
Abstract
Frontotemporal Lobar Degeneration (FTLD) encompasses certain related neurodegenerative disorders which alter personality and cognition. Heterogeneous ribonuclear proteins (hnRNPs) maintain RNA metabolism and changes in their function may underpin the pathogenesis of FTLD. Immunostaining for hnRNP E2 was performed on sections of frontal and temporal cortex with hippocampus from 80 patients with FTLD, stratified by pathology into FTLD-tau and FTLD-TDP type A, B and C subtypes, and by genetics into patients with C9orf72 expansions, MAPT or GRN mutations, or those with no known mutation, and on 10 healthy controls. Semi-quantitative analysis assessed hnRNP staining in frontal and temporal cortex, and in dentate gyrus (DG) of hippocampus, in the different pathology and genetic groups. We find that hnRNP E2 immunostaining detects the TDP-43 positive dystrophic neurites (DN) within frontal and temporal cortex, and the neuronal cytoplasmic inclusions (NCI) seen in DG granule cells, characteristic of patients with Semantic Dementia (SD) and type C TDP-43 pathology, but did not detect TDP-43 or tau inclusions in any of the other pathological or genetic variants of FTLD. Double immunofluorescence for hnRNP E2 and TDP-43 showed most TDP-43 immunopositive DN to contain hnRNP E2. Present findings indicate an association between TDP-43 and hnRNP E2 which might underlie the pathogenetic mechanism of this form of FTLD.
Similar content being viewed by others

Introduction
Frontotemporal Lobar Degeneration (FTLD) is a clinically, pathologically and genetically heterogeneous disorder affecting principally the frontal and temporal lobes of the brain. Three major clinical syndromes are recognised [34]. One syndrome, behavioural variant frontotemporal dementia (bvFTD), is characterised by changes in behaviour and personality and accounts for around 75% of all cases of FTLD, whereas the other two syndromes are disorders of language [34]. Semantic dementia (SD) (also known as semantic variant of primary progressive aphasia (svPPA)) is a disorder characterized by loss of conceptual knowledge of the meaning of words and objects [14, 34], whereas Progressive Non-Fluent Aphasia (PNFA) (also known as nfvPPA) is represented by an inability to construct language such that speech becomes hesitant and stuttering, becoming grammatically and contextually incorrect [14, 34]. The amyotrophic lateral sclerosis (ALS) form of motor neurone disease (MND) is seen in about 15% of patients with bvFTD, but is only rarely combined with either SD or PNFA [32].
Three different pathologies, characterised by abnormal neuronal, and sometimes glial, accumulations of aggregated proteins, are seen. Neuronal intracytoplasmic inclusions (NCI), composed of the microtubule associated protein tau, occur in about 45% cases as neurofibrillary tangle-like structures, or more rounded inclusions known as Pick bodies [33] and termed FTLD-tau [19]. The RNA and DNA binding protein, TDP-43, is present within NCI, neuritic processes (dystrophic neurites, DN) or neuronal intranuclear inclusions (NII) in about 50% of cases [2, 7, 26]. The relative proportions of NCI, DN and NII provide a neuropathological classification of FTLD-TDP subtypes [19]. FTLD-TDP subtype A is applied when NCI and short DN are both commonly present, mostly in outer cortical laminae, type B when NCI present throughout all cortical layers numerically predominate over DN, type C when long thick DN are present throughout all cortical layers and predominate over NCI and type D when NII are most common type of pathological change [19]. Most of the remaining 5% cases show NCI composed of the protein, Fused in Sarcoma (FUS), and are known as FTLD-FUS [19].
TDP-43 and FUS are heterogeneous nuclear riboproteins (hnRNP) [5, 30] and serve as RNA-splicing and transcription regulators, shuttling between nucleus and cytoplasm, thereby controlling cellular levels of protein synthesis. In the nucleus, TDP-43 binding encourages RNA stability, whereas in the cytoplasm it associates with stress granules and non-coding RNAs for post-transcriptional metabolism of RNA and transport. In FTLD-TDP there is a ‘clearing’ of normal physiological TDP-43 from the nucleus with its accumulation within the cytoplasm as NCI, DN or NII. However, the precise mechanism(s) directing this pathological change remain unclear.
Previous studies have pointed to specific interactions between particular hnRNPs and the pathological inclusions of FTLD. For example, we [9] and others [3, 23, 24] have shown that hnRNP A3 is present in the aggregates of dipeptide repeat proteins (DPR) in FTLD patients bearing expansions in C9orf72 gene. Elsewhere, Gami-Patel and colleagues reported the presence of various hnRNPs, but especially hnRNP A1, within NCI in patients with the Neuronal Intermediate Filament Inclusion Body Disease form of FTLD-FUS [12].
TDP-43 is a stress responsive protein, and the TDP-43 aggregates in FTLD-TDP are thought to arise from stress granules [6, 18, 37]. Stress granules are transient cytoplasmic structures composed of mixed protein-RNA complexes, formed in response to cellular stress and believed to act as a sorting station, triaging mRNAs and sequestering transcripts not needed for coping with the stress [10]. Their composition and morphology varies according to the cell and stress type, but are generated by a reversible aggregation of prion-like core components such as the T-cell intracellular antigen-1 (TIA-1) and the poly A binding protein 1 (PABP1) [13] in order to regulate mRNA metabolism and protein translation [1, 18]. Biochemical studies have shown that TDP-43 associates with stress granules by interacting with TIA-1 [18]. Furthermore, using a yeast two-hybrid screen against TIA-1, hnRNP E2 was reported to be present in stress granules and processing bodies in HeLa cells, and an interaction between hnRNP E2 and TIA-1 was demonstrated by double immunofluorescence [11]. Finally, it has been reported that TDP-43 pathological inclusions in patients with bvFTD and FTLD-TDP pathology, and in others with ALS, co-localise with multiple markers of stress granules including TIA-1 [18]. Hence, it seems likely that both TDP-43 and hnRNP-E2 can be incorporated into stress granules under stress conditions.
