
Abstract
Renal cell carcinoma is a lethal disease that is often discovered incidentally. New non-invasive biomarkers are needed to aid diagnosis and treatment. Extracellular vesicles (EVs), membranous vesicles secreted by all cells, are a promising potential source for cancer biomarkers, but new methods are required that are both sensitive and specific for cancer identification. We have developed an EV isolation protocol optimized for kidney tumor and normal kidney tissue that yields a high vesicle concentration, confirmed by nanoparticle tracking analysis (NanoSight) and by nanoscale flow cytometry (NanoFCM). Using Western blot, we confirmed presence of EV markers CD81, CD63, flotillin-1, and absence of cellular debris, calnexin. Transmission electron microscopy images demonstrate intact membranous EVs. This new method improves existing protocols with additional steps to reduce contaminants in the EV product. Characterization of our isolation product confirms successful isolation of EVs with minimal contamination. The particle yields of our protocol are consistent and high as assessed by both standard and novel methods. This optimized protocol will contribute to biomarker discovery and biological studies of EVs in renal cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Renal cell carcinoma (RCC) is the third most common urological cancer in the USA accounting for approximately 73,820 new cases and 14,770 cancer deaths in 2019 [1, 2]. In many cases, RCC patients are asymptomatic at the time of diagnosis [3]. The incidence of so-called “incidentalomas” is correlated to an increased availability of cross-sectional imaging such as ultrasound, CT-scan, and MRI [2]. Although early detection may be beneficial to RCC treatment, one harmful consequence is the overtreatment of benign small renal masses by surgical resection [4]. Non-invasive biomarkers are needed as a tool to aid in the diagnosis and especially the prognosis of RCC. Extracellular vesicles (EVs) have recently emerged as a potential source for biomarkers in many diseases, e.g., Alzheimer’s disease, vascular disease, acute kidney injury, and cancer [5,6,7,8].
Most, if not all, cell types release EVs, a heterogeneous class of membranous particles [9]. EVs can be recovered from all body fluids including plasma and urine [10]. Due to the presence of a bilipid membrane layer, EVs can protect cargo (e.g., protein, RNA, etc.) against harsh environments such as the circulation or urine [11]. Additionally, cells in close or far distance from the originating cell can take up the EVs and their cargo, representing a form of cellular communication [12, 13]. Exosomes and ectosomes are the most researched EV subgroups, distinguished by their size and biogenesis. The diameter of exosomes is 50–150 nm, whereas ectosomes are reported to be 100–1000 nm [10]. The underlying biological pathways leading to the formation of the vesicles differ with exosomes originating from the intracellular endosome [11]. Direct outward budding of the cell membrane forms ectosomes. Although markers found in both types of vesicles are surrogates of the parental cell, the different machineries likely load a different selection of cargos into exosomes than into ectosomes [14].
It has been reported that EV biogenesis pathways are upregulated in cancer cells and thus cancer patients’ plasma contain more EVs compared with healthy donor plasma [13, 15]. EVs have been found to influence cancer progression and are increasingly seen as a source of biomarkers in multiple cancer types, including melanoma, glioblastoma, and prostate cancer [16,17,18,19]. In RCC, however, little work has studied the role of EVs in tumorigenesis or metastasis [11].
A good biomarker is disease-specific, sensitive enough to exclude false-negative patients, and easily reproducible [20]. One major hurdle in identifying RCC-specific EV biomarkers is the lack of tumor specificity in urine- or plasma-derived EVs [11, 20]. The lack of tumor specificity of EVs isolated from blood or urine is a direct consequence of the tumor cells not exclusively secreting EVs [10, 11].
We have adapted an EV isolation method employed for metastatic melanoma and optimized it to isolate EVs from kidney tumor and normal kidney tissue [21]. This protocol demonstrates a high EV yield that is sufficient for downstream analysis. The quality of the isolated EVs was confirmed according to the Minimal Information for Studies of Extracellular Vesicles (MISEV) guidelines [22]. The isolation method can be used to expedite biomarker discovery and biological studies of EVs in renal cancer and has the potential to be modified and applied to other cancer types as well.
