
Abstract
Heterogenous nuclear ribonucleoproteins (hnRNPs) are a complex and functionally diverse family of RNA binding proteins with multifarious roles. They are involved, directly or indirectly, in alternative splicing, transcriptional and translational regulation, stress granule formation, cell cycle regulation, and axonal transport. It is unsurprising, given their heavy involvement in maintaining functional integrity of the cell, that their dysfunction has neurological implications. However, compared to their more established roles in cancer, the evidence of hnRNP implication in neurological diseases is still in its infancy. This review aims to consolidate the evidences for hnRNP involvement in neurological diseases, with a focus on spinal muscular atrophy (SMA), Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), multiple sclerosis (MS), congenital myasthenic syndrome (CMS), and fragile X-associated tremor/ataxia syndrome (FXTAS). Understanding more about hnRNP involvement in neurological diseases can further elucidate the pathomechanisms involved in these diseases and perhaps guide future therapeutic advances.
Similar content being viewed by others
Introduction
Heterogeneous nuclear ribonucleoproteins (hnRNPs) are a family of functionally diverse RNA bindings proteins (RBPs) [1]. Originally named alphabetically from A1 to U, they range from 34 to 120 kDA [2]. Their high involvement in RNA metabolic processes including pre-mRNA processing, splicing, and nucleocytoplasmic shuttling makes them pivotal in the regulation of gene expression [3]. Having a substantial control over post-transcriptional modifications and translation, it is unsurprising that aberrance in hnRNP function can lead to dire functional consequences. While their role in regulating several cellular processes is established, their role in neurological diseases has not been comprehensively investigated. This review aims to consolidate the existing literature of hnRNP abnormalities in various neurological diseases and spur further research in this area.
Structure of hnRNPs
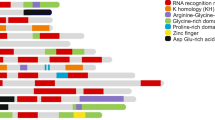
Four evolutionary conserved RNA binding domains (RBD) have been elucidated in hnRNPs. The RNA recognition motif (RRM) is one of the most abundant protein domains in eukaryotes and was first discovered in the U1A protein [4]. It consists of 4 β-sheet (β4β1β3β2) and 2 α-helix (α1α2) domains which fold into a sandwich structure to bind RNAs [5]. Its two consensus sequences which are involved in RNA interaction, RNP1 (Lys/Arg-Gly-Phe/Tyr-Gly/Ala-Phe/Tyr-Val/Ile/Leu-X-Phe/Tyr) and RNP2 (Ile/Val/Leu-Phe/Tyr-Ile/Val/Leu-X-Asn-Leu), are located in β3 and β1 respectively [4]. These motifs, along with distinctive and varied terminal N- and C-sequences, account for the specific affinities of RNA binding [6] (Fig. 1).

RNA binding domains in hnRNPs. RNA-binding domains in hnRNP include RRM (RNA Recognition Motif), KH (K-Homology), and RGG (Arginine-Glycine-Glycine). hnRNPA1 UP1, which spans the first 196 aa at the N-terminus, contains two RRM one in each subdomain of UP1. a RRM contains two α-helices (RRM1: cyan, RRM2: pink), four β-sheets (RRM1: blue, RRM2: purple), and five loops (RRM1: green, RRM2: orange) that order as βαββαβ. b The C-terminal of hnRNPA1 contains another RNA-binding motif known as RGG. The name reflects the abundance of Arg-Gly-Gly tripeptide repeats in the motif. c The KH domain of hnRNPK consists of three α-helices (cyan), three β-sheets (green), and five loops (purple) that fold in the order of βααββα [7,8,9,10,11]
Some hnRNPs contain human K (KH) domains in place of RBMs, named after hnRNP K in which they were first discovered [12]. Different KH domains bind a multitude of molecules; however, they all have a similar structure consisting of three anti-parallel β-strands and three α-helices (βααββα) with surface loops extending from the structure [13], enabling association to RNA molecules [12] (Fig. 1). Some hnRNPs also bind RNA via RGG domains, which are glycine-rich regions interspersed with arginine residues that are integral for RNA binding. The RGG domain also consists of aromatic rings which are responsible for hydrophobic interactions with RNA bases [14]. The glycine residues serve as hinges that allow the proteins to conform to a structure whereby arginine molecules or aromatic rings can come into contact with RNA [14] (Fig. 1).
HnRNPs also possess nuclear localisation sequences and/or nucleocytoplasmic shuttling domains. These ensure hnRNPs are both localized to the nucleus and allow them to be shuttled in order to undertake their cytoplasmic functions [3, 15]. While some hnRNPs can be shuttled in and out of the nucleus, others (hnRNP C and U) localize exclusively to the nucleus [16]. Since most hnRNPs are predominantly nuclear in their steady state and their function is highly dependent on their intracellular location, the integrity of the NLS is of paramount importance [17]. It is noteworthy that NLS defects have been linked to hnRNP instability and abnormal protein distribution [18, 19].
Function of hnRNPs
Gene Regulation
HnRNPs have the ability to both positively and negatively regulate gene expression depending on their binding partner. For instance, hnRNP A binds to the inhibitory subunit of nf-kb alpha, triggering the activation of transcription factor nf-kb [20]. HnRNP K interacts with the promotor of the c-myc gene via the pyrimidine-rich CT region in vivo and in vitro [21, 22] as well as the promotor of eIF4E which both promote transcription [23]. In addition, hnRNP K also binds to the TATA-binding protein and interacts with RNA polymerase II transcription machinery, thus stimulating transcription [24]. Conversely, hnRNP K can also suppress transcription by binding to the zinc finger transcriptional repressor which contains a KRAB-A domain involved in transcriptional repression [25]. Therefore, hnRNPs play key roles in gene regulation and it is likely that hnRNP misregulation may be involved in the pathomechanisms of multiple diseases.
Alternative Splicing
Alternative splicing is a process which leads to great protein diversity and regulates gene expression [26] and the brain is particularly reliant compared to other regions [27, 28]. As a result, the pivotal role of hnRNPs in regulating gene expression and cell metabolism renders the integrity and health of the nervous system highly dependent on their function [29].
Abnormal splicing produces modified or defective proteins which may underlie neurological diseases due to the reliance of the CNS on specific isoforms for vital processes, such as long term potentiation and neurotransmission [30]. HnRNPs are heavily involved in alternative splicing as evidenced by its ability to globally regulate splicing in different cells and tissue types by regulating the availability of its IDR GY-rich motif [31]. Furthermore, iCLIP procedures have revealed 2394 clusters of hnRNP A2/B1 binding sites on 564 genes, including those involved in protein binding, myelination, and neurite projection [32], demonstrating its heavy role in alternative splicing. HnRNP A2/B1 binds to UAGG motifs in a regulatory manner [32], and the glycine-rich domain of hnRNP A1 is involved in splice-site silencing by looping of the RNA between binding elements [33, 34]. HnRNP C is able to crosslink to uridine tracts, regulating alternative splicing [35]. HnRNP F and hnRNP H bind RNA containing G quadruplexes and G tract motifs which result in alternate splicing [29, 36] and hnRNP A binds to splicing factor U2AF [37]. TDP-43, another member of the hnRNP family, is also found to be implicated in many neurodegenerative diseases. Its splicing roles include binding to UG rich regions, non-coding RNAs, 3’ UTRs of mRNAs, and deep intronic binding sites which results in silencing exon inclusion [33]. The critical roles of hnRNPs in mRNA processing and regulating cell metabolism strongly suggest that its aberrance may play a role in neurological diseases and this will be discussed in later sections.
Stress Granule Formation
Stress granules form during periods of cellular stress as a form of energy preservation. The pooling of various RBPs and silenced mRNAs alters the overall levels of functional proteins and are thought to be crucibles of disease [38, 39]. In genetic abnormalities whereby there are expansion repeats, aberrant mRNA can also form toxic aggregates and consequently sequester important factors that regulate gene expression or apoptosis [40, 41]. Accordingly, several RBPs and signaling molecules such as hnRNP A, hnRNP B, TRAF2, RACK1, mTORC1, TDP-43, and FUS have been discovered to be sequestered in stress granules, thereby impairing their usual cell regulatory functions [42,43,44,45,46,47]. Apart from the loss of function due to its sequestration in stress granules, it has been proposed that hnRNPs are also involved in stress granule formation, and that stress granule assembly may be deficient with hnRNP abnormalities. Intrinsically disordered regions (IDR) within hnRNPs were found to be involved in liquid-liquid phase separation, a process which contributes to stress granule assembly [48]. Mutations in IDRs in hnRNP A1 have also been linked to ALS [49], suggesting deficient stress granule formation in ALS. This is an expanding concept and has previously been reviewed in depth [50,51,52].
Cell Cycle Regulation
Often juxtaposed alongside cancer in epidemiological studies, cumulating evidence is beginning to elucidate that beneath the disparate cellular features of cancer and neurodegeneration lies a common underlying pathology of misregulated cell cycle events [53, 54]. For instance, cell death in Alzheimer’s disease (AD) has been postulated to be a result of abnormal cell cycle re-entry which mediates neuronal death [55]. Damaged neurons in the AD brain were also found to possess biomarkers of cell cycle events [56]. Furthermore, persisting genomic instability, possibly due to replication stress and the misregulation of nucleic acid binding proteins, can trigger cellular death [57]. However, despite the roles of hnRNP in cell cycle regulation, much is still unknown about their roles in cell death. For instance, HnRNP A1 is known to be involved in telomere maintenance [58, 59], and its misregulation may result in premature cell death. HnRNP K, a putative cell cycle regulator that interacts with p53 and c-myc and causes cell death and excitotoxicity respectively when misregulated [53, 60], has been disproportionately studied more in cancer and its role in neurodegeneration is still unclear.
