
Abstract
The matricellular protein connective tissue growth factor (CCN2) is considered a faithful marker of fibroblast activation in wound healing and in fibrosis. CCN2 is induced during activation of hepatic stellate cells (HSC). Here, we investigate the molecular basis of CCN2 gene expression in HSC. Fluoroscence activated cell sorting was used to investigate CCN2 expression in HSC in vivo in mice treated with CCl4. CCN2 and TGF-β mRNA expression were assessed by polymerase chain reaction as a function of culture-induced activation of HSC. CCN2 promoter/reporter constructs were used to map cis-acting elements required for basal and TGFβ-induced CCN2 promoter activity. Real-time polymerase chain reaction analysis was used to further clarify signaling pathways required for CCN2 expression in HSC. CCl4 administration in vivo increased CCN2 production by HSC. In vitro, expression of CCN2 and TGF-β mRNA were concommitantly increased in mouse HSC between days 0 and 14 of culture. TGFβ-induced CCN2 promoter activity required the Smad and Ets-1 elements in the CCN2 promoter and was reduced by TGFβ type I receptor (ALK4/5/7) inhibition. CCN2 overexpression in activated HSC was ALK4/5/7-dependent. As CCN2 overexpression is a faithful marker of fibrogenesis, our data are consistent with the notion that signaling through TGFβ type I receptors such as ALK5 contributes to the activation of HSC and hence ALK4/5/7 inhibition would be expected to be an appropriate treatment for liver fibrosis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Hepatic stellate cells (HSC) are the primary target of fibrogenic stimuli in the diseased liver. During fibrogenesis, HSC transition from a resting, vitamin A-rich phenotype to a myofibroblastic phenotype characterized by loss of vitamin A, expression of α-smooth muscle actin, and extracellular matrix production (Gressner and Bachem 1994; Pinzani 1995). Connective tissue growth factor (CCN2), a cysteine-rich matricellular protein, regulates cell adhesion, migration, proliferation, survival, and differentiation via integrin- and heparan sulfate proteoglycan-mediated adhesive signaling (Brigstock 1999; Leask and Abraham 2006). CCN2 has fibrogenic properties in vitro and is characteristically over-expressed in a wide variety of fibrotic lesions, correlating with disease severity (Blom et al. 2002; Dziadzio et al. 2005). Consistent with these observations, CCN2 production is enhanced during the progressive activation of primary HSC in vitro. Moreover, CCN2 is further induced in HSC by the potent pro-fibrotic cytokine TGF-β1 (Paradis et al. 1999; Williams 2000).
The molecular basis underlying CCN2 expression in HSC is unknown. In other cell types, CCN2 expression is controlled primarily at the transcriptional level; therefore elements in its promoter are likely to contribute to its expression in HSC (Blom et al. 2002). Previously, we have analysed the regions in the CCN2 promoter required for its induction in response to TGFβ and endothelin-1 in normal fibroblasts and mesangial cells, and its constitutively elevated activity in fibrotic scleroderma fibroblasts and pancreatic cancer cells (Abraham et al. 2000; Holmes et al. 2001, 2003; Chen et al. 2006; Pickles and Leask 2007). CCN2 induction in response to TGFβ in unactivated fibroblasts and mesangial cells requires Smad and Ets response elements in the CCN2 promoter [Holmes et al. 2001; Chen et al. 2002; Leask et al. 2003; Van Beek et al. 2006]. Smads generally regulate TGFβ signaling, and Smad3 in particular is required for TGFβ to induce CCN2 protein in fibroblasts (Holmes et al. 2001). The Ets family of transcription factors are key regulators of differentiation, hormone responses and tumorigenesis, and Ets-1 in particular is required for TGFβ-induced CCN2 protein expression (Van Beek et al. 2006). Intriguingly, CCN2 overexpression in scleroderma cells is independent of TGFβ signaling via Smads and ALK5, but is dependent on endothelin-1 by the endothelin A and B receptors (Holmes et al. 2001; Chen et al. 2006; Shi-wen et al. 2007). Moreover, CCN2 overexpression in pancreatic cancer cells is independent of both TGFβ and endothelin-1, but is dependent on activated ras/MEK/ERK (Pickles and Leask 2007). Thus although TGFβ can mediate the induction of CCN2 in normal mesenchymal cells, overexpression of CCN2 in pathological states has been shown thus far to be independent of TGFβ.