Methods
Patients and sample collection
The collection of specimens from patients undergoing nephrectomy was preceded by signed written informed consent and approved by the Johns Hopkins Office of Human Subjects Research Institutional Review Board. Kidney tumors and paired normal kidney tissue were collected from four patients that underwent radical nephrectomy (see Online Resource 1, Supplementary Table 1). Because RCC occurs in both sexes, tissues of two female and two male patients were used and selected based on availability of sufficient tissue. In all cases, the AJCC-TNM 8th edition stage group was 3–4 and the histological diagnosis was “clear cell renal cell carcinoma” with “sarcomatoid differentiation in 20%” as an additional finding in two cases [23]. The sampling of RCC and normal kidney was confirmed by histology and histology staining (H&E, see Online Resource 2, Supplementary Fig. 1). Normal kidney tissues and tumor tissues were collected from surgical pathology before formalin fixation (Fig. 1). The tissue was collected in ice-cold phosphate-buffered saline (PBS) and rapidly transferred to the laboratory for further processing. When visible, perirenal fat, capsule, and necrotic parts were removed using fine dissecting scissors (see Online Resource 2, Supplementary Fig. 2). Approximately 1 g of tissue was cut into 2–3 mm pieces. The tissue was transferred to a 50 mL conical tube and PBS was added to a total volume of 50 mL. For a comprehensive list of reagents and materials used, see Online Resource 1, Supplementary Table 2.

Courtesy of T.H. Phelps
Workflow of EV isolation protocol starting from kidney tissue. 1 Sample preparation: normal kidney or kidney tumor is collected in PBS and cut into 2–3 mm fragments. Tissue pieces are collected by centrifugation and the PBS removed by aspiration. 2 Incubation: the tissue pieces are incubated in DMEM with Collagenase D (20 mg/mL) and DNase I (40 U/mL) for 30 min. The tissue is removed from the conditioned media by a 70 µm strainer. 3 Differential centrifugation: non-EVs are precipitated by sequential centrifugation and discarded after each step, retaining the supernatant for subsequent centrifugation steps (three times). 4 Filtration: the supernatant is syringe-filtered with a 0.8 µm filter followed by a 0.45 µm filter. 5 Ultracentrifugation: EVs are isolated from supernatant by ultracentrifugation for 2 h. The supernatant is now aspirated. The EV pellet is retained, washed with PBS, and then precipitated again by 2 h of ultracentrifugation. PBS phosphate-buffered saline, DMEM Dulbecco’s modified Eagle’s medium.

Courtesy of T.H. Phelps
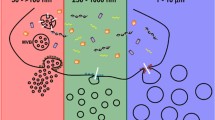
EV isolation protocol steps explained from a particle size perspective. Sequential centrifugation steps with increasing speed (500, 2,000, and 10,000×g) enable exclusion of larger non-EV that would otherwise be co-isolated. Exclusion of live cells, dead cells and apoptotic bodies, and small debris, requires sacrifice of some large EVs within the ectosome range. The filtration steps (800 and 450 nm pore size) reduce chances of co-isolation of particles that are larger but did not precipitate well enough. Finally, ultracentrifugation at 100,000×g pellets the other particles, mostly EVs smaller than 450 nm. For reference, EV sizes are shown: exosomes (green) 40–150 nm and ectosomes (purple) 100–1,000 nm. Non-EV (cells and cellular debris) present in our sample are discarded by these steps. However, the abundance of smaller non-EV can vary between biofluids and require additional separation methods. APT apoptotic bodies, EXO exosome.
Conditioning media and differential centrifugation
The conical tube containing tissue and PBS was centrifuged at 500×g for 5 min. The PBS was aspirated and the tissue pellet was resuspended in 15 mL of serum-free Dulbecco's modified Eagle's medium (DMEM) with Collagenase D (20 mg/mL) and DNase I (40 U/mL). The tissue and media were transferred to a 10 cm cell culture dish and incubated at 37 °C for 30 min. The conditioned media was then strained using a 70 µm Nylon cell strainer (Falcon). Sequential centrifugation steps followed to precipitate and discard: live cells (500×g for 5 min. at 4 °C), apoptotic bodies and dead cells (2000×g for 20 min. at 4 °C), and small cellular debris and larger EVs (10,000×g for 20 min. at 4 °C), as shown in Fig. 2. The supernatant was then filtered twice using hydrophilic polyethersulfone syringe filters (PALL), first pore size 0.80 µm followed by 0.45 µm. The conditioned media was then directly used for EV isolation or stored at − 80 °C. If the media had been stored, it was thawed at 37 °C before use and centrifuged 10 min. at 1000×g to remove precipitated salts.
EV isolation via ultracentrifugation
Ultracentrifugation is the most commonly used EV isolation method [24]. EVs were precipitated in 29.9 mL OptiSeal™ Polypropylene tubes (Beckman Coulter) by 2 h of ultracentrifugation at 100,000×g at 4 °C (Type 70 Ti rotor, adjusted k-factor 90, max. acceleration, max. deceleration, Beckman Coulter) [25, 26]. After the first spin, the supernatant was aspirated. The EV pellet was resuspended and washed with PBS followed by another 2-h ultracentrifugation step at same settings. The EVs were resuspended in 100 µL PBS, collected, and either immediately used or stored at − 80 °C.