Axonal Transport
Due to the unique extensive morphology of neurons and their high dependence on intact intracellular protein transport, axonal defects are often thought to precede neurodegeneration [61,62,63,64]. Neurons also possess intricate and expansive transcriptomic profiles with its axons containing hundreds to thousands of mRNAs, including mRNAs of proteins that regulate gene expression in response to local axonal activity [65]. In addition to the main transport machinery such as dynein, kinesin, and their cargos, RBPs also play an indispensable supportive role in maintaining axonal transport. For instance, hnRNP R co-localizes with beta-actin mRNA in axons and its knockdown impaired axonal growth [66]. Furthermore, hnRNP R interacts with 7SK, a non-coding RNA involved in axon elongation, and reduced levels of hnRNP R depleted 7SK levels and consequently impaired axon growth [67]. HnRNP K was also found to interact with the transcripts of several cytoskeletal genes such as Arp2, tau, and α-internexin-like-neurofilament, all of which are integral for axonogenesis and intracellular transport [68]. HnRNPs are also involved in regulating specific axonal translation. For instance, hnRNP A2/B1 was found to interact with Netrin-1 DCC, which binds a subset of mRNAs involved in cell-cell adhesion and protein targeting, inducing the translation of specific subsets of mRNAs [69, 70]. HnRNP H1, H2, and F are also involved in regulating axonal mRNA regulation [71]. It was found that a combined knockdown reduced levels of axonal Hmgb1 and Nrn1 mRNA levels, and decreased axonal protein synthesis [71]. With the diverse roles of hnRNPs in maintaining neuronal health and the dire consequences when perturbed, it is reasonable to postulate that neurodegeneration can be precipitated by the misregulation of hnRNP in the neuronal parenchyma.
HnRNPs in Neurological Diseases
Mounting evidence points to RNA perturbations as the underlying driving factor of neurodegenerative diseases such as ALS and FTD [72,73,74]. In light of this, there has been a major paradigm shift away from targeting abnormal protein accumulation in neurodegeneration, towards investigating possible RNA disturbances as a precipitating factor for neurological pathogenesis [75] (Table 1). There is emerging evidence of an overlapping RNA pathology in otherwise distinct neurodegenerative diseases, and therefore there is a need to further investigate hnRNPs which tightly govern RNA processing. This may be a hopeful avenue for the development of future effective therapeutic targets.
Spinal Muscular Atrophy
Spinal muscular atrophy (SMA) is an autosomal recessive disease marked by motor neuron death in the anterior horn of the spinal cord. The SMN protein, a protein integral for motor neuron survival, is encoded by the SMN1 gene which is found to be deleted or mutated in SMA [76]. Despite a deficient SMN1, SMA patients possess a paralogous duplicate gene known as SMN2, a gene almost identical to SMN1 except that it excludes exon 7 in its splicing. This transcript produces a truncated non-functional SMN protein [77], resulting in a failure to confer sufficient rescue. As a result, identifying RBPs that regulate the splicing of SMN2 in order to promote the inclusion of exon 7 has been extensively studied as a potential therapeutic target for SMA [78] (Fig. 2). It has been shown that several RBPs regulate the splicing of exon 7, suggesting a possible misregulation of these proteins as a contributor to SMA. For instance, hnRNP G was found to promote the inclusion of exon 7 via direct interaction with Tra2-β1, a splicing factor which induces exon 7 inclusion [79, 80]. In mice, it was also found that hnRNP Q could modulate the splicing inclusion and exclusion of exon 7 depending on the ratio of its isoforms, with overexpression of major isoform Q1 promoting inclusion by direct binding to exon 7 [81]. In addition, not only was hnRNP M overexpression found to facilitate exon 7 inclusion in patient cells through binding an enhancer on exon 7 via recruitment of U2AF65, its knockdown also fostered a splicing environment which excluded exon 7 [82]. However, HnRNP A1 seems to have opposing effects. For instance, C6T transitions in SMN2 genes have been found to create a high affinity exonic silencer binding site for splicing repressor hnRNP A1 [83]. While some postulate high specificity in hnRNP A1 splicing repressor [84], others propose a more general inhibitory role of hnRNP A1 by binding to a common ESS motif spanning the 3′ splice site of exons [85]. It was found that reduction in hnRNP A1 levels correlated with inclusion of exon 7, and a resultant increase in SMN protein [86]. Despite the uncertainty in the precise mechanism of hnRNP A1 repression which warrants further investigation, it is apparent that hnRNP A1 promotes the exclusion of exon 7. One proposed mechanism is that both RRMs in hnRNP A1 bind to the human intronic splicing silencer ISS-N1 as structural disruptions to either or both RRMs can successfully impair exon 7 splicing repression [87].

hnRNPs involvement in spinal muscular atrophy (SMA). Various hnRNPs have been implicated in regulating splicing of the SMA gene. Promoting the inclusion of exon 7 to produce a functional SMA protein or the exclusion of exon 7 which forms a truncated SMA protein.
Apart from splicing regulation, it has been suggested that hnRNPs could also have roles in transporting the SMN protein which is fundamental for neuronal health. While it has been established that SMN proteins are crucial in axonal growth and local synaptic action [88, 89], however, less attention has been placed on the accompanying molecular partners behind it. HnRNP R was found to co-localize with SMN in axons as well as in presynaptic terminals both in vivo and in vitro [90, 91]. HnRNP R also facilitates complex formation with β-actin mRNA [66, 92], suggesting that apart from regulating splicing, hnRNPs also facilitates transport of SMN proteins. Corroborating this, it was found that overexpression of hnRNP R encourages neurite outgrowth in PC12 cells, and that hnRNP R is needed for the binding of SMN to β-actin mRNA [93]. These findings indicate that perturbations to normal hnRNP levels may compromise axonal transport and could serve as another route for neurodegeneration considering the heavy dependence of neurons on intact intracellular transport for localized protein synthesis [66, 92].
Recent advances in the therapeutics of SMA have led to the development of a novel drug Nusinersen, also known as Spinraza, which promotes the expression of full length SMN2 [94,95,96]. Nusinersen is a modified antisense oligonucleotide which binds to the SMN2 pre-messenger RNA, encouraging the inclusion of exon 7, and augmenting the production of functional SMN protein which is deficient in SMA [94,95,96]. Phase 3 trials have found that patients who were on Nusinersen had significant improvements of motor functions as compared to the control group [95]. Clinical success through manipulating the splicing pathway in SMA makes hnRNPs a promising target of therapeutic research with its heavy role in alternative splicing.
Alzheimer’s Disease
AD is a debilitating neurodegenerative disease characterized by neuronal death and abnormal protein aggregates comprising of Aβ and hyperphosphorylated neurofibrillary tau tangles. The initial links between hnRNPs and AD surfaced when it was discovered that hnRNP A1 modulates the splicing of the APP gene, which is the precursor of the Aβ peptide [97, 98]. It was demonstrated that an imbalance in APP isoforms, in particular high concentrations of APP770, potentially elevates Aβ secretion. HnRNP A1, working in concert with SC35, alternatively splices the APP pre-mRNA by binding to hexanucelotide repeat expansions (HREs) within Alu and flanking the introns adjacent to exons 7, thus generating APP695 which is protective against Aβ toxicity [97]. This suggests that increasing hnRNP A levels may ameliorate AD pathology whereas decreased levels may be associated with worsened pathology and the progression of sporadic AD [99].
However, while some evidence suggests a neuroprotective role of elevated levels of hnRNP A1, others present contradictory results. For instance, apart from managing alternative splicing of APP, hnRNP A1 also regulates balance of receptor for advanced glycation end products (RAGE) isoforms mRAGE and esRAGE [100]. It was found that increased relative concentrations of mRAGE, which is linked to AD pathology, were induced by an overexpression of hnRNP A1 [100]. Higher levels of hnRNP A1 are also expressed in peripheral blood mononuclear cells in patients with AD, suggesting that the misregulation of hnRNP A1 along with decreased levels of transcription factor miR-590-3p may be associated with neuronal death [101].
Another angle of neurodegeneration adopts a prion like perspective in which abnormal protein aggregates act like prionoids and exhibit properties such as templating and incessant proliferation [102,103,104,105]. AD is known to be a notorious proteinopathy, and HnRNP A1 was proposed to exhibit prion like properties [106] and may be in part responsible for intercellular prion like spread of proteins resulting diseases such as AD, ALS, and FTD [107].
Unlike the unequivocal role of hnRNP A1 in modulating the pathology of AD, the roles of other hnRNPs are less clear. hnRNP C has been found to promote APP translation by competing with the FMRP gene for the same region [108] and also by stabilizing the APP precursor mRNA by interacting with the 29 nt element at the 3′-untranslated regions [109], suggesting that increased hnRNP C levels promote Aβ secretion. On the other hand, it was found that cAMP regulated APP processing in mechanisms independent of hnRNP C [110]. In addition, a decrease of hnRNP B1 has been shown in the hippocampal regions of AD patients [111], whereas hnRNP B1 levels are relatively preserved in the inferior temporal cortex [112]. Though the mechanism is unclear, hnRNP Q lnRNAs have been found to be crucial in regulating protein folding and aggregation, and were found to be associated with AD [113].
Despite the expanding idea that AD is associated with abnormal re-entry to the cell cycle and that hnRNPs are pivotal in regulating cell cycle events [55, 114,115,116], there has been an underappreciation of the potential underlying link between hnRNP abnormalities and its modulation of cell cycle events and neuronal death. P53, a major cell cycle regulator, gates cells at the G0/G1 and G2/Gm phases and is crucial in maintaining balance between proliferation and apoptosis [53] and hnRNP K is also a necessary cofactor of this regulation [117]. Furthermore, in times of DNA stress and damage, which is typical in AD [118], hnRNP K sumoylation mediates the regulatory mechanisms of p53 [115]. Not only is hnRNP K one of the most important regulators of p53 action, it was discovered earlier that p53 levels are elevated in the temporal cortices of AD patients [119]; hence, it is surprising that hnRNP K misregulation has not been investigated in AD and further suggests a possible hnRNP related pathway of neurodegeneration.
Amyotrophic Lateral Sclerosis and Frontotemporal Dementia
Amyotrophic lateral sclerosis (ALS) is the most common form of motor neuron disease, marked by significant muscle wasting and widespread neurodegeneration of both upper and lower motor neurons. Clinically, patients exhibit spinal symptoms of muscular weakness, spasticity, fasciculations, fatigue and bulbar symptoms of dysphagia, and breathing difficulties [120]. In contrast, frontotemporal dementia (FTD) is characterized by progressive neurodegeneration in the frontal and temporal cortices with accompanying changes in personality and language abilities [121]. Phenotypic heterogeneity is observed in ALS and FTD patients, specifically where ubiquinated TDP-43 pathologies are found [72]. Although pathogenic variants in TARDBP develop TDP-43 pathologies, other familial and sporadic ALS cases also develop TDP-43 pathologies [122]. However, despite the rarity of TARDBP mutations in FTD, those with TDP-43 pathologies account for around 45% of FTD cases [123]. TDP-43 is a DNA/RNA binding protein which is usually concentrated in the nucleus [124]. It contains a nuclear localization signal and nuclear export signal and hence is able to shuttle between the nucleus and cytoplasm [124]. The known functions of TDP-43 are broad, including regulating gene expression and involvement in several RNA processing steps such as pre-mRNA splicing, regulation of mRNA stability, mRNA transport, translation, and the regulation of non-coding RNAs [33, 125, 126]. Notably, the majority of TARDBP mutations are located in the glycine-rich region at the carboxy-terminal region, which is also the region that interacts with other hnRNPs and is heavily involved in pre-mRNA splicing regulation [127]. Pathologically, TDP-43 accumulates in the cytoplasm resulting in a loss of nuclear TDP-43, and it is this loss from the nucleus that has led to proposed mechanisms of disease involving a loss of normal function in the nucleus, a toxic gain of function in the cytoplasm, or both. These mechanisms have been tested in many animal models [128,129,130,131,132,133]. As TDP-43 homeostasis is crucial for normal cellular function, increasing evidence suggests that aberrant TDP-43 regulation may result in disease. Excess TDP-43 in the cytoplasm may lead to formation of inclusion bodies resulting in cellular dysfunction, while nuclear depletion may induce widespread dysregulation of mRNA metabolism, with TDP-43 knockdown shown to cause differential splicing or expression of hundreds of targets [134,135,136].
