amount of sample, extraction solvent’s type and volume, mixing time, and type of cleanup sorbents, were thoroughly investigated. However, only the most important experiments have been presented and described. Additional experiments were attached to Tables 2 and 3 in the supplementary materials.
Optimization of Extraction Conditions
According to available literature data (Kiljanek et al. 2016; Wiest et al. 2011) and to our best knowledge (Kaczyński et al. 2017), acetonitrile appears to be the most common choice due to its ability to extract analytes with different physicochemical properties, such as their molecular weight, boiling point, or polarity. Since certain compounds are sensitive to pH, formic acid is often added in order to obtain a constant pH value of approx. 5. This makes it possible to obtain satisfactory recoveries for acid-sensitive chemical compounds without degrading the base-sensitive compounds. In this study, 0.1% formic acid-acetonitrile, 0.5% formic acid-acetonitrile, and 1% formic acid-acetonitrile were examined to compare their recoveries (Fig. 2a, procedures P1–P3). A lower concentration of formic acid (below 1%) was adverse to the recovery of some compounds (e.g., cadusafos, fenoxycarb, parathion, quinalphos, or tetramethrin). The results indicated in Table S2 show that better recoveries were obtained for all target analytes when the concentration of formic acid in acetonitrile was 1%. In the case of 1% formic acid in acetonitrile, 113 of 132 compounds obtained satisfactory results. In turn, 0.1 and 0.5% formic acid-acetonitrile gave recoveries within the range of 70–120% for the 100 and 106 compounds, respectively.

Effect of concentration of acidifying agents (a), different amounts of sample (b) on extraction efficiency (expressed as recovery) of 132 insecticides at 0.01 mg kg−1
Sample weight was the next parameter tested. Experiments were carried out with the usage of samples weighing 2, 5, and 10 g (Fig. 2b, procedures P3–P5). Satisfactory parameters were obtained for a 5 g sample (for 113 compounds). All recoveries and RSD values have been summarized in Supplementary data - Table S2.
Optimization of Cleanup Conditions
Six dispersive solid-phase extraction (d-SPE) sorbents (chitosan, C8, C18, silica gel, aluminum oxide, Florisil) were chosen to be tested for the ability to remove interfering components and/or fats/lipids from honey samples (Fig. 3a, procedures P6–P15). An analytical portion of 200 mg was used for each sorbent.

Effect of different cleanup sorbents on recovery (a) and matrix effect (b) of 132 insecticides at 0.01 mg kg−1 (procedures P6–P15)
Sorbent C18 is a sorbent based on the reversed phase because of its extreme retentive nature with regard to nonpolar compounds. It is commonly used to remove interfering compounds from various matrices, including honey (Liu et al. 2016). Use of this sorbent at the cleanup stage yielded recoveries within the range of 70–120% for 77 out of 132 tested compounds (Fig. 3a, procedure P6). For 26 insecticides, the recoveries obtained were below 70%, and for 19 compounds, above 120%. Using C8 gave recoveries within the range of 70–120% only for 88 compounds (Fig. 3a, procedure P7). Similar results were obtained for silica gel, except that more compounds (38) had recoveries below 70% and only 7 above 120% (Fig. 3a, procedure P8). Chitosan, a linear polysaccharide consisting of a deacetylated derivative of chitin has recently been successfully applied in pesticide residue analysis (Arias et al. 2014). This material was successfully applied by Arias et al. (2014) to dispersive-SPE cleanup of soil and rice extracts. Use of chitosan (200 mg) gave recoveries in the range of 70–120% for 101 compounds (Fig. 3a, procedure P9). Florisil is a popular sorbent that improved sample cleanup due to interaction of sugars with the polar surface of this sorbent (Kujawski and Namiesnik 2008). Łozowicka et al. (2017) has also reported that Florisil is a universal sorbent for commodities with a high content of pigments, acids, sugars, and organic ingredients. In our study, use of this sorbent gave recoveries within the range of 70–120% for 106 compounds (Fig. 3a, procedure P10). When aluminum oxide was used, satisfactory recoveries were achieved for 99 compounds (Fig. 3a, procedure P11).