Quality control of isolated EVs
Isolated EVs were characterized according to the guidelines set by the International Society for EVs [22]. We have submitted all relevant data of our experiments to the EV-TRACK knowledgebase (EV-TRACK ID: EV190079) [27]. Nanoparticle Tracking Analysis (NTA) and the Flow Nano Analyzer were used to determine particle concentration and particle size distribution. Transmission Electron Microscopy (TEM) assessed the morphology of EVs. Protein content of the EV sample was analyzed by Western blot. Detailed descriptions of the protocols are as follows:
Nanoparticle tracking analysis
Samples were analyzed using the NanoSight NS300 (Malvern Panalytical) according to the manufacturer’s instructions (NanoSight NS300 User Manual, MAN0541-02-EN, 2018) [28]. Briefly, 10 µL of each EV sample was diluted resulting in a particle per frame rate between 20 and 100 particles/frame. After manual adjustments of image focus, particles were distinctly visible. Three 60-s videos were recorded of flowing particles at syringe flow rate 50 using the sCMOS camera (screen gain 3 and camera level 9) and an embedded green 532 nm CW laser. NanoSight software (NTA 3.4 Build 3.4.003) was used to analyze the videos (screen gain 10 and detection threshold 4 gave 10–100 distinct particle cores with less than five false positives). The particle concentrations were normalized for starting tissue weight, conditioned media volume, volume of EV resuspension, and dilution necessary for adequate NTA-reading.
Flow nano analyzer
EV samples were diluted 1:100 and analyzed using the Flow Nano Analyzer (NanoFCM Inc.) according to manufacturer’s protocol [29]. Briefly, the lasers were calibrated using 200 nm control beads (NanoFCM Inc.), which were then analyzed as a reference for particle concentration. Additionally, a mixture of different sized beads (NanoFCM Inc.) was analyzed to set reference for size distribution. PBS was analyzed as background signal and subtracted from the other measurements (see Online Resource 2, Supplementary Fig. 3). The samples were diluted resulting in a particle count within the optimal range of 4,000–14,000. Particle concentration and size distribution were calculated using the NanoFCM software (NanoFCM Profession V1.0) and normalized for starting tissue weight, conditioned media volume, volume of EV resuspension, and dilution necessary for adequate NanoFCM-reading.

Our EV isolation protocol starting from tissue is reproducible based on particle concentrations. a Total particle concentration (in p/mL) of EVs isolated from parallel processed technical replicates of normal kidney tissue (1a–c) measured by NanoSight. b Total particle concentration of normal kidney EVs (1a–c) measured by NanoFCM. c. Total particle concentration of EVs isolated from parallel processed technical replicates of RCC tissue (2a–c) measured by NanoSight. d Total particle concentration of RCC EVs (2a–c) measured by NanoFCM. e Particle size distribution of normal kidney EVs (1a–c) measured by NanoSight. f Particle size distribution of normal kidney EVs (1a–c) measured by NanoFCM. g Particle size distribution of RCC EVs (2a–c) measured by NanoSight. h Particle size distribution of RCC EVs (2a–c) measured by NanoFCM. Data are corrected for starting tissue mass, conditioned media volume, volume of EV resuspension, and dilution necessary for measurement. The numbers in the graphs indicate the mode of particle size distribution
TEM
Two microliters of each EV sample were diluted 1:5 in PBS. A sample was adsorbed to an ultra-thin carbon-coated 400 mesh copper grid that was glow discharged (EMS GloQube™) by floatation for 2 min. Grids were quickly blotted on filter paper and rinsed three times in tris-buffered saline (TBS) for 1 min. The grids were stained in two consecutive drops of 1% uranyl acetate with methylcellulose (filtered twice through 0.22 µm filter). The excessive stain was quickly blotted and aspirated. When completely dry, the grids were imaged using a Philips CM-120 TEM operating at 80 kV with an AMCT XR80 CCD sensor.