Although CCN2 is induced by TGFβ and autocrine TGFβ signaling is associated with HSC activation (Paradis et al. 1999; Williams 2000), it is unclear whether TGFβ signaling mediates the elevated CCN2 expression that occurs during HSC activation. This issue is of particular relevance given that the overexpression of CCN2 in other activated cells is TGFβ independent. To address this question, it is necessary to first identify the mechanism underlying TGFβ-induced CCN2 expression in HSC and then address whether this mechanism is of relevance to the overexpression of CCN2 is activated HSC. The studies reported here address these outstanding issues and provide valuable insights into the fundamentl molecular mechanism underlying HSC activation, and hence into how to control hepatic fibrogenesis.
Materials and methods
CCN2 production in activated mouse HSC in vivo
All animal procedures were approved by Institutional Animal Care and Use Committee of The Research Institute at Nationwide Children’s Hospital, Columbus OH. Male 5-week old Balb/c mice received daily i.m. injections (1 ml/kg) of olive oil or a 1:1 ratio of carbon tetrachloride (CCl4):olive oil for 1 week (Paradis et al. 1999). Mice (n = 3 per group) were sacrificed 2 days later and HSC were isolated as described below. Freshly isolated HSC from CCl4- or oil-treated mice were individually pooled, washed twice in ice-cold PBS, and resuspended in binding buffer (10 mM HEPES, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2). The samples were incubated at 4°C overnight with a 1:1000 dilution of polyclonal CCN2 antiserum raised in rabbits against full-length recombinant CCN2. The samples were incubated at 4°C overnight with a 1:1000 dilution of polyclonal CCN2 antiserum raised in rabbits against full-length recombinant CCN2 (Ball et al. 2003). This antibody, termed “38B”, has been previously used for immunoprecipitation and Western blot and shows absolute specificity for CCN2, with no cross-reactivity with other CCN proteins (Ball et al. 2003 and data not shown). This antibody was selected for cell sorting based on its superior performance in the detection of CCN2-stably transfected Chinese Hamster Ovary cells versus parental cells (Ball et al. 2003), as compared to either non-immune serum or six other CCN2 antibodies produced in the investigators’ laboratory (D.R.B., unpublished observations). After subsequent incubation of the HSC with FITC-conjugated anti-rabbit IgG secondary antibody (1:1000; Invitrogen), the cells were analysed on a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). 104 cells were tested in triplicate per sample and the data were analysed with FlowJo software (Tree Star Inc., USA).
Isolation and culture of HSC
Mouse livers were perfused in situ with sequential 37°C treatments of heparin (100 U), 70 ml HBSS (without Ca2+/Mg2+), 0.5 mg pronase in 40 ml DMEM/F12, and 5 mg collagenase IV in 70 ml DMEM/F12. The livers were then removed, cut into pieces and agitated in 20 ml DMEM containing 0.2 mg DNAse at 37°C for 10 min. The resulting cell suspension was filtered through a metal sieve and HSC were isolated based on their buoyancy using an Optiprep gradient in which the cells were collected in the 11% phase (Iredale 1992). Cell purity (>95%) was determined using phase contrast microscopy and UV excited fluorescence microscopy. Oil-red staining was used to confirm the presence of intracellular lipid droplets. Cell viability (>95%) was determined by trypan blue exclusion For the oil or CCl4-treated animals, HSC were isolated from all 3 animals in each group and pooled for FACS analysis. For in vitro culture experiments, HSC isolated from ten untreated mice were resuspended and grown in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Grand Island, NY, USA) supplemented with 10% fetal calf serum, 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C. Cells used were activated by 14 days of culturing and were placed (3 × 105 cells/well) in 6-well tissue culture plates (Falcon; Becton Dickinson, Franklin Lakes, NJ, USA) for further analysis.