Our results suggest that chitosan, Florisil, C8, and aluminum oxide were effective sorbents for honey (Fig. 3a, procedures P7, P9, P10, and P11). Therefore, various double and triple combinations of these sorbents were tested. The best recoveries were achieved using chitosan/aluminum oxide/C8 (Fig. 3a, procedure P12). Use of this combination gave recoveries within the range of 70–120% for 116 out of 132 tested compounds. Recoveries below 70% were obtained for 12 analytes, and above 120% for 5 compounds. All recoveries and RSD values have been summarized in Supplementary data - Table S2.
Matrix effects (MEs) are a well-known problem in LC-MS analysis and need to be evaluated together with recoveries. Evaluation of the matrix effect was performed by comparing the areas of analytical solution prepared in solvent and in matrix extracts.
ME (%) was evaluated as follows: [(slope of calibration curve in matrix/slope of calibration curve in solvent) − 1] × 100. The matrix effect can be classified into the following categories: (1) negative - high signal suppression (ME > − 50%) and moderate signal suppression (− 50% < ME > − 20); (2) no matrix effect (− 20 < ME > 20%); (3) positive - moderate signal enhancement (20 < ME > 50%) and high signal enhancement (ME > 50%). Our results also suggest that the cleanup step based on the combination of chitosan/aluminum oxicde/C8 also proved to be the best, ensuring signal enhancement and signal suppression for six and eight compounds, respectively. No matrix effect was observed for as many as 119 analytes (Fig. 3b, procedure P12). Usage of chitosan/Florisil and chitosan/aluminum oxicde/C18 during cleanup yielded satisfactory recoveries for almost all of the target compounds in honey. Unfortunately, strong matrix effects were observed (enhancement or suppression of more than ± 50%) for most tested compounds. Thus, the combination of chitosan/aluminum oxicde/C8 was selected as the sorbent for the dispersive SPE cleanup procedure. The % ME values obtained for each insecticide are reported in Supplementary data - Table S3.
There have only been a few studies so far that have described the relationship between the matrix effect and the chemical group. Hajslova and Zrostlikova (2003) reported that the positive matrix effects are stronger for pesticide molecules with specific function groups, i.e., organophosphates, carbamates, imidazoles, and benzimidazoles. According to Anagnostopoulos and Miliadis (2013), who have developed a method enabling determination of 32 different chemical groups of pesticides in samples of olive oil and olives, a considerable impact of the matrix effect was observed for most analytes from such chemical classes as carbamates, triazoles, or neonicotinoids, whereas no significant matrix effect was observed for organophosphate insecticides. In turn, Giacinti et al. (2016) have observed a negative matrix effect for chlorpyrifos of the organophosphate group, boscalid of anilides, and pirimicarb of the carbamate group in their studies on determination of pesticide residues in apple skin samples.
In our study, we examined the four most popular chemical groups of insecticides, namely, organophoshorus, carbamates, pyrethroids, and neonicotinoids. As shown in Fig. 4, signal enhancement was more common than suppression in the case of the pyrethroid and organophosphate insecticide groups. A positive matrix effect was observed for up to 95% of compounds from the group of organophosphate insecticides and, respectively, 43% of compounds from the pyrethroid group. In the case of neonicotinoids, 37% of determined compounds showed a negative matrix effect. On the other hand, in the case of carbamates, just 8% of compounds showed signal enhancement or suppression (92% of compounds did not show any matrix effect).

Distribution of the various chemical groups of analytes according to matrix effect (NEO neonicotinoids, ORG organophoshorus, CAR carbamates, PYR pyrethroids)
Method Validation
Method validation of the newly proposed residue analysis method for honey was conducted. Several validation parameters, including matrix effect, selectivity, linearity and working range, limit of detection (LOD), limit of quantification (LOQ), recovery (accuracy), and repeatability (precision), were determined and are summarized in Table S1, Table S2, and Table S3. Good linearity results with regression coefficients of more than 0.99 were obtained for all the pesticides. The recovery and precision of the method for the 132 targeted insecticides were evaluated by carrying out five consecutive extractions (n = 5) of honey spiked at three concentration levels (0.001, 0.010, and 0.100 mg kg−1). The results were calculated using matrix-matched calibration standards by means of the external calibration method. The relative standard deviations (RSDs) for the three spiked levels were lower than 20%. Full validation data for each insecticide is presented in the supplementary data - Table S4.