Western blot
Membrane and cytosolic fractions of MCF7 cells (HTB-22™, ATCC) were acquired using the Mem-PER™ Plus Membrane Protein Extraction Kit (ThermoFisher Scientific) to serve as positive and negative controls. Samples were lysed using 10 × RIPA buffer (Cell Signaling Technology) with HALT™ Proteinase and Phosphatase Inhibitor Cocktail (ThermoFisher Scientific). Protein concentrations were determined by Pierce™ BCA Protein Assay (ThermoFisher Scientific) according to manufacturer’s instructions. Ten micrograms of EV protein lysate were run on a 12% SDS Mini-PROTEAN® TGX Stain-Free™ Protein Gel (Bio-Rad Laboratories) under non-reducing and denaturing conditions. Each gel contained Chameleon Duo Pre-stained Protein Ladder (LI-COR Biosciences) and positive and negative control lysates, MCF7 membrane, and cytosolic fractions, respectively. The protein was transferred to a nitrocellulose membrane (Trans-Blot® Turbo™ Mini Nitrocellulose, Bio-Rad Laboratories) using the Trans-Blot® Turbo™ Transfer System (Bio-Rad Laboratories). The membrane was blocked in casein blocking buffer (Sigma-Aldrich) for 1 h at room temperature. The membrane was incubated overnight at 4 °C with primary antibodies against CD63 (1:250, ThermoFisher Scientific, Cat. # 10628D), CD81 (1:200, Santa Cruz, Cat. # sc-7637), Flotillin-1 (1:5,000, Abcam, Cat. # ab133497), Calnexin (1:2,000, Abcam, Cat. # ab22595), or GAPDH (1:2,000, Cell Signaling Technology, Cat. # 2118). After three 10-min washes in TBS with 1% Tween-20® (TBS-T), the blots were incubated with secondary antibodies: IRDye® 680RD Goat anti-Mouse IgG, (1:20,000, LI-COR Biosciences, USA, Cat. # 92,668,070) and IRDye® 800CW Goat anti-Rabbit IgG (1:15,000, LI-COR Biosciences, Cat. # 92,632,211). After three 10-min washes in TBS-T, the blots were imaged using the Odyssey® 9120 Infrared Imaging System (LI-COR Biosciences).
Results
Tissue processing and EV isolation protocol is technically reproducible
For the assessment of technical reproducibility of the isolation protocol, normal kidney tissue from one patient was split into three parts that were processed for EV isolation and analyzed separately. Tumor tissue from a second patient was likewise analyzed as three replicates.
After isolation of normal kidney EV (kidney EV), the total particle concentration of the replicates was consistent with less than fourfold differences among replicates (Fig. 3a, b). Particle concentration measured by NanoSight was 1.44 × 1010 ± 2.51 × 109 p/mL (mean ± SD) after normalization. Particle count of identical samples measured by NanoFCM was 1.67 × 1010 ± 6.89 × 109 p/mL (mean ± SD). We found a similar consistency in total particle concentration in replicates of tumor EVs, with differences among replicates less than twofold (Fig. 3c, d). The total particle concentration measured by NanoSight was 1.80 × 1010 ± 3.59 × 109 p/mL (mean ± SD) and by NanoFCM was 1.68 × 1010 ± 4.18 × 109 p/mL (mean ± SD). Additionally, both NanoSight and NanoFCM showed a consistent particle size distribution within the technical replicates of normal and RCC EVs (Fig. 3e–h). Although the measured modes, or peak diameters, of the particle size distribution were consistent within the replicates measured by the same method, there was a difference between the two methods. In identical samples measured by both methods, NanoSight generally estimated the most abundant particle diameter to be larger than NanoFCM did. Additionally, these peaks measured by NanoSight demonstrated a higher variation (131–168 nm, Fig. 3e, f), where the modes of the particle size distribution measured by NanoFCM showed little variation (57–59 nm, Fig. 3g, h).
Successful EV isolation from tissue of multiple patients
We isolated sufficient kidney and RCC EVs from tissue of three patients for downstream analysis. We used NanoSight and NanoFCM to assess matched tissue pairs from three patients (Pts. 2–4). Comparison of particle concentrations of kidney EVs and RCC EVs for each patient demonstrated differences smaller than twofold using both NanoSight and NanoFCM (Fig. 4a, b). For Pt. 2, normalized particle concentrations of kidney EVs were 1.56 × 1010 p/mL and 1.84 × 1010 p/mL by NanoSight and NanoFCM, respectively (Fig. 4a, b). Pt. 2 RCC EV particle counts were calculated as the average of the replicates (Fig. 3c, d) and the particle concentrations were 1.80 × 1010 p/mL and 1.68 × 1010 p/mL measured by NanoSight and NanoFCM, respectively (Fig. 4a, b). Particle concentrations by NanoSight of normal kidney EVs of Pts. 3 and 4 were 2.23 × 109 p/mL and 2.04 × 109 p/mL, respectively (Fig. 4a). Measured by NanoSight, the particle concentrations of RCC EVs of Pts. 3 and 4 were 2.67 × 109 p/mL and 1.19 × 109 p/mL, respectively (Fig. 4a). Measured by NanoFCM, the concentrations of the normal kidney EVs were 8.98 × 109 p/mL of Pt. 3 and 5.64 × 109 p/mL of Pt. 4 (Fig. 4b). Measured by NanoFCM, the concentrations of RCC EVs of Pt. 3 and 4 were 8.61 × 109 p/mL and 3.46 × 109 p/mL, respectively (Fig. 4b). Overall, the measurements by NanoSight and NanoFCM of particle concentrations of identical samples were similar, as no difference was greater than fivefold. The particle size distribution of the pairs was assessed by both methods (Fig. 4c–h). Per patient, the size distribution of normal and RCC EVs were similar, as demonstrated by overlap of the curves. The mode of particle size distribution measured by NanoSight (133–162 nm) varied more than modes measured by NanoFCM (53–59 nm).