The most common genetic cause of ALS and FTD is C9orf72 expansions which produce extensive amounts of GGGGCC repeats [74]. It has been shown that GGGGCC expansions can become highly neurotoxic, correlating with disease severity and are able to sequester otherwise functional RBPs. HnRNP A3 has been identified in neuronal cytoplasmic and intranuclear inclusions in patients with GGGGCC expansion repeats [137] (Fig. 3). HnRNP H and hnRNP F were also found to co-localize with GGGGCC expansion foci in immunoprecipitation studies [138] and western blot analyses [139] respectively. It has been recently suggested that a GGA-rich sequence on hnRNP H may account for its affinity to the GGGGCC expansion repeats and its sequestration in ALS and FTD [140]. HnRNP sequestration by G quadruplexes in the expansion repeats in ALS and FTD suggests that hnRNP implication may be in part responsible for the toxicity incurred by C9orf72 mutations as important RNA processes such as splicing are compromised [141]. In addition, TDP-43, has also been found to be regulated by, co-localize with, or interact with hnRNPs A1, A2/B1, C1/C2, A3, K [127, 142, 143], suggesting a strong association of impaired hnRNP translation machinery and neurological diseases (Fig. 3). Apart from the ability of GGGGCC repeats to sequester important proteins, several other pathogenic mechanisms have been suggested, including loss of function of C9orf72 and generation of toxic dipeptide repeats [144, 145], and these have been reviewed in detail elsewhere [146, 147].

The involvement of hnRNPs in amyotrophic lateral sclerosis and frontotemporal dementia. hnRNPs have been shown to be involved in ALS and FTD in many ways. hnRNPs are able to bind to the hexanucleotide repeats within the nucleus, bind to the dipeptide repeat protein (DPRs) inclusions in the cytoplasm and also to the main pathological inclusions in both diseases TDP-43. In FTLD-FUS, hnRNP proteins have been found to co-localize with FUS in the nucleus and the cytoplasm. It has also been shown that other hnRNP proteins also form patholigcal inclusions without the presence of FUS. FTLD-TDP type C pathology shown distinct inclusions as long twisted neurites which have also been shown to co-localize with hnRNP E2
As hnRNPs are normally localized to the nucleus, cytoplasmic redistribution has been linked to disease pathology and impaired nucleocytoplasmic transport has been suggested to be a common feature of ALS and FTD [49, 148]. For instance, mutations in hnRNP A1 can disrupt its nuclear localization sequence and result in cytoplasmic redistribution and aggregation [149]. It was also found in ALS patients that hnRNP A1, along with TDP-43, had a greater tendency to aggregate in the cytoplasmic inclusions compared to controls [150]. Another hnRNP, E2, also co-localized with TDP-43 in pathological inclusions of the semantic dementia subtype FTD patients [151] (Fig. 3). In FTLD FUS-positive patients, hnRNP A1, D, G, I, and L, which are involved in nuclear transport, have been found to exhibit pathological mislocalisation or accumulated in neuronal cytoplasmic inclusions [152] (Fig. 3). hnRNPs R and Q have also been found to co-deposit with FUS in FTLD-FUS inclusions [153], suggesting that impaired nucleocytoplasmic transport may contribute to disease pathology (Fig. 3).
As in AD, it has also been suggested that ALS proteins such as TDP-43, SOD1, and FUS, exhibit a prion like spread [154]. Accordingly, it was revealed that mutations in the prion like domains in the hnRNP A1 and A2/B1 genes increased the propensity for protein self-aggregation, excessive, fibrilisation, and incorporation into stress granules [155]. Furthermore, it is thought that even though cytoplasmic formation of membrane-less organelles such as stress granules may be advantageous for energy conservation, high concentrations of hnRNPs aggregated within stress granules may accelerate fibrillisation [49].
Multiple Sclerosis
Multiple sclerosis (MS) is an autoimmune disorder of the central nervous system characterized by demyelination; however, the exact target epitope of autoimmunity remains unclear [156]. Focus has been placed on autoimmunity against myelin related targets and it has been shown that antibodies against myelin proteins such as MOG and MBP are not specific to MS and are also elevated in other neurological diseases [157]. As a result, there has been a growing interest in investigating non-myelin targets in contributing to the pathogenesis of MS [158]. HnRNP A1 and hnRNP B1 are highly expressed in neurons [159, 160], and were found at significantly high levels in the CSF of MS patients than in other diseases [161]. Interestingly, their antibodies were found in MS patients. The antibodies generated may have dual roles as they can on one hand impair proper functioning of epitopes and on the other hand directly damage the cells. For instance, MS patients developed antibodies against the M9 epitope of hnRNP A1 where the nuclear localization sequence is found [162, 163]. In line with this, MS patients typically possess genomic single nucleotide variants in the hnRNP A1 gene within the nucleocytoplasmic transport domain TPNO-1, suggesting that impaired hnRNP A1 mediated nucleocytoplasmic transport could be involved in MS pathology [163]. On the other hand, the antibodies generated can also cause a toxic gain of function. It was found that anti-hnRNP A1 antibodies penetrate neuronal cells and alter levels of ATP and apoptotic regulator caspase 3/7, as well as promote a redistribution of hnRNP A1 intracellularly [164]. They also alter transcripts related to hnRNP A1 function, reduce neuronal processes and distort the cytoplasm [162]. Compared to hnRNP A1, less is known about hnRNP B1 despite the elevations of its antibodies in MS patients. Not only were hnRNP B1 antibodies found in the CSF of a large proportion of MS patients examined, the generation of hnRNP B1 antibodies in the CSF was found to be specific only to MS and not found in other neurological diseases [165]. Although not much is known about the precise implications brought about by hnRNP B1 antibodies, its specificity to MS and its potential as a biomarker provides ample basis for further research.
There is also evidence suggesting that hnRNP A1 may be responsible for the phenotypical features in MS. It was found that hnRNP A1 binds to spastin, a gene responsible for hereditary spastic paraparesis, which produces a phenotype that is closely similar to MS [162]. This finding was also replicated by another group which found a hnRNP A1 antibodies in animal models also contributed to neurodegeneration as evidenced by increased localization to stress granules, worsened experimental autoimmune encephalomyelitis (EAE) cases, modifications in phenotype from flaccid to spastic paralysis, and selective degeneration in the cerebellar white matter [166].
Aside from being possible epitopic targets, hnRNPs may also mediate ectopic splicing, causing alterations to levels of RNA and proteins. Lower levels of PRKCA have been shown to increase MS susceptibility [167]. Interestingly, it was found that hnRNP H overexpression mediates the expression of the PRKCA gene by promoting the skipping of exon 3 while hnRNP H silenced cells increased exon 3 inclusion [168], suggesting that hnRNP H abnormalities may be related to MS predisposition.
Congenital Myasthenic Syndrome
Congenital myasthenic syndrome (CMS) consists of genetically inherited diseases in which normal neurotransmission is impaired at the neuromuscular endplate. CMS usually stems from mutations in the muscle nicotinic acetyl choline receptors (AChR) subunits, AChE deficiencies, or inefficient kinetics of AChRs [169]. As in other neurological diseases, the roles of hnRNPs in regulating alternative splicing are pivotal in maintaining transcripts necessary for healthy functioning. A variety of molecules are expressed at the neuromuscular junction and its abnormal expression, highly regulated by RBPs, is impaired in CMS.
The alpha subunit of AChR is encoded by the CHRNA1, and it was discovered that hnRNP H binds to an intronic splicing silencer on CHRNA1 and enhances the skipping of a non-functional P3A exon. The exclusive inclusion of exon P3A is often displayed in CMS causing genetic mutations [170]. From individual patients with CMS, it was revealed that G > A mutations in this region led to a 100-fold decrease in the binding affinity of hnRNP H, suggesting a regulatory role of the alternative splicing of exon P3A [171]. It was later also discovered that a tannic acid induced increase in polypyrimidine tract binding protein could rescue the effects of hnRNP H mediated inclusion by binding close intron 3 on CHRNA1, promoting its exclusion [172]. HnRNP L and hnRNP LL also antagonistically bind the polypyrimidine tract binding protein which modulates P3A splicing. It was found that mutated CHRNA1 generates an otherwise absent binding site for hnRNP LL, a splicing enhancer, and results in the displacement of hnRNP L, a splicing suppressor, which subsequently leads to exclusive P3A inclusion, a pathological feature that is found in CMS [173].
Not only does hnRNP H regulate CHRNA1, it also was found to be involved in the regulation of AChET, AChEH, and AChER, with the former two being expressed at neuromuscular junctions and hematopoietic cells respectively, and the third being rarely expressed [170]. HnRNP H competes against another splice site regulator CstF64 and suppresses cryptic PAS, thereby generating AChET [170]. CstF64 has opposing effects which activates cryptic PAS and its overpowering of hnRNP H generates the other two subtypes AChEH and AChER [170]. This suggests that abnormal hnRNP H activity may result in alterations in AChE levels at the neuromuscular endplate, another feature of CMS.
Another molecule critical in CMS is the muscle specific receptor tyrosine kinase (MuSK), which is involved in the pre-patterning of AChRs, a process required for the high AChR concentration in neuromuscular junctions [174, 175]. The MuSK gene consists of a frizzled cysteine-rich region that is essential for both proper folding as well as the binding of Wnt ligands, important for mediating AChR clustering [176,177,178]. Interestingly, exon 10 of the MuSK gene, which encodes 6 out of 10 of the essential cysteines, is alternatively spliced and functionally skipped in humans but not in mouse. It was revealed that hnRNP C regulates the skipping of exon 10 by binding to the poly-T tract in the exon splicing silencer 5 region, and also mediates hnRNP L and YB-1 binding leading to further additive effects [179].