With this in mind, we sought possible relationships between hnRNP E2 and the TDP-43 pathological inclusions of FTLD since, because of their shared links with stress granules, it is plausible that hnRNP E2 might be a component of the TDP-43 pathological inclusions that form within neuronal cell bodies (NCI) and processes (DN). We find that hnRNP E2 immunostaining co-localises with TDP-43 pathological changes, but only in patients with SD and type C TDP-43 histology. Collectively, present and previous [3, 9, 23, 24] data suggest that although pathological accumulations of TDP-43 or FUS are hallmark characteristics of FTLD, the fundamental biological mechanisms leading to these molecular end-points may differ between pathological subtypes.
Materials and methods
Patients
The study consisted of 2 groups comprising 90 subjects in total. One group had been recruited through Manchester Brain Bank (MBB), 54 with a clinical diagnosis of FTLD (30 males, 24 females; cases #1-54), and 10 healthy control subjects (3 males, 7 females; cases #55-64) (Table 1). The brains of these patients had been consecutively acquired by MBB over the years 1986 to present. All patients were from the North West of England and North Wales, and tissues were obtained through appropriate consenting procedures for the collection and use of the human brain tissues. All patients fulfilled relevant clinical diagnostic criteria [14, 25, 31], having been investigated longitudinally within a specialist dementia clinic using the Manchester Neuropsychological Profile (Man-NP) [35, 36] to determine and characterise the nature of their dementia.
The other group was recruited through Queens Square Brain Bank (QSBB) and comprised 26 patients with a clinical diagnosis of FTLD (12 males, 14 females; cases #65-90) (Table 1). The brains of these patients had been consecutively acquired by QSBB over the years 2004 to present. All patients were from London and South of England, and tissues were obtained through appropriate consenting procedures for the collection and use of the human brain tissues. Again, all patients fulfilled relevant clinical diagnostic criteria [14, 25, 31] having been referred to the Dementia Research Unit, Queen Square, London.
Of the combined 80 FTLD patients, 36 had been clinically diagnosed with bvFTD (17 males, 19 females; cases #1-6,13,14,17-20,23-25,44-54,65,68-75,79), 13 with bvFTD + MND or MND alone (9 males, 4 females; cases #7-10,26-33,76-78), 9 with PNFA (4 males, 5 females; cases #11,12,15,16,21,22,66,67,87) and 20 with SD (12 males, 8 females; cases #34-43, 80-86, 88-90) (Table 1). Pathologically, the FTLD group comprised 30 patients with FTLD-TDP type A (cases #1-5,11-25, 65-74), 18 with FTLD-TDP type B (cases #6-10,26-33,75-79), 21 with FTLD-TDP type C (cases #34-43,80-90), 11 with FTLD-tau (cases #44-54) (Table 1). Furthermore, within the FTLD group there were 16 patients with expansions in C9orf72 (cases #1-10,65-69,75), 10 with GRN mutations (cases #11-19,74), 1 patient with TBK1 mutation (case #70), 11 patients with intronic mutations in MAPT (cases #44-54) and 42 without known mutation (cases #20-43,71-73,76-90) (Table 1).
Histological methods
Standard blocks of frontal (BA 8/9) and temporal (BA21/22) lobe, the latter to include the posterior hippocampus at the level of the geniculate bodies, were cut from the formalin fixed brains. Where possible, and in order to preserve maximum antigenicity, the original blocks taken for diagnosis were employed since these had been cut from brains which had been fixed in formalin for no more than 3-6 months from the time of acquisition. However, it was not possible to do this in all instances, especially in respect of some of the more long-standing cases (ie those acquired before 2007) where this block was no longer available and new blocks had to be cut from 20 cases for the purpose of this study (see Table 1 for those cases where new blocks were cut).
Paraffin sections were cut from these blocks at a thickness of 6 μm. Preliminary titration experiments were performed at dilutions 1:100 to 1:3000 on 5 randomly chosen cases, one from each pathological subgroup, in order to determine optimal specific nuclear and DN/NCI immunostaining for each antibody. When dilutions for hnRNP E2 staining less than what was optimal for nuclear and inclusion body staining were employed, immunostaining for nuclei and DN/NCI was still observed, but less strongly so, as would be expected given the lower antibody concentrations. Subsequently, all antibodies were employed at these optimised dilutions (as specified below) in a standard IHC protocol, as described previously [7,8,9, 22]. The following antibodies were employed: hnRNP E2 (also known as PCBP2) (mouse monoclonal, Santa Cruz, 23G: sc101136, 1:100 and Novus, mouse monoclonal H00005094-M07, 5F12 clone, 1:500), non-phosphorylated TDP-43 (rabbit polyclonal, 10,782-2-AP antibody, Proteintech, Manchester, UK, 1:3000) and phosphorylated TDP (pS409/410-2 antibody, Cosmo Biotech Ltd., Tokyo, Japan, 1:2000) and tau (mouse monoclonal, AT8, Innogenetics, Antwerp, Belgium, 1:750) proteins. Both the Santa Cruz and Novus hnRNP E2 antibodies are raised against recombinant hnRNP E2 of human origin, and on western blotting detect a protein with molecular mass of around 40 kDa. For all antibodies employed, antigen unmasking was performed by pressure cooking in citrate buffer (pH 6.0, 10 mM) over a 30-min period to include warming and cooling times, reaching 123 degrees Celsius for 30 s, and >15 psi pressure.