Conclusions
In this study, a modified QuEChERS method was successfully employed for efficient extraction of 132 insecticides from honey samples. The effectiveness of the cleanup step was tested for various sorbents. Cleanup step with using the chitosan/aluminum oxide/C8 mixture of sorbents turned out to be the best, in term of both the recoveries obtained and matrix effect. Very good analytical results were obtained, including recovery, precision, limit of quantification, and uncertainty. Mean recovery values were within the range of 70–120%. In conclusion, the present procedure proved to be a useful tool for simultaneous determination of insecticide residues in honey.
References
Anagnostopoulos C, Miliadis GE (2013) Development and validation of an easy multiresidue method for the determination of multiclass pesticide residues using GC-MS/MS and LC-MS/MS in olive oil and olives. Talanta 112:1–10. https://doi.org/10.1016/j.talanta.2013.03.051
Arias JLO, Rombaldi C, Caldas SS, Primel EG (2014) Alternative sorbents for the dispersive solid-phase extraction step in quick, easy, cheap, effective, rugged and safe method for extraction of pesticides from rice paddy soils with determination by liquid chromatography tandem mass spectrometry. J Chromatogr A 1360:66–75. https://doi.org/10.1016/j.chroma.2014.07.082
Barganska Z, Slebioda M, Namiesnik J (2013) Pesticide residues levels in honey from apiaries located of northern Poland. Food Control 31:196–201. https://doi.org/10.1016/j.foodcont.2012.09.049
Biever RC, Hoberg JR, Jacobson B, Dionne E, Sulaiman M, McCahon P (2003) ICON rice seed treatment toxicity to crayfish (Procambarus clarkii) in experimental rice paddies. Environ Toxicol Chem 22(1):167–174
Blasco C, Lino CM, Picó Y, Pena A, Font G, Silveira MIN (2004) Determination of organochlorine pesticide residues in honey from the central zone of Portugal and the Valencian community of Spain. J Chromatogr A 1049:155–160. https://doi.org/10.1016/j.chroma.2004.07.049
Chudzinska M, Debska A, Baralkiewicz D (2012) Method validation for determination of 13 elements in honey samples by ICP-MS. Accred Qual Assur 17:65–73. https://doi.org/10.1007/s00769-011-0812-z
Cunha SC, Fernandes JO (2011) Multipesticide residue analysis in maize combining acetonitrile-based extraction with dispersive liquid-liquid microextraction followed by gas chromatography–mass spectrometry. J Chromatogr A 1218:7748–7757. https://doi.org/10.1016/j.chroma.2011.08.066
Ferreira ICFR, Aires E, Barreira JCM, Estevinho LM (2009) Antioxidant activity of Portuguese honey samples: different contributions of the entire honey and phenolic extract. Food Chem 114(4):1438–1443. https://doi.org/10.1016/j.foodchem.2008.11.028
Gbylik-Sikorska M, Sniegocki T, Posyniak A (2015) Determination of neonicotinoid insecticides and their metabolites in honey bee and honey by liquid chromatography tandem mass spectrometry. J Chromatogr B 990:132–140. https://doi.org/10.1016/j.jchromb.2015.03.016
Giacinti G, Raynaud C, Capblancq S, Simon V (2016) Matrix-matching as an improvement strategy for the detection of pesticide residues. J Food Sci 81:1342–1350. https://doi.org/10.1111/1750-3841.13296
Hajslova J, Zrostlikova J (2003) Matrix effects in (ultra)trace analysis of pesticide residues in food and biotic matrices. J Chromatogr A 1(2):181–197. https://doi.org/10.1016/S0021-9673(03)00539-9