Reverse-transcriptase polymerase chain reaction
Total RNA from cultured mouse HSC was extracted at various points using a TRIzol extraction kit (Gibco/BRL) according to the manufacturer’s directions. First-strand cDNA was synthesized using 2 μg of RNA in 20 μl of reaction buffer by reverse transcription using SuperScriptTM II RNAse H- Reverse (Invitrogen) with oligo-dT(18)-primers. Complementary DNA was amplified in a 50 μl total volume reaction comprising 50 mM KCl, 20 mM Tris-HCl (pH 8.0), 10 mM deoxynucleoside triphosphate (dNTP), 1.5 mM MgCl2, 1 U Taq polymerase, and 10 pmol of the following polymerase chain reaction (PCR) primer sets: CTGF: 5′-GCAGGGATCCATGCTCGCCTCCGTCGCA-3′ and 5′-CCAGCGCCGCGAATCTTACGCCATGTCTCC-3′; TGF-β: 5′-CCGGATCCTGTCCAAACTAAGGCTCGC-3′ and 5′-CCTCTAGACCAGTGACGTCAAAAGACAGCC; and β-actin: 5′′-AGCTTGCTGTATTCCCCTCCATCGTG-3′ and 5′′-AATTCGGATGGCTACGTACATGGCTG-3’′. Amplification products were separated by electrophoresis on 1% agarose gel containing ethidium bromide and quantitated by scanning densitometry. The proportion of specific gene product to β-actin product was used for semiquantitive analysis which was determined using an Alpha Innotech (San Leandro, CA, USA) imagaing system.
Transfections and DNA Constructs
Cells were allowed to grow for 24 h at 37°C. Cells were then transfected with Fugene (Roche, Indianapolis, IN, USA) at a ratio of 3 μl:2 μg DNA per well. Cells were transfected (1.75 μg/well) with plasmids containing a CCN2 promoter containing DNA spanning from nucleotides −805 to +17, or otherwise identical constructs containing point mutations in either Smad or Ets sites, fused to a secreted alkaline phosphatase (SEAP) reporter gene (Abraham et al. 2000; Holmes et al. 2001; Leask et al. 2003; Van Beek et al. 2006). To control for transfection efficiency cells were transfected with 0.25 μg of a cytomegalovirus (CMV) promoter-β-galactosidase (CMV-β-gal) reporter gene (Clontech, Palo Alto, CA, USA) construct. Cells were allowed to grow for 48 h in 37°C. Promoter assays were performed with a Phospha-Light kit (Applied Biosystems, Foster City, CA, USA) according to manufacturer’s protocol to detect SEAP reporter expression and β-galactosidase expression was determined by a Galacto-star kit (Applied Biosystems) according to manufacturer’s protocol. After adjusting the SEAP expression values to reflect differences in transfection efficiencies as determined by the β-galactoside expression values, adjusted data was expressed as average values ± standard deviation of at least three replicates and at least two independent trials. Measurement of SEAP levels were obtained from an LMax II 384 luminometer (Molecular Devices, Sunnyvale, CA, USA) and SoftMax Pro 4.7.1 (Molecular Devices, Sunnyvale CA, USA). Statistical tests were done using one-way analysis of variance (ANOVA) and Tukey’s post-hoc test on GraphPad.
Real time RT-PCR
Assays were performed essentially as previously described (Shi-wen et al. 2006; Kennedy et al. 2007; Pala et al. 2008). Cells were plated onto a six well plate and allowed to grow for 24 h at 37°C. Cells were then treated for 18 h with p38 MAP Kinase inhibitor SB203580, MEK/ERK inhibitor U0126, TGFβ type I receptor (ALK4/5/7) inhibitor SB431542 or the protein kinase C (PKC) inhibitor bisindolmaleimide I (all Calbiochem, La Jolla, CA, USA) at a concentration of 10 μM, except SB203580 which was used at 30 μM. Alternatively, cells were serum-starved for 18 h prior to addition of actinomycin D (15 μg/ml; Sigma) for 1 h. Cells were then treated in the presence or absence of TGFβ to 6 h. RNA was harvested using the Trozol (Invitrogen) and used for Real-Time RT-PCR. 25 ng of RNA was reverse transcribed and amplified using TaqMan Assays on Demand (Applied Biosystems) in a 15 μl reaction containing primers for TaqMan Human CCN2 (Applied Biosystems) and 6-carboxyfluroscein labeled TaqMan MGB probe. Reverse Transcriptase One-Step qPCR Mastermix (Applied Biosystems) was added to samples and the ABI Prism 7900 HT sequence detector (Perkin-Elmer-Cetus, Vaudreuil, QC) was used according to manufacturer’s instructions to detect amplified sequences. Samples were run in triplicate, and expression values were standardized to control values from GAPDH primers using the ΔΔC t method (Livak and Schmittgen 2001). Statistical analysis was done using one way ANOVA and Tukey’s post-hoc test on GraphPad.