Double immunohistochemical staining was performed using phosphorylated TDP-43 antibody (pS409/410-2 antibody, Cosmo Biotech Ltd., Tokyo, Japan, 1:2000) and hnRNP E2 (Novus, 5F12, 1:500) antibody to investigate the co-localisation of the two proteins. Sections were cut, pre-treated and incubated in primary antibody as described above. Two Alexa Fluor secondary antibodies (Alexa Fluor 488 and Alexa Fluor 568; Molecular Probes, 1:300, with incubation for 1 hour at room temperature) were used to visualise sites of protein deposition. 4′-6-diamidino-2-phenylindol (DAPI) was used for nuclear counterstaining.
Pathological assessment
Immunostained sections were examined microscopically for the appearance of intracellular distribution of staining within neurones of the temporal cortex, dentate gyrus and CA4 region of the hippocampus. These regions were chosen since it was known from previous work [7, 33] that the temporal cortex and dentate gyrus of the hippocampus are involved in all forms of FTLD-TDP or tau pathology in those patients with MAPT mutation [29]. Moreover, the CA4 region of the hippocampus was included because this is one of the principal regions affected by DPR pathology in patients with expansions in C9orf72 [8, 22, 23]. The degree of neuronal nuclear and/or cytoplasmic hnRNP E2 immunostaining in each region was scored semi-quantitatively [9] at an objective magnification of ×25 (overall microscope magnification of ×250) employing the following rating scale:
-
0 = No staining present.
-
0.5 = rare (ie 1-5) cells per section showing weak nuclear and/or cytoplasmic staining.
-
1 = few (1-5) cells showing weak nuclear and/or cytoplasmic staining per ×250 microscope field.
-
2 = moderate number (5-10) cells showing moderate nuclear and/or cytoplasmic staining per ×250 microscope field.
-
3 = more than 10 cells showing strong nuclear and/or cytoplasmic staining per ×250 microscope field.
The severity of hnRNP E2-immunoreactive, and TDP-43-immunoreactive, inclusion body immunostaining (ie NCI and DN) in frontal and temporal cortex, and in granule cells of the dentate gyrus of the hippocampus, was separately graded at an objective magnification of ×25 (overall microscope magnification of ×250), employing the following rating scale:
-
0 = no inclusions present.
-
0.5 = rare (ie 1-5 inclusions per section).
-
1 = few (ie 1-5 inclusions per ×250 microscope field).
-
2 = moderate (ie 5-10 inclusions per ×250 microscope field).
-
3 = many (ie 10-50 inclusions per ×250 microscope field).
-
4 = very many (ie more than 50 inclusions per ×250 microscope field).
Scoring of staining was performed by a single observer (DMAM) blinded to clinical, histopathological and genetic status. Previous use of this particular scoring system has shown robust agreement in assessments when employed by both highly and lesser experienced observers [9].
Double immunolabelled sections were viewed with a Leica TCS4D confocal microscope using a 3-channel scan head and argon/krypton laser.
Statistical analysis
Rating data was entered into an excel spreadsheet and analyzed using Statistical Package for Social Sciences (SPSS) software (version 17.0). The 80 FTLD patients were stratified according to genetic and pathological subtype for statistical analysis of the effect of each mutation and underlying pathology on the degree and pattern of hnRNP E2 staining. Comparisons of semi-quantitative scores for the intensity of neuronal hnRNP E2 immunostaining in nucleus and cytoplasm of neurones of the frontal and temporal cortex, and dentate gyrus of the hippocampus, with respect to pathological type or genetic mutation, were performed using Kruskal-Wallis test with post-hoc Mann-Whitney test where Kruskal-Wallis yielded a significant difference between antibody staining scores. Comparisons of semi-quantitative scores for the severity of hnRNP E2- and TDP-43-immunoreactive inclusions in frontal and temporal cortex, and dentate gyrus of the hippocampus, with respect to pathological subtype, were also performed using Kruskal-Wallis test. Comparison of scores for the severity of hnRNP E2- and TDP-43-immunoreactive inclusions in the same cases was made using Wilcoxon matched pairs test. Group comparisons of age at onset, age at death, post mortem delay interval and duration of illness were made using ANOVA with post hoc Tukey test. In all instances, significance levels were set at p < 0.05.
Results
Demographic comparisons
Comparison of the 4 FTLD pathology patient groups showed significant differences in mean age at onset of disease (F3,76 = 4.9, p = 0.004), mean age at death (F3,76 = 7.0, p < 0.001) and duration of illness did differ (F3,76 = 8.3, p < 0.001). Patients with FTLD-tau had an earlier age at onset than those with FTLD-TDP type A (p = 0.002), type B (p = 0.016) and type C (p = 0.012) pathology (Table 2) and an earlier age at death than those with FTLD-TDP type A (p = 0.014) and type C (p < 0.001) pathology, but not those with FTLD-TDP type B (p = 0.449) pathology. None of the 3 FTLD-TDP groups differed from each other in terms of age at onset, though patients with FTLD-TDP type B pathology died at an earlier age than those with FTLD-TDP type C pathology (p = 0.015). Patients with FTLD-TDP type B had a shorter disease duration than those with FTLD-TDP type C (p = 0.001) and those with FTLD-tau (p = 0.040); those with FTLD-TDP type A pathology also had a shorter disease duration than those with FTLD-TDP type C (p = 0.006), but there were no other differences between the other sub-types (Table 2). The healthy control group was also significantly older at death (p < 0.001) than each of the FTLD subgroups (Table 2).