Collagen Q, encoded by the COLQ gene, is necessary for the anchoring of AChE to NMJ endplates [180] and mutations in COLQ are associated with AChE deficiency, a defect often evident in CMS [181]. HnRNP H suppresses the splicing of exon 16 in COLQ and results in aberrant skipping, and is antagonistically modulated by SRSF1, a splicing enhancer that promotes its inclusion [180, 182].
Whereas the exact roles of P3A and exon 10 alternative splicing have yet to be elucidated, the disease-causing effects of the missplicing of these proteins which are subjected to hnRNP regulation provide a strong basis for further investigation.
Fragile X-Associated Tremor/Ataxia Syndrome
Fragile X-associated tremor/ataxia syndrome (FXTAS) is an adult onset neurodegenerative disorder and has phenotypic characteristics that include motor and cognitive impairment. It is predominant in males and involves the expansion of CGG repeats in the fragile X mental retardation gene FMR1 [183]. As in other neurological disorders with expansion repeats, the CGG repeats sequester important RBPs, thus depriving the cell of the normal function of these RBPs and thereby contributing to the pathomechanisms of the disease. HnRNP A2 binds to the CGG repeats in fly models [184] and is found in cytoplasmic inclusions in FXTAS post mortem brains [185]. Furthermore, it was found that hnRNP A2 overexpression could suppress the rough eye phenotype caused by rCGG [184], suggesting that rCGG sequestration of hnRNP A2 may be involved in the pathogenesis of FXTAS since its over expression could rescue the neurodegenerative phenotype. It was also found that misspliced hnRNP A2/B1, which is prevalent in CGG expression, could be corrected by TDP-43 and resulted in a corresponding reduction of rCGG induced damage [186].
Not only does sequestration of hnRNP A2 diminish its normal cellular functions, it was also revealed that hnRNP A2 can mediate neurodegeneration through interaction with its binding partners. It was shown that miRNA miR-277 could mediate CGG neurodegeneration as overexpression of miR-277 increased neuronal toxicity whereas decreased expression of miR-277 suppressed neurodegeneration [187]. Coincidently, miR-277 is also regulated by hnRNP A2 [187], suggesting that sequestration of hnRNP A2 by CGG repeats could further result in ectopic miR-277 levels, resulting in a loop which further perpetuates rCGG mediated neurodegeneration. The neurodegenerative effects of rCGG are mediated by retrotransposon activation, and it was discovered that hnRNP A2 has the ability to suppress the toxicity of a particular retrotransposon, Gypsy, by binding to HP1, a retrotransposon silencer [188].
It has also been suggested that the CGG repeats are constantly expanding dynamic structures and may recruit more proteins at later stages. For instance, it was found that in later stages of the disease where CGG aggregates have expanded significantly, sam-68 mediated hnRNP G inclusions were also found [189]. This suggest that the rCGG sequences themselves may not recruit proteins directly, but does so indirectly by protein-protein interactions with inclusions that were recruited earlier. This is noteworthy since hnRNPs are known to have an expanding repertoire of binding partners; hence, more information about hnRNP mediated sequestration of other proteins into inclusion bodies may shed light onto the pathomechanisms of disease.
Conclusion
This review provides a summary of the existing evidence of aberrant hnRNP profiles in various neurological diseases, specifically SMA, AD, ALS, FTD, MS, CMS, and FXTAS. Our understanding of hnRNPs in these diseases is still in its infancy, and there remains much that has yet to be uncovered. Human genome studies have revealed that 40–60% of our genome relies extensively on alternative splicing to confer functional diversification upon our otherwise limited gene repertoire [190]. With the viability and normal functioning of cells being heavily subjected to intact and tightly regulated splicing mechanisms, it would be expected that RBPs dysfunction can serve as major contributors to disease pathology [191].
However, the array of hnRNP mediated interactions, as well as binding partners, are far from being fully elucidated, and better understanding in the structural morphology of hnRNPs and their interacting partners may provide further insight into their roles in health and disease. Its implication in various neurological diseases highly suggests that hnRNPs possess an underappreciated role in disease pathology. The role of hnRNPs in neurological disease has been a largely overlooked area and more research may serve as a promising platform for the development of novel therapeutic targets.
References
Dreyfuss G, Matunis MJ, Pinol-Roma S, Burd CG (1993) hnRNP proteins and the biogenesis of mRNA. Anna Rev Biochem 62:289–321
Piñol-Roma S, Choi YD, Matunis MJ, Dreyfuss G (1988) Immunopurification of heterogeneous nuclear ribonucleoprotein particles reveals an assortment of RNA-binding proteins. Genes Dev 2:215–227. https://doi.org/10.1101/gad.2.2.215
Piñol-Roma S, Dreyfuss G (1992) Shuttling of pre-mRNA binding proteins between nucleus and cytoplasm. Nature. 355:730–732. https://doi.org/10.1038/355730a0
Maris C, Dominguez C, Allain FHT (2005) The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J 272:2118–2131. https://doi.org/10.1111/j.1742-4658.2005.04653.x
Nagai K, Oubridge C, Jessen TH, Li J, Evans PR (1990) Crystal structure of the RNA-binding domain of the U1 small nuclear ribonucleoprotein a. Nature. 348:515–520. https://doi.org/10.1038/348515a0
Avis JM, Allain FHT, Howe PWA, Varani G, Nagai K, Neuhaus D (1996) Solution structure of the N-terminal RNP domain of U1A protein: The role of C-terminal residues in structure stability and RNA binding. J Mol Biol 257:398–411. https://doi.org/10.1006/jmbi.1996.0171
Xu RM, Jokhan L, Cheng X, Mayeda A, Krainer AR (1997) Crystal structure of human UP1, the domain of hnRNP A1 that contains two RNA-recognition motifs, structure. https://doi.org/10.1016/S0969-2126(97)00211-6.
Shamoo Y, Krueger U, Rice LM, Williams KR, Steitz TA (1997) Crystal structure of the two RNA binding domains of human hnRNP A1 at 1.75 A resolution. Nat Struct Biol. https://doi.org/10.1038/nsb0397-215.
Baber JL, Libutti D, Levens D, Tjandra N (1999) High precision solution structure of the C-terminal KH domain of heterogeneous nuclear ribonucleoprotein K, a c-myc transcription factor. J Mol Biol. https://doi.org/10.1006/jmbi.1999.2818.
Masuzawa T, Oyoshi T, Roles of the RGG domain and RNA recognition motif of nucleolin in G-quadruplex stabilization, ACS Appl Mater Interfaces (2020). https://doi.org/10.1021/acsomega.9b04221.
Gui X, Luo F, Li Y, Zhou H, Qin Z, Liu Z, Gu J, Xie M, Zhao K, Dai B, Shin WS, He J, He L, Jiang L, Zhao M, Sun B, Li X, Liu C, Li D (2019) Structural basis for reversible amyloids of hnRNPA1 elucidates their role in stress granule assembly. Nat Commun . https://doi.org/10.1038/s41467-019-09902-7.
Siomi H, Matunis MJ, Michael WM, Dreyfuss G (1993) The pre-mRNA binding K protein contains a novel evolutionary conserved motif. Nucleic Acids Res 21:1193–1198. https://doi.org/10.1093/nar/21.5.1193
Makeyev AV, Liebhaber SA (2002) The poly(C)-binding proteins: A multiplicity of functions and a search for mechanisms. RNA. 8:265–278. https://doi.org/10.1017/S1355838202024627
Kiledjian M, Dreyfuss G (1992) Primary structure and binding activity of the hnRNP U protein: Binding RNA through RGG box. EMBO J 11:2655–2664. https://doi.org/10.1002/j.1460-2075.1992.tb05331.x
Dreyfuss G, Kim VN, Kataoka N (2002) Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol 3:195–205. https://doi.org/10.1038/nrm760
Chaudhury A, Chander P, Howe PH (2010) Heterogeneous nuclear ribonucleoproteins (hnRNPs) in cellular processes: Focus on hnRNP E1’s multifunctional regulatory roles. RNA. 16:1449–1462. https://doi.org/10.1261/rna.2254110
Geuens T, Bouhy D, Timmerman V (2016) The hnRNP family: Insights into their role in health and disease. Hum Genet 135:851–867. https://doi.org/10.1007/s00439-016-1683-5
Vance C, Scotter EL, Nishimura AL, Troakes C, Mitchell JC, Kathe C, Urwin H, Manser C et al (2013) ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum Mol Genet 22:2676–2688. https://doi.org/10.1093/hmg/ddt117
Hutchins EJ, Belrose JL, Szaro BG (2016) A novel role for the nuclear localization signal in regulating hnRNP K protein stability in vivo. Biochem Biophys Res Commun 478:772–776. https://doi.org/10.1016/j.bbrc.2016.08.023
Hay DC, Kemp GD, Dargemont C, Hay RT (2001) Interaction between hnRNPA1 and IkappaBalpha is required for maximal activation of NF-kappaB-dependent transcription. Mol Cell Biol 21:3482–3490. https://doi.org/10.1128/MCB.21.10.3482-3490.2001
Takimoto M, Tomonaga T, Matunis M, Avigan M, Krutzsch H, Dreyfuss G, Levens D (1993) Specific binding of heterogeneous ribonucleoprotein particle protein K to the human c-myc promoter, in vitro. J Biol Chem 268:18249–18258
Tomonaga T, Levens D (1995) Heterogeneous nuclear ribonucleoprotein K is a DNA-binding transactivator. J Biol Chem 270:4875–4881. https://doi.org/10.1074/jbc.270.9.4875
Lynch M, Chen L, Ravitz MJ, Mehtani S, Korenblat K, Pazin MJ, Schmidt EV (2005) hnRNP K binds a core polypyrimidine element in the eukaryotic translation initiation factor 4E (eIF4E) promoter, and its regulation of eIF4E contributes to neoplastic transformation. Mol Cell Biol 25:6436–6453. https://doi.org/10.1128/MCB.25.15.6436-6453.2005
Michelotti EF, Michelotti GA, Aronsohn AI, Levens D (1996) Heterogeneous nuclear ribonucleoprotein K is a transcription factor. Mol Cell Biol 16:2350–2360. https://doi.org/10.1074/jbc.270.9.4875
Denisenko ON, O’Neill B, Ostrowski J, Van Seuningen I, Bomsztyk K (1996) Zik1, a transcriptional repressor that interacts with the heterogeneous nuclear ribonucleoprotein particle K protein. J Biol Chem 271:27701–27706. https://doi.org/10.1074/jbc.271.44.27701
Graveley BR (2001) Alternative splicing: Increasing diversity in the proteomic world. Trends Genet 17:100–107. https://doi.org/10.1016/S0168-9525(00)02176-4
Yeo G, Holste D, Kreiman G, Burge CB (2004) Variation in alternative splicing across human tissues. Genome Biol 5:R74. https://doi.org/10.1186/gb-2004-5-10-r74
Conlon EG, Manley JL (2017) RNA-binding proteins in neurodegeneration: Mechanisms in aggregate. Genes Dev 31:1509–1528. https://doi.org/10.1101/gad.304055.117
Huang H, Zhang J, Harvey SE, Hu X, Cheng C (2017) RNA G-quadruplex secondary structure promotes alternative splicing via the RNA-binding protein hnRNPF. Genes Dev 31:2296–2309. https://doi.org/10.1101/gad.305862.117
Grabowski PJ, Black DL (2001) Alternative RNA splicing in the nervous system. Prog Neurobiol 65:289–308. https://doi.org/10.1016/S0301-0082(01)00007-7
S. Gueroussov, R.J. Weatheritt, D. O’Hanlon, Z.Y. Lin, A. Narula, A.C. Gingras, B.J. Blencowe, Regulatory expansion in mammals of multivalent hnRNP assemblies that globally control alternative splicing, Cell. (2017). https://doi.org/10.1016/j.cell.2017.06.037.