Results
CCN2 production in mouse HSC is associated with the activated phenotype
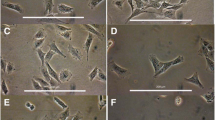
Since CCl4 causes liver injury and HSC activation in vivo, this approach was used to explore CCN2 production by mouse HSC. As shown in Fig. 1A, 92% of the HSC isolated from the livers of CCl4-treated mice were CCN2-positive whereas only 12% of the HSC from oil-treated mice were CCN2-positive. Cultured HSC from non-treated mice, contained cytoplasmic oil droplets on days 1–2 that were visible under phase contrast and when stained with oil red (Fig. 1B). By day 7, the droplets were no longer present and the cells had undergone dramatic phenotypic changes, appearing myofibroblastic and positive for αSMA expression (Fig. 1B and data not shown). Whereas freshly isolated HSC exhibited very low expression of CCN2, the amount of CCN2 mRNA increased as a function of time in culture and was correlated with the autonomous transformation of the cultured cells from a quiecent to activated phenotype (Fig. 1C). Culture-induced HSC activation was also associated with a concommitant increase in TGF-β expression (Fig. 1C). Collectively, these data showed that activation of mouse HSC either in vivo or in vitro is accompanied by elevated CCN2 production.

CCN2 expression in activated mouse HSC. A FACS analysis of CCN2-positive HSC from Balb/c mice receiving oil or CCl4 administration. HSC from each treatment group (n = 3) were isolated and a total of 10,000 cells were counted. The absolute number of CCN2-positive cells is expressed as a percent of the total number counted. B Characterization of cultured mouse HSC. Primary cultures of mouse HSC on day 1 (a, b) or day 2 (c) were poorly spread and contained oil droplets that stained with oil red. By day 7 (d), cells had lost their oil droplets, had numerous cytoplasmic extensions, and were αSMA-positive (not shown). Scale bars: 100 μm. C Reverse-transcriptase PCR analysis of CCN2 or TGF-β mRNA expression in mouse HSC during culture-induced activation. Data from each time point shown in the inset were scanned and the signal intensity was determined relative to that of β-actin. Data shown in A–C were typical of three or more independent analyses
Basal and TGFβ-induced CCN2 expression in HSC depends on transcription
To begin to probe the mechanism underlying the overexpression of CCN2 in 14 day culture-activated HSC, we assessed if autocrine TGFβ signaling could contribute to this process. First, we ascertained whether transcriptional regulation was likely to play a role. To perform this assay, we serum-starved culture-activated HSC for 18 h, and cells were treated in the presence or absence of TGFβ for 6 h. Prior to addition of TGFβ, cells were treated for 1 h in the presence or absence of actinomycin D, which blocks de novo transcription. RNAs were harvested and subjected to real-time PCR analysis to detect CCN2 mRNA. We found that actinomycin D reduced basal CCN2 mRNA expression in HSC (Fig. 2). Moreover, actinomycin D blocked TGFβ-induced CCN2 mRNA expression (Fig. 2). Thus transcriptional regulation contributes to both basal and TGFβ-induced CCN2 expression in HSC.

Transcription is required for basal and TGFβ-induced CCN2 mRNA in HSC. Culture-activated HSC were serum-starved for 18 h prior to addition of actinomycin D (15 μg/ml, Act) for 1 h. Cells were then incubated for 6 h in the presence or absence of TGFβ1 (4 ng/ml; TGF). RNAs were harvested and subjected to real-time PCR analysis with primers recognizing CCN2 or 18S. Relative CCN2 expression was calcutated using the ΔΔC t method. Fold increase relative to control, untreated samples is shown. Assays were performed in triplicate. Standard deviation was less than 10%. Whereas TGFβ increases CCN2 mRNA in HSC, this induction is blocked by actinomycin D (TGF vs. TGF/Act,*p < 0.05). Moreover, basal CCN2 expression is reduced in the presence of actinomycin D (control vs. Act, *p < 0.05)
Basal and TGFβ-induced CCN2 expression in HSC depends on ALK5 and Smad and Ets elements in the CCN2 promoter
To begin to evaluate the hypothesis that TGFβ signaling was responsible for the elevation of CCN2 expression during HSC activation and to begin to determine which promoter elements contributed to CCN2 overexpression in HSC, we first ascertained the elements required for TGFβ-induced CCN2 expression. HSC were transfected with CCN2 promoter-SEAP reporter constructs containing regions spanning between −805 to +17, or otherwise identical constructs in which Smad or Ets-1 sites were mutated (Holmes et al. 2001; Leask et al. 2003; Van Beek et al. 2006; Pickles and Leask 2007). Mutation of either the Smad or the Ets-1 site significantly reduced the ability of the CCN2 promoter to respond to TGFβ (Fig. 3). Similarly, addition of the TGFβ type I receptor (ALK4/5/7) inhibitor SB431542 blocked the ability of TGFβ to induce the CCN2 promoter. Thus, the induction of the CCN2 promoter by TGFβ in HSC required ALK5, and the Smad and Ets-1 sites.