Comparison of the 4 FTLD genetic patient groups also showed significant differences in mean age at onset of disease (F3,75 = 5.1, p = 0.003) and mean age at death (F3,75 = 5.0, p = 0.003), though duration of illness did not differ significantly (F3,75 = 1.2, p = 0.329). Patients with MAPT mutation had an earlier age at onset than those with GRN mutation (p = 0.050) and those without known mutation (p = 0.001), but not those with C9orf72 expansion (p = 0.088). The other groups did not significantly differ from each other. Mean age at death was significantly earlier in MAPT than the no mutation group (p = 0.002), but otherwise there were no significant differences between all other groups (Table 2).
There were no significant differences between mean post mortem intervals for each pathological (F4,83 = 1.0, p = 0.401) or genetic (F4,73 = 1.6, p = 0.181) group (see Table 2), nor were there any significant correlations between each pathological measure and post mortem interval (p = 0.351-0.993).
TDP-43 immunostaining
TDP-43- and tau-immunostaining was employed to classify the 80 FTLD patients into their respective pathological subgroups (FTLD-TDP subtypes A, B or C and FTLD-tau) according to the form and distribution of the TDP-43 or tau-immunoreactive inclusions (NCI, DN) present (see [19] for criteria). Median scores (with interquartile range) from TDP-43 immunostaining of pathological inclusions and neuronal nuclei and cytoplasm, derived from semi-quantitative scoring are shown in Table 3. As expected, given the inclusion of FTLD-tau group, the degree of TDP-43 inclusion body immunostaining (irrespective of whether these were in the form of NCI or DN, or both) differed between the 4 FTLD pathological groups in both the frontal (χ2 = 34.1, p < 0.001) and temporal (χ2 = 36.8, p < 0.001) cortex, and in the dentate gyrus (χ2 = 35.3, p < 0.001) with all 3 FTLD-TDP subtypes differing significantly from FTLD-tau group (p < 0.001 in every instance). However, there were no significant differences in the overall degree of TDP-43 immunostaining between FTLD-TDP type A, type B or type C subgroups.
hnRNP E2 immunostaining
When using Santa Cruz hnRNP E2 antibody, 58/80 FTLD patients, and 8/10 controls, showed a variable level of cellular staining within nerve cells of the frontal and temporal cortex, and in granule cells of the dentate gyrus of hippocampus. When present, this was usually observed in both nucleus and cytoplasm, and ranged in intensity from very weak to strong. Conversely, 22 FTLD patients and 2 controls showed no nuclear or cytoplasmic staining at all in any region examined. Twenty of these cases had been stored for long periods (in excess of 10 years) in formalin fixation before blocks had been taken for this study, whereas the brains of those cases based on diagnostic blocks had been stored for a shorter time in formalin fixation before blocking (3-6 months), and all these, except 4 cases, showed variable degrees of nuclear and cytoplasmic staining. There did not appear to be any pathological or genetic group preference for presence/absence of this kind of staining. Hence, some degree of positive (nuclear or cytoplasmic, or both) staining was seen in 22/30 (73%) patients with FTLD-TDP type A histology, 11/18 (61%) patients with FTLD-TDP type B, 16/21 (76%) patients with FTLD-TDP type C, 9/11 (81%) patients with FTLD-tau and 8/10 (80%) controls. According to FTLD genetics, some positive degree of positive staining was seen in 9/16 (56%) patients with C9orf72 expansion, 7/10 (70%) patients with GRN mutation, 32/42 (76%) patients with no mutation and 9/11 (80%) patients with MAPT mutation. There were no significant differences between the proportions of FTLD patients showing some degree of hnRNP E2 immunostaining when stratified either by pathological (χ2 = 2.09, p = 0.718) or genetic (χ2 = 1.98, p = 0.577) groupings.
Median scores (with interquartile range) from hnRNP E2 immunostaining of pathological inclusions and neuronal nuclei and cytoplasm, derived from semi-quantitative scoring, for each of the pathological groups are shown in Table 3. Statistical analysis showed no significant differences in scores for nuclear and cytoplasmic hnRNP E2 staining in frontal cortex (χ2 = 5.8, p = 0.120 and χ2 = 2.5, p = 0.479, respectively), temporal cortex (χ2 = 6.7, p = 0.080 and χ2 = 3.6, p = 0.312, respectively) or dentate gyrus (χ2 = 5.7, p = 0.128 and χ2 = 2.7, p = 0.444, respectively). Similarly, no significant differences in scores for nuclear and cytoplasmic hnRNP E2 staining in the 4 genetic groups were seen in frontal cortex (χ2 = 4.0, p = 0.400 and χ2 = 1.1, p = 0.891, respectively), temporal cortex (χ2 = 3.6, p = 0.463 and χ2 = 1.1, p = 0.889, respectively) or dentate gyrus (χ2 = 4.5, p = 0.338 and χ2 = 1.2, p = 0.882, respectively).
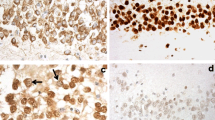
DN with morphology akin to that seen on TDP-43 immunostaining (Fig. 1a and b) were seen on hnRNP E2 immunostaining (Fig. 1d and e) in the frontal (Fig. 1a and c) and temporal (Fig. 1b and e) cortex of 15/21 FTLD-TDP type C cases. In the same cases similar rounded, solid-appearing NCI seen on TDP-43 immunostaining in dentate gyrus granule cells (Fig. 1c) were also seen on hnRNP E2 immunostaining (Fig. 1f). Only FTLD-TDP type C cases had pathological inclusions labelled by hnRNP E2.There was no immunolabelling by hnRNP E2 of TDP-43 or tau inclusions in FTLD-TDP type A or type B cases or those with FTLD-tau. Interestingly, there was no apparent loss of physiological immunostaining of the nucleus for hnRNP E2 in those cells of the dentate gyrus bearing NCI (Fig. 1f), in contrast to that seen with TDP-43 where there was loss of normal nuclear staining in those cells containing TDP-43 immunoreactive NCI (Fig. 1c).