Martinez FJ, Pratt GA, Van Nostrand EL, Batra R, Huelga SC, Kapeli K, Freese P, Chun SJ, Ling K, Gelboin-Burkhart C, Fijany L, Wang HC, Nussbacher JK, Broski SM, Kim HJ, Lardelli R, Sundararaman B, Donohue JP, Javaherian A, Lykke-Andersen J, Finkbeiner S, Bennett CF, Ares M, Burge CB, Taylor JP, Rigo F, Yeo GW (2016) Protein-RNA networks regulated by normal and ALS-associated mutant HNRNPA2B1 in the nervous system, Neuron. https://doi.org/10.1016/j.neuron.2016.09.050.
Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, König J, Hortobágyi T et al (2011) Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. https://doi.org/10.1038/nn.2778
Black DL (2003) Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. https://doi.org/10.1146/annurev.biochem.72.121801.161720
König J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM et al (2010) ICLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol. https://doi.org/10.1038/nsmb.1838
Xiao X, Wang Z, Jang M, Nutiu R, Wang ET, Burge CB (2009) Splice site strength-dependent activity and genetic buffering by poly-G runs. Nat Struct Mol Biol 16:1094–1100. https://doi.org/10.1038/nsmb.1661
Tavanez J, Madl T, Kooshapur H, Sattler M, Valcárcel J (2012) HnRNP A1 proofreads 3′ splice site recognition by U2AF. Mol Cell 45:314–329. https://doi.org/10.1016/j.molcel.2011.11.033
Dewey CM, Cenik B, Sephton CF, Johnson BA, Herz J, Yu G (2012) TDP-43 aggregation in neurodegeneration: Are stress granules the key? Brain Res 1462:16–25. https://doi.org/10.1016/j.brainres.2012.02.032
Ash PEA, Vanderweyde TE, Youmans KL, Apicco DJ, Wolozin B (2014) Pathological stress granules in Alzheimer’s disease. Brain Res 1584:52–58. https://doi.org/10.1016/j.brainres.2014.05.052
Buchan JR, Parker R (2009) Eukaryotic stress granules: The ins and outs of translation. Mol Cell 36:932–941. https://doi.org/10.1016/j.molcel.2009.11.020
King OD, Gitler AD, Shorter J (2012) The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res 1462:61–80. https://doi.org/10.1016/j.brainres.2012.01.016
Bowden HA, Dormann D (2016) Altered mRNP granule dynamics in FTLD pathogenesis. J Neurochem:112–133. https://doi.org/10.1111/jnc.13601
Guil S, Long JC, Caceres JF (2006) hnRNP A1 relocalization to the stress granules reflects a role in the stress response. Mol Cell Biol 26:5744–5758. https://doi.org/10.1128/MCB.00224-06
Kim WJ, Back SH, Kim V, Ryu I, Jang SK (2005) Sequestration of TRAF2 into stress granules interrupts tumor necrosis factor signaling under stress conditions. Mol Cell Biol 25:2450–2462. https://doi.org/10.1128/MCB.25.6.2450-2462.2005
Arimoto K, Fukuda H, Imajoh-Ohmi S, Saito H, Takekawa M (2008) Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat Cell Biol 10:1324–1332. https://doi.org/10.1038/ncb1791
Takahara T, Maeda T (2012) Transient sequestration of TORC1 into stress granules during heat stress. Mol Cell 47:242–252. https://doi.org/10.1016/j.molcel.2012.05.019
Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, Clutario KM, Ling SC et al (2012) Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci 15:1488–1497. https://doi.org/10.1038/nn.3230
Schmidt HB, Rohatgi R (2016) In vivo formation of vacuolated multi-phase compartments lacking membranes. Cell Rep. https://doi.org/10.1016/j.celrep.2016.06.088
Taylor JP, Brown RH, Cleveland DW (2016) Decoding ALS: From genes to mechanism. Nature. 539:197–206. https://doi.org/10.1038/nature20413
Hondele M, Heinrich S, Rios P. De Los, Weis K (2020) Membraneless organelles: phasing out of equilibrium, Emerg. Top Life Sci. ETLS201901. doi: https://doi.org/10.1042/ETLS20190190.
Y.G. Zhao, H. Zhang, Phase separation in membrane biology: the interplay between membrane-bound organelles and membraneless condensates., Dev. Cell. (2020). https://doi.org/10.1016/j.devcel.2020.06.033.
de Oliveira GAP, Cordeiro Y, Silva JL, Vieira TCRG (2019) Liquid-liquid phase transitions and amyloid aggregation in proteins related to cancer and neurodegenerative diseases, Adv. Protein Chem. Struct. Biol. https://doi.org/10.1016/bs.apcsb.2019.08.002.
Lanni C, Racchi M, Memo M, Govoni S, Uberti D (2012) P53 at the crossroads between cancer and neurodegeneration. Free Radic Biol Med 52:1727–1733. https://doi.org/10.1016/j.freeradbiomed.2012.02.034
Zhang Q, Guo S, Zhang X, Tang S, Shao W, Han X, Wang L, Du Y (2015) Inverse relationship between cancer and Alzheimer’s disease: A systemic review meta-analysis. Neurol Sci 36:1987–1994. https://doi.org/10.1007/s10072-015-2282-2
Yang Y, Geldmacher DS, Herrup K (2001) DNA replication precedes neuronal cell death in Alzheimer’s disease. J Neurosci 21:2661–2668
Yurov YB, Vorsanova SG, Iourov IY (2011) The DNA replication stress hypothesis of Alzheimer’s disease. Sci World J 11:2602–2612. https://doi.org/10.1100/2011/625690
Weaver BAA, Cleveland DW (2005) Decoding the links between mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell death. Cancer Cell 8:7–12. https://doi.org/10.1016/j.ccr.2005.06.011
Ding J, Hayashi MK, Zhang Y, Manche L, Krainer AR, Xu RM (1999) Crystal structure of the two-RRM domain of hnRNP A1 (UP1) complexed with single-stranded telomeric DNA. Genes Dev 13:1102–1115. https://doi.org/10.1101/gad.13.9.1102
LaBranche H, Dupuis S, Ben-David Y, Bani MR, Wellinger RJ, Chabot B (1998) Telomere elongation by hnRNP A1 and a derivative that interacts with telomeric repeats and telomerase. Nat Genet 19:199–202. https://doi.org/10.1038/575
Grandori C, Cowley SM, James LP, Eisenman RN (2000) The Myc/max/mad network and the transcriptional control of cell behavior. Annu Rev Cell Dev Biol 16:653–699. https://doi.org/10.1146/annurev.cellbio.16.1.653
Chevalier-Larsen E, Holzbaur EL (2006) Axonal transport and neurodegenerative disease. Biochim Biophys Acta 1762:1094–1108. https://doi.org/10.1016/j.bbadis.2006.04.002
Morfini GA, Burns M, Binder LI, Kanaan NM, LaPointe N, Bosco DA, Brown RH, Brown H et al (2009) Axonal transport defects in neurodegenerative diseases. J Neurosci 29:12776–12786. https://doi.org/10.1523/JNEUROSCI.3463-09.2009
Millecamps S, Julien JP (2013) Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci 14:161–176. https://doi.org/10.1038/nrn3380
Hares K, WIlkins A (2017) Axonal transport proteins as biomarkers of neurodegeneration? Biomark Med 11:589–591
von Kügelgen N, Chekulaeva M (2020) Conservation of a core neurite transcriptome across neuronal types and species, Wiley Interdiscip. Rev. RNA. e1590
Glinka M, Herrmann T, Funk N, Havlicek S, Rossoll W, Winkler C, Sendtner M (2010) The heterogeneous nuclear ribonucleoprotein-R is necessary for axonal β-actin mRNA translocation in spinal motor neurons. Hum Mol Genet 19:1951–1966. https://doi.org/10.1093/hmg/ddq073
Briese M, Saal-Bauernschubert L, Ji C, Moradi M, Ghanawi H, Uhl M, Appenzeller S, Backofen R, Sendtner M (2018) hnRNP R and its main interactor, the noncoding RNA 7SK, coregulate the axonal transcriptome of motoneurons., PNAS. Published
Liu Y, Szaro BG (2011) hnRNP K post-transcriptionally co-regulates multiple cytoskeletal genes needed for axonogenesis. Development 138:3079–3090. https://doi.org/10.1242/dev.066993
Koppers M, Cagnetta R, Shigeoka T, Wunderlich LC, Vallejo-Ramirez P, Lin JQ, Zhao S, Jakobs MA, Dwivedy A, Minett MS, Bellon A, Kaminski CF, Harris WA, Flanagan JG, Holt CE (2019) Receptor-specific interactome as a hub for rapid cue-induced selective translation in axons, Elife. 8
Pushpalatha KV, Besse F (2019) Local translation in axons: When membraneless RNP granules meet membrane-bound organelles. Front Mol Biosci 6:129
Lee SJ, Oses-Prieto JA, Kawaguchi R, Sahoo PK, Kar AN, Rozenbaum M, Oliver D, Chand S et al (2018) hnRNPs interacting with mRNA localization motifs define axonal RNA regulons. Mol Cell Proteomics 17:2091–2106
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. https://doi.org/10.1126/science.1134108
Neumann M, Kwong LK, Sampathu DM, Trojanowski JQ, Lee VMY (2007) TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: Protein misfolding diseases without amyloidosis. Arch Neurol 64:1388–1394. https://doi.org/10.1001/archneur.64.10.1388
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NCA et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72:245–256. https://doi.org/10.1016/j.neuron.2011.09.011