Activity of CCN2 promoter constructs in HSC: elements required for TGFβ response. HSC (14 days in culture) were seeded into 12 well plates and transfected with CCN2 promoter/SEAP reporter constructs, as described in Methods. Experiments were performed in quadruplicate, at least twice. SEAP reporter activity (after adjustment for differences among samples in transfection efficiency as revealed by cotransfected CMV-β-gal reporter construct) was calculated. Fold increase in response to TGFβ for each consctruct and treatment was then calculated. Average fold increase ± standard deviation is shown. Post-transfection, cells were serum-starved for 18 h prior to addition of TGFβ1 (4 ng/ml) for an additional 24 h. When indicated, cells were pre-treated for 45 min with 10 μM ALK4/5/7 inhibitor prior to addition of TGFβ1. Whereas TGFβ induced a promoter/reporter construct contaning nucleotides spanning −805 to +17 CCN promoter (FL), mutation of the Smad or Ets element within this promoter fragment or addition of ALK4/5/7 inhibitor (ALKi) blocked this induction (*p < 0.05)
To further investigate the specific promoter elements involved in CCN2 overexpression in HSC and to probe the involvement of autocrine TGFβ signaling in this process, cells were transfected with CCN2 promoter-SEAP reporter constructs containing the wild-type promoter sequence, or point mutations to either the Smad or Ets-1 sites. Point mutation of either the Smad or Ets-1 sites resulted in a significant decrease in CCN2 promoter activity (Fig. 4). Moreover, ALK5 inhibition also reduced activity of the wild-type CCN2 promoter (Fig. 4). These results suggest that activation of CCN2 expression in HSC is due at least in part to activated TGFβ signaling.

Activity of CCN2 promoter constructs in HSC: elements required for basal promoter activity HSC (14 days in culture) were seeded into 12 well plates and transfected with CCN2 promoter/SEAP reporter constructs, as described in Methods. SEAP reporter activity (after adjustment for differences among samples in transfection efficiency as revealed by cotransfected CMV-β-gal reporter construct) was calculated. Experiments were performed in quadruplicate, at least twice. Relative light units (averages ± standard deviation) are shown. Mutation of the Smad or Ets element or addition of ALK4/5/7 inhibitor also reduced basal CCN2 promoter activity (*p < 0.05)
To confirm these data and to determine which signaling pathways contributed to CCN2 overexpression in HSC, cells were treated with inhibitors to p38 (SB 203580, p38i), MEK/ERK (U0126, ERKi), ALK4/5/7 (ALKi) or PKC (bisindolmaleimide I, PKCi) at concentrations previously demonstrated to selectively block action of the respective targets. Real-time RT-PCR analysis determined that while all inhibitors somewhat reduced CCN2 mRNA expression, inhibition of either ALK5 and PKC resulted in a potent (∼70%) reduction of CCN2 mRNA expression in HSC (Fig. 5). Collectively, these data suggest that overexpression of CCN2 in HSC is due, at least in part, to autocrine TGFβ signaling acting through ALK5 and the Smad and Ets-1 sites in the CCN2 promoter.