Dystrophic neurites seen on TDP-43 immunostaining (arrowed in a and b) were similarly seen on hnRNP E2 immunostaining (arrowed in d and e), in frontal (a and d) and temporal (b and e) cortex in FTLD-TDP type C cases. In the same cases rounded, solid-appearing NCI (arrowed) seen on TDP-43 immunostaining in dentate gyrus granule cells (c) were also seen in TDP-43 immunostaining (f). Immunoperoxidase-haematoxylin; microscope magnification ×400
There were no clinical or neuropathological differences between the 15 hnRNP E2 positive cases (cases #37,40-43,81-90) and the 6 hnRNP E2 negative cases (cases #34-36,38,39,80). Moreover, there were no differences in age at onset (p = 0.542), age at death (p = 0.966), duration of illness (p = 0.405) or post mortem interval (p = 0.285) between the 15 positive and 6 negative cases. The only difference between the two groups was that the negative FTLD-TDP type C cases employed tissue blocks that had been newly cut for this study from long-standing (before 2007) archived fixed tissues as the original diagnostic blocks were no longer available for study. All other cases employed the original diagnostic blocks cut after 3-6 months fixation. Furthermore, within those FTLD cases bearing expansions in C9orf72 gene, there was no immunostaining of the TDP-43-negative, p62-positive DPR inclusions within neurones of frontal and temporal cortex, or dentate gyrus granule cells and CA4 neurones of the hippocampus.
Consequently, semi-quantitative analysis within 5 pathological groups showed a significant difference in scores for hnRNP E2 positive inclusions in the frontal cortex (χ2 = 46.6, p < 0.001), temporal cortex (χ2 = 50.6, p < 0.001) and dentate gyrus (χ2 = 50.7, p < 0.001), with the number of inclusions in both areas being significantly different in FTLD-TDP type C cases than all other pathological subtypes (p < 0.001 in every instance). This result would be expected given the observations that only in FTLD-TDP type C cases were there pathological inclusions immunostained by hnRNP E2.
The results of immunostaining with the Novus hnRNP E2 antibody appeared broadly similar to those obtained with Santa Cruz antibody on all cases of FTLD-TDP type C, and selected cases of the other pathological subtypes, where both antibodies were employed, both with respect to the intensity of neuronal staining and severity of inclusion body staining. Comparison of semi-quantitative scores for the intensity of nuclear and cytoplasmic staining, and inclusion body staining, on all 21 cases of FTLD-TDP type C showed that Novus antibody immunostained nuclei less intensely than Santa Cruz antibody (frontal cortex p = 0.008, temporal cortex p = 0.005; dentate gyrus p = 0.005), but there were no significant differences between the antibodies for level of cytoplasmic staining (p = 0.617, p = 0.120 and p = 0.104, respectively). There were no significant differences between scores for inclusion body staining with both antibodies for frontal cortex (p = 0.054) and temporal cortex (p = 0.655) with only a trend for scores in dentate gyrus to be greater (p = 0.023) for Novus antibody.
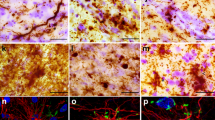
In general, the number of DN or NCI visualised on hnRNP E2 immunostaining appeared fewer than those seen in TDP-43 immunostaining (compare Fig. 1a with d, and b with e and c with f). Indeed, comparison of scores for TDP-43- and hnRNP E2- immunostaining of DN in FTLD-TDP type C cases showed that the number of DN immunostained for hnRNP E2 was significantly less than that immunostained for TDP-43 in frontal cortex (p = 0.004), temporal cortex p = 0.002) and dentate gyrus (p = 0.010). Consequently, double immunofluorescence labelling for TDP-43 and hnRNP E2 (using either the Santa Cruz or the Novus antibody) was performed on selected cases of FTLD-TDP type C and showed a good degree of colocalisation between the two proteins (Fig. 2).

Immunofluorescence for TDP43 (red) and hnRNP E2 (green) in the temporal cortex (a,b) and dentate gyrus of the hippocampus (c,d) of FTLD cases. TDP43 and hnRNP E2 (merge) co-localize in both neuropil threads (a,b) and neuronal inclusions (c,d). Microscope magnification: ×200 (a,c); ×630 (c,d).
Discussion
In the present study, we have investigated the pattern of hnRNP E2 immunostaining across a range of clinical, pathological and genetic forms of FTLD. We found no significant changes in the amount, or intraneuronal distribution pattern, of neuronal hnRNP E2 immunostaining in any of the clinical, pathological or genetic FTLD subgroups. On the other hand, a strong immunostaining of DN and NCI, resembling that seen on TDP-43 immunostaining, was seen in 15/21 cases of SD with FTLD-TDP type C histology. No immunostaining of NCI, DN or NII for hnRNP E2 was seen in any of the other histological forms of FTLD-TDP, or in cases of FTLD-tau. However, it is not clear why only 15/21 cases of SD showed hnRNP E2-immunoreactive DN and NCI, despite there being plentiful TDP-43-immunoreactive DN and NCI present in the 6 other negatively staining cases. Long term storage of tissues in formalin rather than variations in clinical or pathological features, or differences in post mortem interval, is most likely responsible for the lack of hnRNP E2 immunostaining of DN and NCI, since it was necessary to prepare new samples from these 6 cases from archived fixed tissues as original freshly cut tissue blocks were no longer available for study. The complete lack of neuronal nuclear or cytoplasmic hnRNP E2 immunostaining in these same 6 cases supports this interpretation.