Jovanov P, Guzsvány V, Franko M, Lazić S, Sakač M, Šarić B, Banjac V (2013) Multi-residue method for determination of selected neonicotinoid insecticides in honey using optimized dispersive liquid-liquid microextraction combined with liquid chromatography-tandem mass spectrometry. Talanta 111:125–133. https://doi.org/10.1016/j.talanta.2013.02.059
Jovanov P, Guzsvány V, Franko M, Lazić S, Sakač M, Milovanović I, Nedeljković N (2014) Development of multiresidue DLLME and QuEChERS based LC–MS/MS method for determination of selected neonicotinoid insecticides in honey liqueur. Food Res Int 55:11–19. https://doi.org/10.1016/j.foodres.2013.10.031
Kaczyński P, Hrynko I, Łozowicka B (2017) Evolution of novel sorbents for effective clean-up of honeybee matrix in highly toxic insecticide LC/MS/MS analysis. Ecotoxicol Environ Saf 139:124–131. https://doi.org/10.1016/j.ecoenv.2017.01.033
Kasiotis KM, Anagnostopoulos C, Anastasiadou P, Machera K (2014) Pesticide residues in honeybees, honey and bee pollen by LC–MS/MS screening: reported death incidents in honeybees. Sci Total Environ 485:633–642. https://doi.org/10.1016/j.scitotenv.2014.03.042
Kiljanek T, Niewiadowska A, Semeniuk S, Gaweł M, Borzęcka M, Posyniak A (2016) Multi-residue method for the determination of pesticides and pesticide metabolites in honeybees by liquid and gas chromatography coupled with tandem mass spectrometry—honeybee poisoning incidents. J Chromatogr A 1435:100–114. https://doi.org/10.1016/j.chroma.2016.01.045
Kujawski MW, Namiesnik J (2008) Challenges in preparing honey samples for chromatographic determination of contaminants and trace residues. Trends Anal Chem 27:785–793. https://doi.org/10.1016/j.trac.2008.07.004
Li M, Zhang J, Li Y, Peng B, Zhou W, Gao H (2013) Ionic liquid-linked dual magnetic microextraction: a novel and facile procedure for the determination of pyrethroids in honey samples. Talanta 107:81–87. https://doi.org/10.1016/j.talanta.2012.12.056
Liu H, Zhang M, Guo Y, Qiu H (2016) Solid-phase extraction of flavonoids in honey samples using carbamate-embedded triacontyl-modified silica sorbent. Food Chem 204:56–61. https://doi.org/10.1016/j.foodchem.2016.02.102
Łozowicka B (2013) The development, validation and application of a GC-dual detector (NPD-ECD) multi-pesticide residue method for monitoring bee poisoning incidents. Ecotoxicol Environ Saf 97:210–222. https://doi.org/10.1016/j.ecoenv.2013.07.010
Łozowicka B, Mojsak P, Kaczyński P, Konecki R, Borusiewicz A (2017) The fate of spirotetramat and dissipation metabolites in Apiaceae and Brassicaceae leaf-root and soil system under greenhouse conditions estimated by modified QuEChERS/LC–MS/MS. Sci Total Environ 603–604:178–184. https://doi.org/10.1016/j.scitotenv.2017.06.046
Naggar YA, Codling G, Vogt A, Naiem E, Mona M, Seif A, Giesy JP (2015) Organophosphorus insecticides in honey, pollen and bees (Apis mellifera L.) and their potential hazard to bee colonies in Egypt. Ecotoxicol Environ Saf 114:1–8. https://doi.org/10.1016/j.ecoenv.2014.12.039
Orso D, Floriano L, Ribeiro LC, Bandeira NM, Prestes OD, Zanella R (2016) Simultaneous determination of multiclass pesticides and antibiotics in honey samples based on ultra-high performance liquid chromatography-tandem mass spectrometry. Food Anal Methods 9:1638–1653. https://doi.org/10.1007/s12161-015-0339-8
Paradis D, Bérail G, Bonmatin JM, Belzunces LP (2014) Sensitive analytical methods for 22 relevant insecticides of 3 chemical families in honey by GC–MS/MS and LC–MS/MS. Anal Bioanal Chem 406:621–633. https://doi.org/10.1007/s00216-013-7483-z