Successful EV isolation of tissue from multiple patients (n = 3) measured by two methods. a Total particle concentration (in p/mL) of EVs isolated from normal kidney tissue and RCC of 3 patients (Pt. 2–4) measured by NanoSight. b Total particle concentration of EVs isolated from normal kidney tissue and RCC of 3 patients (Pt. 2–4) measured by NanoFCM. c–e Particle size distribution of normal kidney EVs and RCC EVs of Pt. 2–4 measured by NanoSight. f–h Particle size distribution of normal kidney EVs and RCC EVs of Pt. 2–4 measured by NanoFCM. Data are corrected for starting tissue mass, conditioned media volume, volume of EV resuspension, and dilution necessary for measurement. The numbers in the graphs indicate the mode of particle size distribution
Protein analyses confirm the presence of CD63, CD81, and flotillin-1 in EV samples
Kidney EVs and RCC EVs of all patients were assessed for three established EV protein markers (CD63, CD81, and flotillin-1) and one non-EV marker (calnexin) to demonstrate EV purity. Calnexin is a protein associated with the endoplasmatic reticulum and could be released by cells suffering mechanical damage (i.e., during tissue preparation). All EV samples, including technical replicates of normal kidney EVs (1a–c) and tumor EVs (2a–c) as well as EVs isolated from paired tissue of multiple patients (Pts. 2–4), were positive for flotillin-1, CD63, and CD81, indicating the presence of EV proteins (Fig. 5a, b). Furthermore, all of the samples were negative for calnexin.

Protein analyses confirm EV proteins are present in samples while cell debris marker is absent. Western blots of flotillin-1, CD63, CD81, and calnexin of a EVs isolated from replicates of normal kidney tissue (kidney EV 1a–c) and kidney tumor (RCC EV 1a–c); b EVs isolated from 3 different patients’ kidney tissue pairs (normal and tumor). MCF7 membrane and cytosolic protein fractions served as positive and negative controls, respectively. GAPDH staining was used as a loading control for the cellular fractions. Pt. 2 normal kidney was ran on the same gel as the replicates in a. The image of the controls and RCC EV 2c were repeated for clarity. Kid normal kidney EVs, RCC kidney tumor EVs
TEM confirms presence of membranous vesicles in our EV samples
On TEM images of negatively stained EV samples, we observed cup-shaped particles in different sizes (Fig. 6a, b). The cup shape indicates an intact bilipid membranous vesicle, but dehydrated and, therefore, not perfectly spherical. These EVs are present in all our EV samples isolated from RCC and normal kidney tissue. In some of the images, protein aggregates are visible (Fig. 6b).

Transmission electron microscopy images confirm presence of negative-stained EVs, seen as cup-shaped vesicles. a Representative widefield and close-up of normal kidney EVs. b Representative widefield and close-up of RCC EVs. White arrows indicate small EVs. Black scale bar represents 500 nm. White scale bar represents 100 nm
Discussion
The variation of EV isolation protocols used by different laboratories leads to heterogeneous and irreproducible results [22, 30]. The complexity of the various studied biological samples contributes to this, as well as the length of isolation protocols with many steps. Therefore, the standardization of protocols is necessary.