Interestingly, while incorporation of TDP-43 into NCI and DN was associated with loss of normal physiological TDP-43 immunostaining, there was no apparent loss of nuclear hnRNP E2 immunostaining in those cells of the dentate gyrus of the hippocampus containing hnRNP E2 immunoreactive NCI. Although, by analogy with TDP-43, this loss might have been anticipated to have occurred, this cannot be necessarily assumed to be the case, since while some hnRNP E2 protein will be binding to TDP inclusions during the formation/evolution of NCI (and DN), at the same time the remainder will still be available to participate in its normal physiological role (and generating nuclear staining) up until the time when the cell dies. It therefore cannot be assumed that all hnRNP E2 protein will be mislocalised into the cytoplasm, as is the case with TDP-43.
Present data therefore indicate an association between hnRNP E2 and TDP-43 pathology but only in this pathological form of FTLD. Indeed, double labelling immunofluorescence showed that most TDP-43 immunoreactive DN were immunoreactive for hnRNP E2 protein. In a previous study [9], we showed that DN and NCI in SD did not contain other hnRNPs such as hnRNP A1, A2/B1 or A3 suggesting that incorporation of hnRNP E2 into DN is not simply due to a passive recruitment of hnRNP E2 protein, along with other hnRNPs, into the aggregating protein conglomerate. It is not clear why co-localization was not complete, but this again could be due to technical reasons such as antigen preservation or accessibility. Alternatively, it might indicate that hnRNP E2 protein is incorporated into pre-existing TDP-43 pathological inclusions at later points in time, with those inclusions that are hnRNP E2 negative not having had sufficient time to accrue enough hnRNP E2 protein to be detectable by immunohistochemistry.
The results of this study imply an important role for hnRNP E2 in the pathogenesis of SD. hnRNP E1 and E2 are the most highly expressed and well characterised isoforms of hnRNP E proteins in human tissues with 89% amino acid homology. They belong to the hnRNP K protein family and have a triple hnRNP K (KH) domain, designated KH1, KH2 and KH3, through which they can bind both poly(rC) regions and low rC mRNAs [20, 27, 28]. They can shuttle between nucleus and cytoplasm and participate in the regulation of mRNA stability and translation [20, 27]. It is believed that hnRNP E1 is encoded by an intronless gene that is a product of a retrotransposition event of a fully processed minor isoform of hnRNP E2 [21]. Each of the KH domains is able to interact independently with a target RNA sequence which gives this protein a potentially high number of complex specific RNA interactions. Woolaway and colleagues demonstrated that depletion of either hnRNP E1 or hnRNP E2 lead to increased production of HIV-1 structural proteins, whereas overexpression of hnRNP E1, but not hnRNP E2, inhibited expression of Rev.-dependent RNAs encoding gp120 and p24 [38]. Other work has demonstrated a higher affinity of hnRNP E1 for hnRNP D than hnRNP E2 [17], and hnRNP E1 and hnRNP E2 have differential responses to hypoxic stress [39]. Both hnRNP E1 and hnRNP E2 can regulate BC200 RNA-mediated translation inhibition but not through the same control mechanism [16]. Therefore, despite the high degree of sequence similarities between hnRNP E1 and hnRNP E2 isoforms, they each have distinct non-redundant cellular functions.
Little is known concerning any specific role for hnRNP E2 within the nervous system in health or neurodegenerative disease, beyond that of control of mRNA stability and mRNA translation. Broderick and coworkers reported that hnRNP E2 can bind to exon 10 of MAPT and activate/regulate alternative splicing [4]. Mis-splicing of exon 10 is a cause of that form of FTLD-tau, known as Frontotemporal Dementia with parkinsonism linked to chromosome 17 (FTDP-17) in which there is increased use of this splice site leading to an imbalance in the ratio of 3-repeat and 4-repeat tau isoforms, in favour of 4-repeat tau, with aggregation of the excess 4-repeat tau into neurofibrillary tangle-like structures [15, 29]. In the present study we investigated 11 cases of FTDP-17 associated with intronic mutations affecting exon 10, but did not find any changes in neuronal hnRNP E2 immunoreactivity or binding of hnRNP E2 to aggregated tau, implying that the functional disturbances leading to increased splicing of exon 10 in this form of FTLD are not mediated by changes in hnRNP E2. Such a conclusion would be supported by the lack of tauopathy in patients with SD where hnRNP E2 pathological changes are associated with TDP-43 proteinopathy instead.
To our knowledge, there have been no previous reports documenting any direct functional or pathological association between hnRNP E2 protein and TDP-43, and the mechanism leading to binding of hnRNP E2 to DN and NCI in patients with SD remains unclear. It is plausible that this scenario could underpin the presence of hnRNP E2 and TDP-43 in DN in patients with SD. However, it is not clear why, in the present report, hnRNP E2 was not also seen in the TDP-43 pathological inclusions of other forms of FTLD-TDP (ie FTLD-TDP types A and B) when these have also been shown to contain markers of stress granules such as TIA-1 [18].The absence of hnRNP E2 in the TDP-43 pathological inclusions of other forms of FTLD-TDP points to a disease mechanism which is specific to patients with SD and FTLD-TDP type C pathology, and one not shared by other forms of FTLD-TDP despite all pathological forms being linked by the same pathological ‘end product’.
Conclusions
In the present study we have shown that a high proportion of TDP-43-positive DN in patients with SD contain hnRNP E2 protein; no other histological forms of FTLD-TDP showed this association, nor were NCI in FTLD-tau hnRNP E2-immunoreactive despite evidence that hnRNP E2 may function as a modulator of alternative splicing of MAPT. The association between hnRNP E2 and TDP-43 in DN in SD, when taken in conjunction with previous findings showing specific interactions between hnRNP A1 and FUS-positive NCI [12] and hnRNP A3 and DPR in C9orf72 expansion carriers [3, 9, 23, 24], suggests that specific changes in different hnRNPs might underlie each pathological form of FTLD. The exact nature of how these proteins (hnRNP E2, TDP) might interact is outside the scope of the present study and requires further work, employing expression studies, western blotting or pull-down methodologies, for example, to support the present argument that an increase in hnRNP E2 protein in NCI is specific to FTLD-TDP type C.