The effect of signal transduction inhibitors on CCN2 mRNA expression in HSC. HSC (14 days in culture) were Incubated for 24 h with: DMSO (control); ALK4/5/7 inhibitor (Alki, 10 μM); the MEK inhibitor U0126 (meki, 10 μM); the p38 Inhibitor SB 203580 (p38i, 30 μM) or the PKC inhibitor Bisindolylmaleimide I (PKCi, 10 μM). RNAs were harvested and subjected to real time PCR analysis with primers recognizing CCN2 and 18S. Relative expression (to 18S) was calculated and adjusted to control (DMSO) alone treated values usingthe ΔΔC t method. Average expression values (three replicates) is shown. The standard deviation was less that 5% (*p < 0.05). Blocking the TGFβ type I ALK4/5/7 receptor (Alki) and PKC (PKCi) potently reduces basal CCN2 mRNA expression
Discussion
CCN2 is over-expressed in fibrotic diseases, including those of the liver; indeed, CCN2 expression is a reliable marker of the severity of fibrosis (Blom et al. 2002; Leask and Abraham 2006). CCN2 appears to be a key contributor to the fibrogenic phenotype of HSC in that CCN2 promotes proliferation, adhesion, and collagen production of this cell type through integrins and HSPGs (Gao et al. 2004; Gao and Brigstock 2004). Through its role as a pro-adhesive matricellular molecule, CCN2 can also amplify adhesive signaling emanating from other pro-fibrotic ligands such as fibronectin and TGFβ, and pro-inflammatory ligands such as TNFα (Shi-wen et al. 2006; Chen et al. 2004, 2007). Moreover, CCN2-deficient mouse embryonic fibroblasts show impairment of a myriad of fibrogenic activity, inclusing cell adhesion, matrix contraction, and gene expression (Shi-wen et al. 2006; Kennedy et al. 2007; Pala et al. 2008). Thus CCN2 is a promising marker of fibrosis and also represents an excellent direct anti-fibrotic target (Blom et al. 2002; Leask and Abraham 2006). As such, CCN2 is expressed in activated HSC and is induced by TGFβ in this cell type. However, until this report there was no proposed mechanism for CCN2 induction and overexpression in activated HSC. Moreover, although TGFβ can mediate the induction of CCN2 in normal mesenchymal cells, TGFβ through the Smad/ALK5 pathway does not appear to mediate the overexpression of CCN2 in activated myofibroblasts (Holmes et al. 2001; Chen et al. 2006). As there is currently no sufficient treatment for liver fibrosis, obtaining a greater understanding of the mechanisms underlying HSC activation, such as the mechanism underlying the overexpression of CCN2, is crucial in order to identify novel potential drug targets and to evaluate the possibility of targeting the TGFβ receptor ALK5 to combat fibrosis.
In this report we probe the mechanism underlying the regulation of CCN2 expression in HSC. Basal and TGFβ-induced expression in activated HSC relied at least on part on gene transcription, as basal CCN2 mRNA expression was reduced and TGFβ-induced CCN2 mRNA expression was ablated in the presence of actinomycin D. It has been shown previously in normal fibroblasts and mesangial cells that CCN2 is induced by TGF-β acting through the Smad 3/4 pathway and an Ets element (Leask et al. 2003; Chen et al. 2002; Van Beek et al. 2006). In this report we show that such a mechanism also operates in HSC, in that the increased activity of the CCN2 promoter in response TGFβ was diminished by mutation of the Smad and Ets elements. We extend our data in other cell systems to show that, in HSC, ALK4/5/7 inhibition can block the TGFβ-induction of the CCN2 promoter and mRNA. For the first time, however, we present data indicating that activated TGFβ signaling can contribute to CCN2 overexpression in myofibroblasts by showing that in activated HSC, mutation of the Ets and Smad elements and addition of an ALK4/5/7 inhibitor reduced the activity of the CCN2 promoter in this cell type. Moreover, ALK4/5/7 inhibition reduced CCN2 mRNA expression in this cell type. Collectively, these results support the notion that activated TGFβ signaling may contribute to HSC activation. Conversely, in scleroderma fibroblasts in which CCN2 overexpression is TGFβ/Smad/ALK5-independent but ET-1 dependent and in pancreatic cancer cells CCN2 expression is independent of both TGFβ and ET-1 (Holmes et al. 2001; Chen et al. 2006; Pickles and Leask 2007). Our results indicate that, CCN2 overexpression in HSC can be antagonized by ALK4/5/7 inhibition and support the notion that activated TGFβ signaling via the ALK4/5/7 receptor may play a key role HSC activation. Given that CCN2 overexpression is a faithful marker of the fibrogenic phenotype, ALK4/5/7 inhibition may be an appropriate method of attenuating liver fibrosis.