Rial-Otero R, Gaspar EM, Moura I, Capelo JL (2007) Chromatographic-based methods for pesticide determination in honey: an overview. Talanta 71:503–514. https://doi.org/10.1016/j.talanta.2006.05.033
Sekhar PR, Savithri Y, Kishore S, Doss PJ, Rao KJ (2009) Effect of cypermethrin on total carbohydrates, glycogen, pyruvate and lactic acid contents in liver and kidney tissues of albino rat. Asian J Environ Sci 4(1):24–28
Shendy AH, Al-Ghobashy MA, Mohammed MN, Gad Alla SA, Lofty HM (2016) Simultaneous determination of 200 pesticide residues in honey using gas chromatography-tandem mass spectrometry in conjunction with streamlined quantification approach. J Chromatogr A 1427:142–160. https://doi.org/10.1016/j.chroma.2015.11.068
Tette PAS, Guidi LR, de Abreu Gloria MB, Fernandes C (2016a) Pesticides in honey: a review on chromatographic analytical methods. Talanta 149:124–141. https://doi.org/10.1016/j.talanta.2015.11.045
Tette PAS, da Silva Oliveira FA, Pereira EN, Silva G, de Abreu Glória MB, Fernandes C (2016b) Multiclass method for pesticides quantification in honey by means of modified QuEChERS and UHPLC-MS/MS. Food Chem 211:130–139. https://doi.org/10.1016/j.foodchem.2016.05.036
Wang H, Hu L, Li W, Lu R, Zhang S, Zhou W, Gao H (2016) A rapid and simple pretreatment method for benzoylurea insecticides in honey samples using in-syringe dispersive liquid–liquid microextraction based on the direct solidification of ionic liquids. J Chromatogr A 1471:60–67. https://doi.org/10.1016/j.chroma.2016.10.027
Wiest L, Buleté A, Giroud B, Frattaa C, Amic S, Lambert O, Pouliquen H, Arnaudguilhem C (2011) Multi-residue analysis of 80 environmental contaminants in honeys, honeybees and pollens by one extraction procedure followed by liquid and gas chromatography coupled with mass spectrometric detection. J Chromatogr A 1218:5743–5756. https://doi.org/10.1016/j.chroma.2011.06.079
Zacharis CK, Rotsias I, Zachariadis PG, Zotos A (2012) Dispersive liquid-liquid microextraction for the determination of organochlorine pesticides residues in honey by gas chromatography-electron capture and ion trap mass spectrometric detection. Food Chem 134(3):1665–1672. https://doi.org/10.1016/j.foodchem.2012.03.073
Zheng S, Chen B, Qiu X, Chen M, Ma Z, Yu X (2016) Distribution and risk assessment of 82 pesticides in Jiulong River and estuary in South China. Chemosphere 144:1177–1192. https://doi.org/10.1016/820j.chemosphere.2015.09.050
Zhou J, Yao L, Li Y, Chen L, Wu L, Zhao J (2014) Floral classification of honey using liquid chromatography–diode array detection–tandem mass spectrometry and chemometric analysis. Food Chem 145:9. https://doi.org/10.1016/j.foodchem.2013.08
Funding
This study has been performed consistently with statutory topics (grant number SB-01 “Development and implementation of novel methods for the identification of residues of plant protection products in plant, biological and environmental materials” funded by the Ministry of Science and Higher Education, Poland.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Izabela Hrynko declares that she has no conflict of interest. Bożena Łozowicka declares that she has no conflict of interest. Piotr Kaczyński declares that he has no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Not applicable.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hrynko, I., Łozowicka, B. & Kaczyński, P. Liquid Chromatographic MS/MS Analysis of a Large Group of Insecticides in Honey by Modified QuEChERS. Food Anal. Methods 11, 2307–2319 (2018). https://doi.org/10.1007/s12161-018-1208-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-018-1208-z