References
Anderson P, Kedersha N (2008) Stress granules: the Tao of RNA triage. Trends Biochem Sci 33:141–150
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611
Boeynaems S, Bogaert E, Michiels E, Gijselinck I, Sieben A, Jovičić A, De Baets G, Scheveneels W, Steyaert J, Cuijt I, Verstrepen KJ, Callaerts P, Rousseau F, Schymkowitz J, Cruts M, Van Broeckhoven C, Van Damme P, Gitler AD, Robberecht W, Van Den Bosch L (2016) Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci Rep 6:20877
Broderick J, Wang J, Andreadis A (2004) Heterogeneous nuclear ribonucleoprotein E2 binds to tau exon 10 and moderately activates its splicing. Gene 331:107–114
Chaudhury A, Chander P, Howe PH (2010) Heterogeneous nuclear ribonucleoproteins (hnRNPs) in cellular processes: Focus on hnRNP E1's multifunctional regulatory roles. RNA 16:1449–1462
Colombrita C, Zennaro E, Fallini C, Weber M, Sommacal A, Buratti E, Silani V, Ratti A (2009) TDP-43 is recruited to stress granules in conditions of oxidative insult. J Neurochem 111:1051–1061
Davidson Y, Kelley T, Mackenzie IR, Pickering-Brown SM, Du Plessis D, Neary D, Snowden JS, Mann DMA (2007) Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol 113:521–533
Davidson Y, Barker H, Robinson AC, Troakes C, Smith B, Al-Sarraj S, Shaw C, Rollinson S, Masuda-Suzukake M, Hasegawa M, Pickering-Brown S, Snowden JS, Mann DMA (2014) Brain distribution of dipeptide repeat proteins in Frontotemporal Lobar Degeneration and Motor Neurone Disease associated with expansions in C9ORF72. Acta Neuropathol Comm 2:70
Davidson YS, Flood L, Robinson AC, Nihei Y, Mori K, Rollinson S, Richardson A, Benson BC, Jones M, Snowden JS, Pickering-Brown S, Haass C, Lashley T, Mann DMA (2017) Heterogeneous ribonuclear protein A3 (hnRNP A3) is present in dipeptide repeat protein containing inclusions in Frontotemporal Lobar Degeneration and Motor Neurone disease associated with expansions in C9orf72 gene. Acta Neuropathol Commun 5:31
Dewey CM, Ceni B, Sephton CF, Johnson BA, Herz J, Yu G (2012) TDP-43 aggregation in neurodegeneration: are stress granules the key? Brain Res 1462:16–25
Fujimura K, Kano F, Murata M (2008) Identification of PCBP2, a facilitator of IRES-mediated translation, as a novel constituent of stress granules and processing bodies. RNA 14:425–431
Gami-Patel P, Bandopadhyay R, Brelstaff J, Revesz T, Lashley T (2016) The presence of heterogeneous nuclear ribonucleoproteins in Frontotemporal Lobar degeneration with FUS-positive inclusions. Neurobiol Aging 46:192–203
Gilks N, Kedersha N, Ayodele M, Shen L, Stoecklin G (2014) Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol Biol Cell 15:5383–5398
Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, Ogar JM, Rohrer JD, Black S, Boeve BF, Manes F, Dronkers NF, Vandenberghe R, Rascovsky K, Patterson K, Miller BL, Knopman DS, Hodges JR, Mesulam MM, Grossman M (2011) Classification of primary progressive aphasia and its variants. Neurology 76:1006–1014
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden M, Pickering-Brown SM, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaf E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski JQ, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd P, Hayward N, JBJ K, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann DM, Lynch T, Heutink P (1998) Association of missense and 5′–splice-site mutation in tau with inherited dementia FTDP-17. Nature 393:702–705
Jang S, Shin H, Lee J, Kim Y, Bak G, Lee Y (2017) Regulation of BC200 RNA-mediated translation inhibition by hnRNP E1 and E2. FEBS Lett 591:393–405
Kiledjian M, Wang X, Liebhaber SA (1995) Identification of two KH domain proteins in the alpha-globin mRNP stability complex. EMBO J 14:4357–4364
Liu-Yesucevitz L, Bilgutay A, Zhang YJ, Vanderweyde T, Citro A, Mehta T, Zaarur N, McKee A, Bowser R, Sherman M, Petrucelli L, Wolozin B (2010) Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One 5:e13250
Mackenzie IRA, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, Perry RH, Trojanowski JQ, Mann DMA, Lee VM-Y (2011) A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 122:111–113
Makeyev AV, Liebhaber SA (2002) The poly(C)-binding proteins: a multiplicity of functions and a search for mechanisms. RNA 8:265–278
Makeyev AV, Chkheidze AN, Liebhaber SA (1999) A set of highly conserved RNA-binding proteins, alpha CP-1 and alpha CP2, implicated inmRNA stabilization, are co-expressed from an intronless gene and its intron-containing paralog. J Biol Chem 274:24849–24857
Mann DMA, Rollinson S, Robinson A, Callister J, Snowden JS, Gendron T, Petrucelli L, Masuda-Suzukake M, Hasegawa M, Davidson YS, Pickering-Brown S (2013) Dipeptide repeat proteins are present in the p62 positive inclusions in patients with Frontotemporal Lobar Degeneration and Motor Neurone Disease associated with expansions in C9ORF72. Acta Neuropathol Comm 1:68
Mori K, Lammich S, Mackenzie IRA, Forne I, Zilow S, Kretzschmar H, Edbauer D, Jannssens J, Kleinberger G, Cruts M, Herms J, Neumann M, Van Broekhoven C, Arzberger T, Haass C (2013) hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP-43 negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol 125:413–423