Renoux AJ, Todd PK (2012) Neurodegeneration the RNA way. Prog Neurobiol 97:173–189. https://doi.org/10.1016/j.pneurobio.2011.10.006
Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C et al (1995) Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 80:155–165. https://doi.org/10.1016/0092-8674(95)90460-3
Lorson CL, Hahnen E, Androphy EJ, Wirth B (1999) A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci 96:6307–6311. https://doi.org/10.1073/pnas.96.11.6307
Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR (2008) Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet 82:834–848. https://doi.org/10.1016/j.ajhg.2008.01.014
Hofmann Y, Wirth B (2002) hnRNP-G promotes exon 7 inclusion of survival motor neuron (SMN) via direct interaction with Htra2-beta1. Hum Mol Genet 11:2037–2049. https://doi.org/10.1093/hmg/11.17.2037
Moursy A, Allain FHT, Cléry A (2014) Characterization of the RNA recognition mode of hnRNP G extends its role in SMN2 splicing regulation. Nucleic Acids Res 42:6659–6672. https://doi.org/10.1093/nar/gku244
Chen H-H, Chang J-G, Lu R-M, Peng T-Y, Tarn W-Y (2008) The RNA binding protein hnRNP Q modulates the utilization of exon 7 in the survival motor neuron 2 (SMN2) Gene. Mol Cell Biol 28:6929–6938. https://doi.org/10.1128/MCB.01332-08
Cho S, Moon H, Loh TJ, Oh HK, Cho S, Choy HE, Song WK, Chun JS et al (2014) HnRNP M facilitates exon 7 inclusion of SMN2 pre-mRNA in spinal muscular atrophy by targeting an enhancer on exon 7. Biochim Biophys Acta - Gene Regul Mech 1839:306–315. https://doi.org/10.1016/j.bbagrm.2014.02.006
Kashima T, Manley JL (2003) A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet 34:460–463. https://doi.org/10.1038/ng1207
Kashima T, Rao N, David CJ, Manley JI (2007) hnRNP A1 functions with specificity in repression of SMN2 exon 7 splicing. Hum Mol Genet 16:3149–3159. https://doi.org/10.1093/hmg/ddm276
Koed Doktor T, Schroeder LD, Vested A, Palmfeldt J, Andersen HS, Gregersen N, Andresen BS (2011) SMN2 exon 7 splicing is inhibited by binding of hnRNP A1 to a common ESS motif that spans the 3′ splice site. Hum Mutat 32:220–230. https://doi.org/10.1002/humu.21419
Baek J, Jeong H, Ham Y, Jo YH, Choi M, Kang M, Son B, Choi S, Ryu HW, Kim J, Shen H, Sydara K, Lee SW, Kim SY, Han SB, Oh SR, Cho S (2019) Improvement of spinal muscular atrophy via correction of the SMN2 splicing defect by Brucea javanica (L.) Merr. extract and Bruceine D, Phytomedicine. https://doi.org/10.1016/j.phymed.2019.153089.
Beusch I, Barraud P, Moursy A, Cléry A, Allain FHT (2017) Tandem hnRNP A1 RNA recognition motifs act in concert to repress the splicing of survival motor neuron exon 7, Elife. 6. https://doi.org/10.7554/eLife.25736.
Ruiz R, Casañas JJ, Torres-Benito L, Cano R, Tabares L (2010) Altered intracellular Ca2+ homeostasis in nerve terminals of severe spinal muscular atrophy mice. J Neurosci 30:849–857. https://doi.org/10.1523/JNEUROSCI.4496-09.2010
Kariya S, Obis T, Garone C, Akay T, Sera F, Iwata S, Homma S, Monani UR (2014) Requirement of enhanced survival motoneuron protein imposed during neuromuscular junction maturation. J Clin Invest 124:785–800. https://doi.org/10.1172/JCI72017
Rossoll W, Kröning A-K, Ohndorf U-M, Steegborn C, Jablonka S, Sendtner M (2002) Specific interaction of Smn, the spinal muscular atrophy determining gene product, with hnRNP-R and gry-rbp/hnRNP-Q: A role for Smn in RNA processing in motor axons? Hum Mol Genet 11:93–105. https://doi.org/10.1093/hmg/11.1.93
Dombert B, Sivadasan R, Simon CM, Jablonka S, Sendtner M (2014) Presynaptic localization of SMN and hnRNP R in axon terminals of embryonic and postnatal mouse motoneurons. PLoS One. 9. https://doi.org/10.1371/journal.pone.0110846.
De Vos KJ, Grierson AJ, Ackerley S, Miller CCJ (2008) Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci 31:151–173. https://doi.org/10.1146/annurev.neuro.31.061307.090711
Rossoll W, Jablonka S, Andreassi C, Kröning AK, Karle K, Monani UR, Sendtner M (2003) Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of β-actin mRNA in growth cones of motoneurons. J Cell Biol 163:801–812. https://doi.org/10.1083/jcb.200304128
Bennett Frank C, Krainer AR, Cleveland DW (2019) Antisense oligonucleotide therapies for neurodegenerative diseases. Annu Rev Neurosci. https://doi.org/10.1146/annurev-neuro-070918-050501
Mercuri E, Darras BT, Chiriboga CA, Day JW, Campbell C, Connolly AM, Iannaccone ST, Kirschner J et al (2018) Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med. https://doi.org/10.1056/NEJMoa1710504
Chiriboga CA, Swoboda KJ, Darras BT, Iannaccone ST, Montes J, De Vivo DC, Norris DA, Bennett CF, Bishop KM (2016) Results from a phase 1 study of nusinersen (ISIS-SMN Rx) in children with spinal muscular atrophy, Neurology. https://doi.org/10.1212/WNL.0000000000002445.
Donev R, Newall A, Thome J, Sheer D (2007) A role for SC35 and hnRNPA1 in the determination of amyloid precursor protein isoforms. Mol Psychiatry 12:681–690. https://doi.org/10.1038/sj.mp.4001971
Bekenstein U, Soreq H (2013) Heterogeneous nuclear ribonucleoprotein A1 in health and neurodegenerative disease: From structural insights to post-transcriptional regulatory roles. Mol Cell Neurosci 56:436–446. https://doi.org/10.1016/j.mcn.2012.12.002
Berson A, Barbash S, Shaltiel G, Goll Y, Hanin G, Greenberg DS, Ketzef M, Becker AJ et al (2012) Cholinergic-associated loss of hnRNP-A/B in Alzheimer’s disease impairs cortical splicing and cognitive function in mice. EMBO Mol Med 4:730–742. https://doi.org/10.1002/emmm.201100995
Liu XY, Li HL, Bin Su J, Ding FH, Zhao JJ, Chai F, Li YX, Cui SC et al (2015) Regulation of RAGE splicing by hnRNP A1 and Tra2β-1 and its potential role in AD pathogenesis. J Neurochem 133:187–198. https://doi.org/10.1111/jnc.13069
Villa C, Fenoglio C, De Riz M, Clerici F, Marcone A, Benussi L, Ghidoni R, Gallone S et al (2011) Role of hnRNP-A1 and miR-590-3p in neuronal death: Genetics and expression analysis in patients with Alzheimer disease and frontotemporal lobar degeneration. Rejuvenation Res 14:275–281. https://doi.org/10.1089/rej.2010.1123
Jarrett JT, Lansbury PT (1993) Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell. 73:1055–1058. https://doi.org/10.1016/0092-8674(93)90635-4
Goedert M, Clavaguera F, Tolnay M (2010) The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci 33:317–325. https://doi.org/10.1016/j.tins.2010.04.003
Frost B, Diamond MI (2010) Prion-like mechanisms in neurodegenerative diseases. Nat Rev Neurosci 11:155–159. https://doi.org/10.1038/nrn2786
Brundin P, Melki R, Kopito R (2010) Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol 11:301–307. https://doi.org/10.1038/nrm2873
Zearfoss NR, Johnson ES, Ryder SP (2013) hnRNP A1 and secondary structure coordinate alternative splicing of mag. RNA 19:948–957. https://doi.org/10.1261/rna.036780.112
Jean-Philippe J, Paz S, Caputi M (2013) hnRNP A1: The Swiss Army knife of gene expression. Int J Mol Sci 14:18999–19024. https://doi.org/10.3390/ijms140918999
Lee EK, Kim HH, Kuwano Y, Abdelmohsen K, Srikantan S, Subaran SS, Gleichmann M, Mughal MR et al (2010) HnRNP C promotes APP translation by competing with FMRP for APP mRNA recruitment to P bodies. Nat Struct Mol Biol 17:732–739. https://doi.org/10.1038/nsmb.1815
Rajagopalan LE, Westmark CJ, Jarzembowski JA, Malter JS (1998) HnRNP C increases amyloid precursor protein (APP) production by stabilizing APP mRNA. Nucleic Acids Res 26:3418–3423. https://doi.org/10.1093/nar/26.14.3418
Rivera D, Fedele E, Marinari UM, Pronzato MA, Ricciarelli R (2015) Evaluating the role of hnRNP-C and FMRP in the cAMP-induced APP metabolism. BioFactors. 41:121–126. https://doi.org/10.1002/biof.1207
Mizukami K, Ishikawa M, Iwakiri M, Ikonomovic MD, Dekosky ST, Kamma H, Asada T (2005) Immunohistochemical study of the hnRNP A2 and B1 in the hippocampal formations of brains with Alzheimer’s disease. Neurosci Lett 386:111–115. https://doi.org/10.1016/j.neulet.2005.05.070
Ishikawa M, Mizukami K, Iwakiri M, Kamma H, Ikonomovic MD, Dekosky ST, Asada T (2004) Immunohistochemical study of hnRNP B1 in the postmortem temporal cortices of patients with Alzheimer’s disease. Neurosci Res 50:481–484. https://doi.org/10.1016/j.neures.2004.08.013
Ashraf GM, Ganash M, Athanasios A (2019) Computational analysis of non-coding RNAs in Alzheimer’s disease. Bioinformation. https://doi.org/10.6026/97320630015351.