Abbreviations
- ALK:
-
activin linked kinase
- CMV:
-
cytomegalovirus
- CTGF:
-
connective tissue growth factor
- FACS:
-
fluorescence activated cell sorter
- HSC:
-
Hepatic stellate cells
- HSPG:
-
heparan sulfate containing proteoglycans
- PBS:
-
phosphate buffered saline
- PKC:
-
protein kinase C
- SEAP:
-
secreted enhanced alkaline phosphatase
- TGFβ:
-
Transforming growth factor β
References
Abraham DJ, Shiwen X, Black CM, Sa S, Xu Y, Leask A (2000) Tumor necrosis factor alpha suppresses the induction of connective tissue growth factor by transforming growth factor-beta in normal and scleroderma fibroblasts. J Biol Chem 275:15220–15225. doi:10.1074/jbc.275.20.15220
Ball DK, Moussad EA, Rageh MAE, Kemper SA, Brigstock DR (2003) Establishment of a recombinant expression system for connective tissue growth factor (CTGF) that models CTGF processing in utero. Reproduction 125:271–284. doi:10.1530/rep.0.1250271
Blom IE, Goldschmeding R, Leask A (2002) Gene regulation of connective tissue growth factor: new targets for antifibrotic therapy? Matrix Biol 21:473–482. doi:10.1016/S0945-053X(02)00055-0
Brigstock DR (1999) The connective tissue growth factor/cysteine-rich 61/nephroblastoma overexpressed (CCN) family. Endocr Rev 20:189–206. doi:10.1210/er.20.2.189
Chen Y, Blom IE, Sa S, Goldschmeding R, Abraham DJ, Leask A (2002) CTGF expression in mesangial cells: involvement of SMADs, MAP kinase and PKC. Kidney Int 62:1149–1159
Chen Y, Abraham DJ, Shi-Wen X, Pearson JD, Black CM, Lyons KM et al (2004) CCN2 (connective tissue growth factor) promotes fibroblast adhesion to fibronectin. Mol Biol Cell 15:5635–5646. doi:10.1091/mbc.E04-06-0490
Chen Y, Shi-wen X, Eastwood M, Black CM, Denton CP, Leask A et al (2006) Contribution of activin receptor-like kinase 5 (transforming growth factor beta receptor type I) signaling to the fibrotic phenotype of scleroderma fibroblasts. Arthritis Rheum 54:1309–1316. doi:10.1002/art.21725
Chen CC, Young JL, Monzon RI, Chen N, Todorović V, Lau LF (2007) Cytotoxicity of TNFalpha is regulated by integrin-mediated matrix signaling. EMBO J 26:1257–1267. doi:10.1038/sj.emboj.7601596
Dziadzio M, Usinger W, Leask A, Abraham D, Black CM, Denton D et al (2005) N-terminal CTGF is marker of fibrosis for the connective tissue disease scleroderma. QJM 98:485–492. doi:10.1093/qjmed/hci078
Gao R, Brigstock DR (2004) Connective tissue growth factor (CCN2) induces adhesion of rat activated hepatic stellate cells by binding of its C-terminal domain to integrin alpha(v)beta(3) and heparan sulfate proteoglycan. J Biol Chem 279:8848–8855. doi:10.1074/jbc.M313204200
Gao R, Ball DK, Perbal B, Brigstock DR (2004) Connective tissue growth factor induces c-fos gene activation and cell proliferation through p44/42 MAP kinase in primary rat hepatic stellate cells. J Hepatol 40:431–438. doi:10.1016/j.jhep.2003.11.012
Gressner AM, Bachem MG (1994) Cellular communications and cell-matrix interactions in the pathogenesis of fibroproliferative diseases: liver fibrosis as a paradigm. Ann Biol Clin (Paris) 52:205–226
Holmes A, Abraham DJ, Sa S, Shiwen X, Black CM, Leask A (2001) CTGF and SMADs, maintenance of scleroderma phenotype is independent of SMAD signaling. J Biol Chem 276:10594–10601. doi:10.1074/jbc.M010149200
Holmes A, Abraham DJ, Chen Y, Denton C, Shi-wen X, Black CM et al (2003) Constitutive connective tissue growth factor expression in scleroderma fibroblasts is dependent on Sp1. J Biol Chem 278:41728–41733. doi:10.1074/jbc.M305019200