The concept of studying EVs secreted directly by tissue is relatively novel. In 2017, Vella et al. used frozen human brain tissue to study EVs [31]. More recently, Jang et al. studied EVs from metastatic human melanoma and Jingushi et al. isolated EVs from human kidney tumors [21, 32]. Although Jingushi et al. reported successful EV isolation from RCC, we found it necessary to optimize available protocols to reduce non-EV contaminants to acceptable levels. EV isolation from clear cell renal cell carcinoma (ccRCC) tissue, the most common histological RCC subtype, is particularly challenging due to the intracellular lipids that give the ccRCC cell its white, “clear” appearance under a microscope. Release of these lipids during tissue processing, i.e., cutting, can result in high contamination [33, 34]. If liberated lipids were present, they disrupted the sample processing by floating during centrifugation steps that are performed to discard precipitated non-EVs (Fig. 1, step 3). When these larger non-EV contaminants are left present in supernatant during low-speed differential centrifugation, they will be co-isolated with the EVs during ultracentrifugation. Therefore, we included additional steps in our isolation protocol to reduce contamination while maintaining sufficient EV yield for downstream analysis. We included enzymatic tissue digestion with Collagenase D and DNase I to prevent cluster formation due to free DNA that both require incubation at 37 °C instead of the 4 °C used in other kidney processing methods. In addition, we used multiple low-speed centrifugation steps to increase chance of precipitating debris and to allow more opportunities to remove floating lipids by careful aspiration. We only performed the aspiration when necessary, i.e., when the lipids were visible and had turned the clear-conditioned media into an emulsion. Finally, after the multiple centrifugation steps, we added two filtration steps to further reduce the presence of lipids, leading to macroscopic reduction in the turbidity of the tissue-conditioned media (see Online Resource 2, Supplementary Fig. 4). Especially in the case of a lipid-rich tumor, up to 25% of conditioned media is lost by aspiration and clogged filters. For consistency, we normalized all particle counts by the volume of media actually used for ultracentrifugation.
The particle concentration measurements of technical replicates (normal kidney or RCC tissue) by NanoSight and NanoFCM were consistent with differences of less than tenfold, indicating robust reproducibility of isolation procedures. To our knowledge, this is the first report of using NanoFCM to assess EVs isolated from kidney (tumor) tissue or any tissue. We found that NanoFCM has some advantages over NanoSight, such as lower required input sample volume and higher sensitivity. Where the NanoSight uses an algorithm to calculate particle diameter, NanoFCM uses a mixture of reference beads to calculate the size. In our data, the NanoFCM calculated particle size distributions more consistently than NanoSight.
Using both methods, we observed a difference in particle concentration among the patients. These likely demonstrate true biological differences, because differences within each patient (between normal and RCC EVs) are less than twofold, while differences among patients varied more (1.5 to 10-fold). With these methods, we consider differences less than tenfold as similar. Patient-to-patient variability may be technical (e.g., processing time from excision to incubation start) or biological (e.g., metabolic state and kidney function). Previous reports showed increased EV concentrations in cancer patients’ plasma compared with healthy donors, depending on tumor burden [15]. In tissue, we found no differences in concentration between normal kidney EVs and RCC EVs from the same patient, as differences were not greater than twofold in favor of normal kidney EVs. Potential reasons that the RCC EVs concentrations were lower include cell numbers, tissue vitality, and sampling bias. We normalized particle counts by 1 g of tissue, which most likely translates into a difference between cell numbers of used tumor and normal tissues. Tissue vitality influences EV secretion, as non-vital tissue secretes fewer small EVs and more apoptotic bodies. The tumor tissue often has small sections of necrosis and hemorrhage, especially in high stage RCC that we carefully attempted to exclude from further processing. However, standardization of sampling location is difficult. Within the heterogenic tumor tissue, it is easier to miss necrotic areas by macroscopic assessment leading to a sampling bias.
Conclusions
In this study, we optimized a protocol to isolate EVs from kidney and RCC tissue. This new method improves previously reported protocols with additional steps to reduce contaminants in the EV product. Characterization of our isolation product confirms successful isolation of EVs with minimal contamination. The particle yield of our protocol is consistent and high as assessed by both standard and novel methods. We expect that our protocol can contribute to biological studies in kidney cancer and can aid the discovery of liquid biopsy RCC biomarkers.
Abbreviations
- ccRCC:
-
Clear cell renal cell carcinoma
- DMEM:
-
Dulbecco's modified Eagle’s medium
- EVs:
-
Extracellular vesicles
- H&E:
-
Hematoxylin and eosin
- MISEV:
-
Minimal information for studies of extracellular vesicles
- NTA:
-
Nanoparticle tracking analysis
- PBS:
-
Phosphate-buffered saline
- RCC:
-
Renal cell carcinoma
- TBS:
-
Tris-buffered saline
- TEM:
-
Transmission electron microscopy
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. https://doi.org/10.3322/caac.21551.
Capitanio U, Bensalah K, Bex A, Boorjian SA, Bray F, Coleman J, et al. Epidemiology of renal cell carcinoma. Eur Urol. 2019;75(1):74–84. https://doi.org/10.1016/j.eururo.2018.08.036.