Herrup K (2004) Divide and die: Cell cycle events as triggers of nerve cell death. J Neurosci 24:9232–9239. https://doi.org/10.1523/JNEUROSCI.3347-04.2004
Lee SW, Lee MH, Park JH, Kang SH, Yoo HM, Ka SH, Oh YM, Jeon YJ et al (2012) SUMOylation of hnRNP-K is required for p53-mediated cell-cycle arrest in response to DNA damage. EMBO J 31:4441–4452. https://doi.org/10.1038/emboj.2012.293
Yang F, Yi F, Han X, Du Q, Liang Z (2013) MALAT-1 interacts with hnRNP C in cell cycle regulation. FEBS Lett 587:3175–3181. https://doi.org/10.1016/j.febslet.2013.07.048
Moumen A, Masterson P, O’Connor MJ, Jackson SP (2005) hnRNP K: An HDM2 target and transcriptional coactivator of p53 in response to DNA damage. Cell 123:1065–1078. https://doi.org/10.1016/j.cell.2005.09.032
Mecocci P, MacGarvey U, Beal MF (1994) Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann Neurol 36:747–751. https://doi.org/10.1002/ana.410360510
Kitamura Y, Shimohama S, Kamoshima W, Matsuoka Y, Nomura Y, Taniguchi T (1997) Changes of p53 in the brains of patients with Alzheimer’s disease. Biochem Biophys Res Commun 232:418–421. https://doi.org/10.1006/bbrc.1997.6301
Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC (2011) Amyotrophic lateral sclerosis, 942–955. Lancet 377. https://doi.org/10.1016/S0140-6736(10)61156-7
McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ (2001) Clinical and pathological diagnosis of frontotemporal dementia: Report of the work group on Frontotemporal dementia and Pick’s disease. Arch Neurol 58:1803–1809. https://doi.org/10.1001/archneur.58.11.1803
Boylan K (2015) Familial amyotrophic lateral sclerosis. Neurol Clin. https://doi.org/10.1016/j.ncl.2015.07.001
Lashley T, Rohrer JD, Mead S, Revesz T (2015) Review: An update on clinical, genetic and pathological aspects of frontotemporal lobar degenerations. Neuropathol Appl Neurobiol. https://doi.org/10.1111/nan.12250
Ayala YM, Zago P, D’Ambrogio A, Xu YF, Petrucelli L, Buratti E, Baralle FE (2008) Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci. https://doi.org/10.1242/jcs.038950
Buratti E, Baralle FE (2010) The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol. https://doi.org/10.4161/rna.7.4.12205.
Ratti A, Buratti E (2016) Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J Neurochem. https://doi.org/10.1111/jnc.13625
Buratti E, Brindisi A, Giombi M, Tisminetzky S, Ayala YM, Baralle FE (2005) TDP-43 binds heterogeneous nuclear ribonucleoprotein a/B through its C-terminal tail: An important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem 280:37572–37584. https://doi.org/10.1074/jbc.M505557200
Ash PEA, Zhang YJ, Roberts CM, Saldi T, Hutter H, Buratti E, Petrucelli L, Link CD (2010) Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum Mol Genet. https://doi.org/10.1093/hmg/ddq230.
Kabashi E, Lin L, Tradewell ML, Dion PA, Bercier V, Bourgouin P, Rochefort D, Bel Hadj S, Durham HD, Vande Velde C, Rouleau GA, Drapeau P (2009) Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum Mol Genet. https://doi.org/10.1093/hmg/ddp534.
Liachko NF, Guthrie CR, Kraemer BC (2010) Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans model of TDP-43 proteinopathy. J Neurosci. https://doi.org/10.1523/JNEUROSCI.2911-10.2010.
Stallings NR, Puttaparthi K, Luther CM, Burns DK, Elliott JL (2010) Progressive motor weakness in transgenic mice expressing human TDP-43. Neurobiol Dis. https://doi.org/10.1016/j.nbd.2010.06.017
Wils H, Kleinberger G, Janssens J, Pereson S, Joris G, Cuijt I, Smits V, Ceuterick-De Groote C et al (2010) TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.0912417107
Xu YF, Zhang YJ, Lin WL, Cao X, Stetler C, Dickson DW, Lewis J, Petrucelli L (2011) Expression of mutant TDP-43 induces neuronal dysfunction in transgenic mice. Mol Neurodegener. https://doi.org/10.1186/1750-1326-6-73.
Highley JR, Kirby J, Jansweijer JA, Webb PS, Hewamadduma CA, Heath PR, Higginbottom A, Raman R, Ferraiuolo L, Cooper-Knock J, Mcdermott CJ, Wharton SB, Shaw PJ, Ince PG (2014) Loss of nuclear TDP-43 in amyotrophic lateral sclerosis (ALS) causes altered expression of splicing machinery and widespread dysregulation of RNA splicing in motor neurones. Neuropathol Appl Neurobiol. https://doi.org/10.1111/nan.12148.
Colombrita C, Onesto E, Buratti E, de la Grange P, Gumina V, Baralle FE, Silani V, Ratti A (2015) From transcriptomic to protein level changes in TDP-43 and FUS loss-of-function cell models. Biochim Biophys Acta - Gene Regul Mech. https://doi.org/10.1016/j.bbagrm.2015.10.015.
Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, Burberry A, Steinbaugh MJ, Gamage KK, Kirchner R, Moccia R, Cassel SH, Chen K, Wainger BJ, Woolf CJ, Eggan K (2019) ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci. https://doi.org/10.1038/s41593-018-0300-4.
Mori K, Lammich S, Mackenzie IRA, Forné I, Zilow S, Kretzschmar H, Edbauer D, Janssens J et al (2013) HnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol 125:413–423. https://doi.org/10.1007/s00401-013-1088-7
Lee YB, Chen HJ, Peres JN, Gomez-Deza J, Attig J, Štalekar M, Troakes C, Nishimura AL et al (2013) Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep 5:1178–1186. https://doi.org/10.1016/j.celrep.2013.10.049
Haeusler AR, Donnelly CJ, Periz G, Simko EAJ, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC et al (2014) C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 507:195–200. https://doi.org/10.1038/nature13124
Nahalka J (2019) The role of the protein–RNA recognition code in neurodegeneration, Cell Mol Life Sci. https://doi.org/10.1007/s00018-019-03096-3.
Conlon EG, Lu L, Sharma A, Yamazaki T, Tang T, Shneider NA, Manley JL (2016) The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. Elife. 5. https://doi.org/10.7554/eLife.17820.
Freibaum BD, Chitta RK, High AA, Taylor JP (2010) Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J Proteome Res 9:1104–1120. https://doi.org/10.1021/pr901076y
Moujalled D, James JL, Yang S, Zhang K, Duncan C, Moujalled DM, Parker SJ, Caragounis A et al (2015) Phosphorylation of hnRNP K by cyclin-dependent kinase 2 controls cytosolic accumulation of TDP-43. Hum Mol Genet 24:1655–1669. https://doi.org/10.1093/hmg/ddu578
Lee KH, Zhang P, Kim HJ, Mitrea DM, Sarkar M, Freibaum BD, Cika J, Coughlin M, Messing J, Molliex A, Maxwell BA, Kim NC, Temirov J, Moore J, Kolaitis RM, Shaw TI, Bai B, Peng J, Kriwacki RW, Taylor JP (2016) C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles, cell. https://doi.org/10.1016/j.cell.2016.10.002.
Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y, Lin S, Shneider NA, Monaghan J, Pandey UB, Pasinelli P, Ichida JK, Trotti D (2014) Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate invitro and invivo neuronal death, neuron. https://doi.org/10.1016/j.neuron.2014.12.010.
Balendra R, Isaacs AM (2018) C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat Rev Neurol. https://doi.org/10.1038/s41582-018-0047-2.
Leko MB, Župunski V, Kirincich J, Smilović D, Hortobágyi T, Hof PR, Šimić G (2019) Molecular mechanisms of neurodegeneration related to C9orf72 hexanucleotide repeat expansion. Behav Neurol. https://doi.org/10.1155/2019/2909168
Jovičić A, Paul JW, Gitler AD (2016) Nuclear transport dysfunction: A common theme in amyotrophic lateral sclerosis and frontotemporal dementia. J Neurochem:134–144. https://doi.org/10.1111/jnc.13642
Liu Q, Shu S, Wang RR, Liu F, Cui B, Guo XN, Lu CX, Li XG et al (2016) Whole-exome sequencing identifies a missense mutation in hnRNPA1 in a family with flail arm ALS. Neurology. 87:1763–1769. https://doi.org/10.1212/WNL.0000000000003256
Honda H, Hamasaki H, Wakamiya T, Koyama S, Suzuki SO, Fujii N, Iwaki T (2015) Loss of hnRNPA1 in ALS spinal cord motor neurons with TDP-43-positive inclusions. Neuropathology. 35:37–43. https://doi.org/10.1111/neup.12153
Davidson YS, Robinson AC, Flood L, Rollinson S, Benson BC, Asi YT, Richardson A, Jones M et al (2017) Heterogeneous ribonuclear protein E2 (hnRNP E2) is associated with TDP-43-immunoreactive neurites in semantic dementia but not with other TDP-43 pathological subtypes of Frontotemporal lobar degeneration. Acta Neuropathol Commun 5:54. https://doi.org/10.1186/s40478-017-0454-4
Gami-Patel P, Bandopadhyay R, Brelstaff J, Revesz T, Lashley T (2016) The presence of heterogeneous nuclear ribonucleoproteins in frontotemporal lobar degeneration with FUS-positive inclusions. Neurobiol Aging 46:192–203. https://doi.org/10.1016/j.neurobiolaging.2016.07.004
Gittings LM, Foti SC, Benson BC, Gami-Patel P, Isaacs AM, Lashley T (2019) Heterogeneous nuclear ribonucleoproteins R and Q accumulate in pathological inclusions in FTLD-FUS. Acta Neuropathol Commun. https://doi.org/10.1186/s40478-019-0673-y.