Iredale JPG (1992) Murphy, G. Hembry, R.M. Friedman S.L. and Arthur, M.J. Human hepatic lipocytes synthesize tissue inhibitor of metalloproteinases-1. Implications for regulation of matrix degradation in liver. J Clin Invest 90:282–287. doi:10.1172/JCI115850
Kennedy L, Liu S, Shi-Wen X, Chen Y, Eastwood M, Carter DE et al (2007) CCN2 is necessary for the function of mouse embryonic fibroblasts. Exp Cell Res 313:952–964. doi:10.1016/j.yexcr.2006.12.006
Leask A, Abraham DJ (2006) All in the CCN family. J Cell Sci 119:4803–4810. doi:10.1242/jcs.03270
Leask A, Holmes A, Black CM, Abraham DJ (2003) Connective tissue growth factor gene regulation. Requirements for its induction by transforming growth factor-beta 2 in fibroblasts. J Biol Chem 278:13008–13015. doi:10.1074/jbc.M210366200
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 25:402–408. doi:10.1006/meth.2001.1262
Pala D, Kapoor M, Woods A (2008) Kennedy, L., Liu, S., Chen, S., Bursell, L., Lyons, K., Carter, D., Beier, F, Leask, A. FAK/src suppresses early chondrogenesis: Central role of CCN2. J Biol Chem 283:9239–9247. doi:10.1074/jbc.M705175200
Paradis V, Dargere D, Vidaud M, De Gouville AC, Huet S, Martinez V et al (1999) Expression of connective tissue growth factor in experimental rat and human liver fibrosis. Hepatology 30:968–976. doi:10.1002/hep.510300425
Pickles M, Leask A (2007) Analysis of the CCN2 promoter in PANC-1 cells: regulation by ras/MEK/ERK. J. Cell Commun. Signal 1:85–90. doi:10.1007/s12079-007-0008-9
Pinzani M (1995) Novel insights into the biology and physiology of the Ito cell. Pharmacol Ther 66:387–412. doi:10.1016/0163-7258(94)00072-B
Shi-wen X, Stanton LA, Kennedy L, Pala D, Chen Y, Howat SL et al (2006) CCN2 is necessary for adhesive responses to transforming growth factor-beta1 in embryonic fibroblasts. J Biol Chem 281:10715–10726. doi:10.1074/jbc.M511343200
Shi-wen X, Renzoni EA, Kennedy L, Howat S, Chen Y, Pearson JD (2007) Bou-Gharios, G, Dashwood, MR, du Bois, RM, Black, CM, Denton, CP, Abraham, DJ, Leask, A. Endogenous endothelin-1 signaling contributes to type I collagen and CCN2 overexpression in fibrotic fibroblasts. Matrix Biol 26:625–632. doi:10.1016/j.matbio.2007.06.003
Van Beek JP, Kennedy L, Rockel JS, Bernier SM (2006) Leask, A. The induction of CCN2 involves Ets-1. Arthritis Res Ther 8:R36. doi:10.1186/ar1890
Williams EJ (2000) Gaca, M.D. Brigstock, D.R. Arthur M.J. and Benyon R.C., Increased expression of connective tissue growth factor in fibrotic human liver and in activated hepatic stellate cells. J Hepatol 32:754–761. doi:10.1016/S0168-8278(00)80244-5
Acknowledgements
This work was supported by grants from the Canadian Institutes of Health Research, the Canadian Foundation for Innovation and the National Institute of Health (AA 016003). A.L is a New Investigator of the Arthritis Society (Scleroderma Society of Ontario) and a recipient of an Early Researcher Award. DP was a NORTH Undergraduate Student and the recipient of an OGSST Graduate Fellowship. We thank C. Lawrencia for technical assistance with data shown in Fig. 1.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Leask, A., Chen, S., Pala, D. et al. Regulation of CCN2 mRNA expression and promoter activity in activated hepatic stellate cells. J. Cell Commun. Signal. 2, 49–56 (2008). https://doi.org/10.1007/s12079-008-0029-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12079-008-0029-z