Novara G, Ficarra V, Antonelli A, Artibani W, Bertini R, Carini M, et al. Validation of the 2009 TNM version in a large multi-institutional cohort of patients treated for renal cell carcinoma: are further improvements needed? Eur Urol. 2010;58(4):588–95. https://doi.org/10.1016/j.eururo.2010.07.006.
Johnson DC, Vukina J, Smith AB, Meyer AM, Wheeler SB, Kuo TM, et al. Preoperatively misclassified, surgically removed benign renal masses: a systematic review of surgical series and United States population level burden estimate. J Urol. 2015;193(1):30–5. https://doi.org/10.1016/j.juro.2014.07.102.
Cosin-Tomas M, Antonell A, Llado A, Alcolea D, Fortea J, Ezquerra M, et al. Plasma miR-34a-5p and miR-545-3p as early biomarkers of Alzheimer's disease: potential and limitations. Mol Neurobiol. 2017;54(7):5550–62. https://doi.org/10.1007/s12035-016-0088-8.
Kanhai DA, Visseren FL, van der Graaf Y, Schoneveld AH, Catanzariti LM, Timmers L, et al. Microvesicle protein levels are associated with increased risk for future vascular events and mortality in patients with clinically manifest vascular disease. Int J Cardiol. 2013;168(3):2358–63. https://doi.org/10.1016/j.ijcard.2013.01.231.
Zhou H, Cheruvanky A, Hu X, Matsumoto T, Hiramatsu N, Cho ME, et al. Urinary exosomal transcription factors, a new class of biomarkers for renal disease. Kidney Int. 2008;74(5):613–21. https://doi.org/10.1038/ki.2008.206.
D'Souza-Schorey C, Clancy JW. Tumor-derived microvesicles: shedding light on novel microenvironment modulators and prospective cancer biomarkers. Genes Dev. 2012;26(12):1287–99. https://doi.org/10.1101/gad.192351.112.
Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: artefacts no more. Trends Cell Biol. 2009;19(2):43–51. https://doi.org/10.1016/j.tcb.2008.11.003.
Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200(4):373–83. https://doi.org/10.1083/jcb.201211138.
Dong L, Zieren RC, Wang Y, de Reijke TM, Xue W, Pienta KJ. Recent advances in extracellular vesicle research for urological cancers: From technology to application. Biochim Biophys Acta Rev Cancer. 2019;1871(2):342–60. https://doi.org/10.1016/j.bbcan.2019.01.008.
Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9(8):581–93. https://doi.org/10.1038/nri2567.
Corrado C, Raimondo S, Chiesi A, Ciccia F, De Leo G, Alessandro R. Exosomes as intercellular signaling organelles involved in health and disease: basic science and clinical applications. Int J Mol Sci. 2013;14(3):5338–666. https://doi.org/10.3390/ijms14035338.
Lasser C, Jang SC, Lotvall J. Subpopulations of extracellular vesicles and their therapeutic potential. Mol Aspects Med. 2018;60:1–14. https://doi.org/10.1016/j.mam.2018.02.002.
Whiteside TL. Tumor-derived exosomes and their role in cancer progression. Adv Clin Chem. 2016;74:103–41. https://doi.org/10.1016/bs.acc.2015.12.005.
Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560(7718):382–6. https://doi.org/10.1038/s41586-018-0392-8.
Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10(12):1470–6. https://doi.org/10.1038/ncb1800.
Nilsson J, Skog J, Nordstrand A, Baranov V, Mincheva-Nilsson L, Breakefield XO, et al. Prostate cancer-derived urine exosomes: a novel approach to biomarkers for prostate cancer. Br J Cancer. 2009;100(10):1603–7. https://doi.org/10.1038/sj.bjc.6605058.
McKiernan J, Donovan MJ, O'Neill V, Bentink S, Noerholm M, Belzer S, et al. A novel urine exosome gene expression assay to predict high-grade prostate cancer at initial biopsy. JAMA Oncol. 2016;2(7):882–9. https://doi.org/10.1001/jamaoncol.2016.0097.
Witwer KW. Circulating microRNA biomarker studies: pitfalls and potential solutions. Clin Chem. 2015;61(1):56–63. https://doi.org/10.1373/clinchem.2014.221341.
Jang SC, Crescitelli R, Cvjetkovic A, Belgrano V, Olofsson Bagge R, Sundfeldt K, et al. Mitochondrial protein enriched extracellular vesicles discovered in human melanoma tissues can be detected in patient plasma. J Extracell Vesicles. 2019;8(1):1635420. https://doi.org/10.1080/20013078.2019.1635420.
Thery C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for extracellular vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750. https://doi.org/10.1080/20013078.2018.1535750.
Amin MB, Edge SB. AJCC cancer staging manual. Cham: Springer; 2017.