Mori K, Nihei Y, Arzberger T, Zhou Q, Mackenzie IR, Hermann A, Hanisch F, German Consortium for Frontotemporal Lobar Degeneration, Bavarian Brain Banking Alliance, Kamp F, Nuscher B, Orozco D, Edbauer D, Haass C (2016) Reduced hnRNPA3 increases C9orf72 repeat RNA levels and dipeptide-repeat protein deposition. EMBO Rep 17:1314–1325
Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J, Benson DF (1998) Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51:1546–1554
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Ostareck-Lederer A, Ostareck DH (2004) Control of mRNA translation and stability in haematopoietic cells: the function of hnRNPs K and E1/E2. Biol Cell 96:407–411
Ostareck-Lederer A, Ostareck DH, Hentze MW (1998) Cytoplasmic regulatory functions of the KH domain proteins hnRNPs K and E1/E2. TIBS 23:409–411
Pickering-Brown SM, Richardson AMT, Snowden JS, McDonagh AM, Burns A, Braude W, Baker M, Liu W-K, Yen S-H, Hardy J, Hutton M, Davies Y, Allsop D, Craufurd D, Neary D, Mann DMA (2002) Inherited frontotemporal dementia in 9 British families associated with intronic mutations in the tau gene. Brain 125:732–751
Piñol-Roma S, Choi YD, Matunis MJ, Dreyfuss G (1988) Immunopurification of heterogeneous nuclear ribonucleoprotein particles reveals an assortment of RNA-binding proteins. Genes Dev 2:215–227
Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, van Swieten JC, Seelaar H, Dopper EG, Onyike CU, Hillis AE, Josephs KA, Boeve BF, Kertesz A, Seeley WW, Rankin KP, Johnson JK, Gorno-Tempini ML, Rosen H, Prioleau-Latham CE, Lee A, Kipps CM, Lillo P, Piguet O, Rohrer JD, Rossor MN, Warren JD, Fox NC, Galasko D, Salmon DP, Black SE, Mesulam M, Weintraub S, Dickerson BC, Diehl-Schmid J, Pasquier F, Deramecourt V, Lebert F, Pijnenburg Y, Chow TW, Manes F, Grafman J, Cappa SF, Freedman M, Grossman M, Miller BL (2011) Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134:2456–2471
Saxon JA, Harris JM, Thompson JC, Jones M, Richardson AMT, Langheinrich T, Neary D, DMA M, Snowden JS (2017) Semantic dementia, progressive non-fluent aphasia and their association with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry (in press)
Shi J, Shaw CL, Richardson AMT, Bailey K, Tian J, Varma AR, Neary D, Snowden JS, Mann DMA (2005) Histopathological changes underlying frontotemporal lobar degeneration with clinicopathological correlation. Acta Neuropathol 110:501–512
Snowden JS, Neary D, Mann DMA (2005) Frontotemporal dementia. Lancet Neurol 4:771–780
Snowden JS, Thompson JC, Stopford CL, Richardson AMT, Gerhard A, Neary D, Mann DMA (2011) The clinical diagnosis of early-onset dementias: diagnostic accuracy and clinicopathological relationships. Brain 135:693–708
Thompson JC, Stopford CL, Snowden JS, Neary D (2005) Qualitative neuropsychological performance characteristics in frontotemporal dementia and Alzheimer’s disease. J Neurol Neurosurg Psychiatry 76:920–927
Wang IF, Wu LS, Chang HY, Shen CK (2008) TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J Neurochem 105:797–806
Woolaway K, Asai K, Emili A, Cochrane A (2007) hnRNP E1 and E2 have distinct roles in modulating HIV-1 gene expression. Retrovirology 4:28
Zhu Y, Sun Y, Mao XO, Jin KL, Greenberg DA (2002) Expression of poly(C)-binding proteins is differentially regulated by hypoxia and ischaemia in cortical neurons. Neuroscience 110:191–198
Acknowledgements
We acknowledge the support of the Manchester Brain Bank by Alzheimer’s Research UK and Alzheimer’s Society through their funding of the Brains for Dementia Research (BDR) Programme. Manchester Brain Bank also receives Service Support costs from Medical Research Council. This work was supported by Medical Research Council of UK, grant number G0701441. TL is funded by an Alzheimer’s Research UK senior fellowship. BCB and YTA are supported by the Leonard Wolfson Centre for Experimental Neurology. The Queen Square Brain Bank is supported by the Reta Lila Weston Institute for Neurological Studies and the Progressive Supranuclear Palsy (Europe) Association.
Authors’ contributions
YD and LF performed all immunohistochemical staining, AR provided technical support and carried out all statistical analyses. SR and SPB provided genetic analyses. BCB, YTA and TL provided technical help with the double immunolabeling work, advice concerning the London cases and assisted with writing of the paper. AR, MJ and JSS helped with patient characterization and review of the manuscript. DMAM conceived the study, performed the semiquantitative assays, prepared the figures and wrote and revised the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Davidson, Y.S., Robinson, A.C., Flood, L. et al. Heterogeneous ribonuclear protein E2 (hnRNP E2) is associated with TDP-43-immunoreactive neurites in Semantic Dementia but not with other TDP-43 pathological subtypes of Frontotemporal Lobar Degeneration. acta neuropathol commun 5, 54 (2017). https://doi.org/10.1186/s40478-017-0454-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-017-0454-4