Maniecka Z, Polymenidou M (2015) From nucleation to widespread propagation: A prion-like concept for ALS. Virus Res 207:94–105. https://doi.org/10.1016/j.virusres.2014.12.032
Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B et al (2013) Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495:467–473. https://doi.org/10.1038/nature11922
Steinman L (1996) Multiple sclerosis: A coordinated immunological attack against myelin in the central nervous system. Cell. 85:299–302. https://doi.org/10.1016/S0092-8674(00)81107-1
Reindl M, Linington C, Brehm U, Egg R, Dilitz E, Deisenhammer F, Poewe W, Berger T (1999) Antibodies against the myelin oligodendrocyte glycoprotein and the myelin basic protein in multiple sclerosis and other neurological diseases: A comparative study. Brain. 122:2047–2056. https://doi.org/10.1093/brain/122.11.2047
Levin MC, Lee S, Gardner LA, Shin Y, Douglas JN, Cooper C (2013) Autoantibodies to non-myelin antigens as contributors to the pathogenesis of multiple sclerosis. J Clin Cell Immunol. 4. https://doi.org/10.4172/2155-9899.1000148.
Kamma H, Portman DS, Dreyfuss G (1995) Cell type-specific expression of hnRNP proteins. Exp Cell Res 221:187–196. https://doi.org/10.1006/excr.1995.1366
Kamma H, Horiguchi H, Wan L, Matsui M, Fujiwara M, Fujimoto M, Yazawa T, Dreyfuss G (1999) Molecular characterization of the hnRNP A2/B1 proteins: Tissue-specific expression and novel isoforms. Exp Cell Res 246:399–411. https://doi.org/10.1006/excr.1998.4323
Yukitake M, Sueoka E, Sueoka-Aragane N, Sato A, Ohashi H, Yakushiji Y, Saito M, Osame M et al (2008) Significantly increased antibody response to heterogeneous nuclear ribonucleoproteins in cerebrospinal fluid of multiple sclerosis patients but not in patients with human T-lymphotropic virus type I-associated myelopathy/tropical spastic paraparesis. J Neuro-Oncol 14:130–135. https://doi.org/10.1080/13550280701883840
Lee S, Xu L, Shin Y, Gardner L, Hartzes A, Dohan FC, Raine C, Homayouni R et al (2011) A potential link between autoimmunity and neurodegeneration in immune-mediated neurological disease. J Neuroimmunol 235:56–69. https://doi.org/10.1016/j.jneuroim.2011.02.007
Lee S, Levin M (2014) Novel somatic single nucleotide variants within the RNA binding protein hnRNP A1 in multiple sclerosis patients, F1000Research. https://doi.org/10.12688/f1000research.4436.2.
Douglas JN (2013) Antibodies to an intracellular antigen penetrate neuronal cells and cause deleterious effects. J Clin Cell Immunol 04:1–7. https://doi.org/10.4172/2155-9899.1000134
Sueoka E, Yukitake M, Iwanaga K, Sueoka N, Aihara T, Kuroda Y (2004) Autoantibodies against heterogeneous nuclear ribonucleoprotein B1 in CSF of MS patients. Ann Neurol 56:778–786. https://doi.org/10.1002/ana.20276
Douglas JN, Gardner LA, Salapa HE, Lalor SJ, Lee S, Segal BM, Sawchenko PE, Levin MC (2016) Antibodies to the RNA-binding protein hnRNP A1 contribute to neurodegeneration in a model of central nervous system autoimmune inflammatory disease. J. Neuroinflammation. 13. https://doi.org/10.1186/s12974-016-0647-y.
Saarela J, Kallio SP, Chen D, Montpetit A, Jokiaho A, Choi E, Asselta R, Bronnikov D et al (2006) PRKCA and multiple sclerosis: Association in two independent populations. PLoS Genet 2:0364–0375. https://doi.org/10.1371/journal.pgen.0020042
Paraboschi EM, Rimoldi V, Soldà G, Tabaglio T, Dall’Osso C, Saba E, Vigliano M, Salviati A et al (2014) Functional variations modulating PRKCA expression and alternative splicing predispose to multiple sclerosis. Hum Mol Genet 23:6746–6761. https://doi.org/10.1093/hmg/ddu392
Engel AG, Shen X-M, Selcen D, Sine SM (2015) Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. Lancet Neurol 14:420–434. https://doi.org/10.1016/S1474-4422(14)70201-7
Ohno K, Rahman MA, Nazim M, Nasrin F, Lin Y, Takeda JI, Masuda A (2017) Splicing regulation and dysregulation of cholinergic genes expressed at the neuromuscular junction. J Neurochem 142:64–72. https://doi.org/10.1111/jnc.13954
Masuda A, Shen XM, Ito M, Matsuura T, Engel AG, Ohno K (2008) hnRNP H enhances skipping of a nonfunctional exon P3A in CHRNA1 and a mutation disrupting its binding causes congenital myasthenic syndrome. Hum Mol Genet 17:4022–4035. https://doi.org/10.1093/hmg/ddn305
Bian Y, Masuda A, Matsuura T, Ito M, Okushin K, Engel AG, Ohno K (2009) Tannic acid facilitates expression of the polypyrimidine tract binding protein and alleviates deleterious inclusion of CHRNA1 exon P3A due to an hnRNP H-disrupting mutation in congenital myasthenic syndrome. Hum Mol Genet 18:1229–1237. https://doi.org/10.1093/hmg/ddp023
Rahman MA, Masuda A, Ohe K, Ito M, Hutchinson DO, Mayeda A, Engel AG, Ohno K (2013) HnRNP L and hnRNP LL antagonistically modulate PTB-mediated splicing suppression of CHRNA1 pre-mRNA. Sci Rep 3. https://doi.org/10.1038/srep02931.
DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL et al (1996) The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell 85:501–512. https://doi.org/10.1016/S0092-8674(00)81251-9
Kim N, Burden SJ (2008) MuSK controls where motor axons grow and form synapses. Nat Neurosci 11:19–27. https://doi.org/10.1038/nn2026
Zhou H, Glass DJ, Yancopoulos GD, Sanes JR (1999) Distinct domains of MuSK mediate its abilities to induce and to associate with postsynaptic specializations. J Cell Biol 146:1133–1146. https://doi.org/10.1083/jcb.146.5.1133
Roszmusz E, Patthy A, Trexler M, Patthy L (2001) Localization of disulfide bonds in the frizzled module of Ror1 receptor tyrosine kinase. J Biol Chem 276:18485–18490. https://doi.org/10.1074/jbc.M100100200
Gordon LR, Gribble KD, Syrett CM, Granato M (2012) Initiation of synapse formation by Wnt-induced MuSK endocytosis. Development. 139:1023–1033. https://doi.org/10.1242/dev.071555
Nasrin F, Rahman MA, Masuda A, Ohe K, Takeda JI, Ohno K (2014) HnRNP C, YB-1 and hnRNP L coordinately enhance skipping of human musk exon 10 to generate a wnt-insensitive musk isoform. Sci Rep 4. https://doi.org/10.1038/srep06841.
Kimbell LM, Ohno K, Engel AG, Rotundo RL (2004) C-terminal and heparin-binding domains of collagenic tail subunit are both essential for anchoring acetylcholinesterase at the synapse. J Biol Chem 279:10997–11005. https://doi.org/10.1074/jbc.M305462200
Ohno K, Ohkawara B, Ito M, Engel AG (2014) Molecular genetics of congenital myasthenic syndromes, in: ELS.
Rahman MA, Azuma Y, Nasrin F, Takeda JI, Nazim M, Bin Ahsan K, Masuda A, Engel AG, Ohno K (2015) SRSF<inf>1</inf> and hnRNP H antagonistically regulate splicing of COLQ exon 16 in a congenital myasthenic syndrome Sci Rep 5. https://doi.org/10.1038/srep13208.
Leehey MA (2009) Fragile X-associated tremor/ataxia syndrome: clinical phenotype, diagnosis, and treatment. J Investig Med 57(8):830–836. https://doi.org/10.2310/JIM.0b013e3181af59c4
Sofola OA, Jin P, Qin Y, Duan R, Liu H, de Haro M, Nelson DL, Botas J (2007) RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG Premutation repeat-induced neurodegeneration in a drosophila model of FXTAS. Neuron. 55:565–571. https://doi.org/10.1016/j.neuron.2007.07.021
Iwahashi CK, Yasui DH, An HJ, Greco CM, Tassone F, Nannen K, Babineau B, Lebrilla CB et al (2006) Protein composition of the intranuclear inclusions of FXTAS. Brain. 129:256–271. https://doi.org/10.1093/brain/awh650
He F, Krans A, Freibaum BD, Paul Taylor J, Todd PK (2014) TDP-43 suppresses CGG repeat-induced neurotoxicity through interactions with HnRNP A2/B1. Hum Mol Genet 23:5036–5051. https://doi.org/10.1093/hmg/ddu216
Tan H, Poidevin M, Li H, Chen D, Jin P (2012) MicroRNA-277 modulates the neurodegeneration caused by fragile X premutation rCGG repeats. PLoS Genet 8. https://doi.org/10.1371/journal.pgen.1002681.
Tan H, Qurashi A, Poidevin M, Nelson DL, Li H, Jin P (2012) Retrotransposon activation contributes to fragile X premutation rCGG-mediated neurodegeneration. Hum Mol Genet 21:57–65. https://doi.org/10.1093/hmg/ddr437
Sellier C, Rau F, Liu Y, Tassone F, Hukema RK, Gattoni R, Schneider A, Richard S et al (2010) Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J 29:1248–1261. https://doi.org/10.1038/emboj.2010.21
Modrek B, Lee C (2002) A genomic view of alternative splicing. Nat Genet 30:13–19. https://doi.org/10.1038/ng0102-13
Lemmens R, Moore MJ, Al-Chalabi A, Brown RH, Robberecht W (2010) RNA metabolism and the pathogenesis of motor neuron diseases. Trends Neurosci 33:249–258. https://doi.org/10.1016/j.tins.2010.02.003
Acknowledgments
TL is supported by an Alzheimer’s Research UK senior fellowship. We thank Alexander Bampton for producing the figures for the manuscript. The Queen Square Brain Bank is supported by the Reta Lila Weston Institute of Neurological Studies, UCL Institute of Neurology.
Author information
Authors and Affiliations
Contributions
YL and TL undertook a literature review and drafted the initial manuscript and drafted the figures. All authors were involved in producing the figures and editing the manuscript.
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Low, YH., Asi, Y., Foti, S.C. et al. Heterogeneous Nuclear Ribonucleoproteins: Implications in Neurological Diseases. Mol Neurobiol 58, 631–646 (2021). https://doi.org/10.1007/s12035-020-02137-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-02137-4