Gardiner C, Di Vizio D, Sahoo S, Thery C, Witwer KW, Wauben M, et al. Techniques used for the isolation and characterization of extracellular vesicles: results of a worldwide survey. J Extracell Vesicles. 2016;5:32945. https://doi.org/10.3402/jev.v5.32945.
Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006;30:3–22. https://doi.org/10.1002/0471143030.cb0322s30.
Cvjetkovic A, Lotvall J, Lasser C. The influence of rotor type and centrifugation time on the yield and purity of extracellular vesicles. J Extracell Vesicles. 2014;3(1):23111. https://doi.org/10.3402/jev.v3.23111.
Consortium E-T, Van Deun J, Mestdagh P, Agostinis P, Akay O, Anand S, et al. EV-TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nat Methods. 2017;14(3):228–32. https://doi.org/10.1038/nmeth.4185.
Gardiner C, Ferreira YJ, Dragovic RA, Redman CW, Sargent IL. Extracellular vesicle sizing and enumeration by nanoparticle tracking analysis. J Extracell Vesicles. 2013. https://doi.org/10.3402/jev.v2i0.19671.
Tian Y, Ma L, Gong M, Su G, Zhu S, Zhang W, et al. Protein profiling and sizing of extracellular vesicles from colorectal cancer patients via flow cytometry. ACS Nano. 2018;12(1):671–80. https://doi.org/10.1021/acsnano.7b07782.
Coumans FAW, Brisson AR, Buzas EI, Dignat-George F, Drees EEE, El-Andaloussi S, et al. Methodological guidelines to study extracellular vesicles. Circ Res. 2017;120(10):1632–48. https://doi.org/10.1161/CIRCRESAHA.117.309417.
Vella LJ, Scicluna BJ, Cheng L, Bawden EG, Masters CL, Ang CS, et al. A rigorous method to enrich for exosomes from brain tissue. J Extracell Vesicles. 2017;6(1):1348885. https://doi.org/10.1080/20013078.2017.1348885.
Jingushi K, Uemura M, Ohnishi N, Nakata W, Fujita K, Naito T, et al. Extracellular vesicles isolated from human renal cell carcinoma tissues disrupt vascular endothelial cell morphology via azurocidin. Int J Cancer. 2018;142(3):607–17. https://doi.org/10.1002/ijc.31080.
Reuter VE, Tickoo SK. Differential diagnosis of renal tumours with clear cell histology. Pathology. 2010;42(4):374–83. https://doi.org/10.3109/00313021003785746.
Du W, Zhang L, Brett-Morris A, Aguila B, Kerner J, Hoppel CL, et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat Commun. 2017;8(1):1769. https://doi.org/10.1038/s41467-017-01965-8.
Acknowledgements
The authors thank Tina Wlajnitz for obtaining voluntary informed consent from patients. We thank Barbara Smith, microscopy specialist of the Microscope Facility at Johns Hopkins University School of Medicine, for acquiring TEM images. We thank Timothy H. Phelps, Professor and Medical Illustrator at Johns Hopkins University School of Medicine, for generating illustrations. We thank Dr. Kenneth W. Witwer, Associate Professor of Molecular and Comparative Pathobiology, Neurology, and Cellular and Molecular Medicine, for his feedback. Special thanks to Jan Lötvall for inspirational words. Finally, we thank all members of the Pienta Laboratory for their feedback
Funding
RCZ was supported by the Stichting Cure for Cancer foundation, Amsterdam, the Netherlands. LD was supported by the Shanghai Natural Science Fund exploration project (17ZR1447400), the Shanghai Jiao Tong University medicine-engineering cross project (YG2016QN56) and the Key Laboratory of Specialty Fiber Optics and Optical Access Network of Shanghai University. The Pienta Laboratory was supported by the Prostate Cancer Foundation, the Patrick C. Walsh Prostate Cancer Research Fund, and the William and Carolyn Stutt Research Fund. KJP was supported by NCI Grants U54CA143803, CA163124, CA093900, and CA143055. SRA was supported by the Patrick C. Walsh Prostate Cancer Research Fund and the Prostate Cancer Foundation.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Richard Zieren, and Liang Dong. The first draft of the manuscript was written by Richard Zieren and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
All procedures performed in studies were in accordance with the ethical standards of the Johns Hopkins Office of Human Subjects Research Institutional Review Board.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zieren, R.C., Dong, L., Pierorazio, P.M. et al. Extracellular vesicle isolation from human renal cancer tissue. Med Oncol 37, 28 (2020). https://doi.org/10.1007/s12032-020-1346-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-020-1346-1