
Abstract
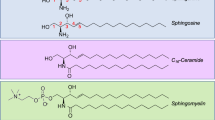
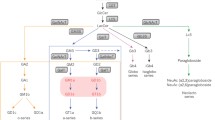
Sphingolipidoses constitute a large subgroup of lysosomal storage disorders (LSDs). Many of them are associated with a progressive neurodegeneration. As is the case for LSDs in general, most sphingolipidoses are caused by deficiencies in lysosomal hydrolases. However, accumulation of sphingolipids can also result from deficiencies in proteins involved in the transport or posttranslational modification of lysosomal enzymes, transport of lipids, or lysosomal membrane proteins required for transport of lysosomal degradation end products. The accumulation of sphingolipids in the lysosome together with secondary changes in the concentration and localization of other lipids may cause trafficking defects of membrane lipids and proteins, affect calcium homeostasis, induce the unfolded protein response, activate apoptotic cascades, and affect various signal transduction pathways. To what extent, however, these changes contribute to the pathogenesis of the diseases is not fully understood. Currently, there is no cure for sphingolipidoses. Therapies like enzyme replacement, pharmacological chaperone, and substrate reduction therapy, which have been shown to be efficient in non-neuronopathic LSDs, are currently evaluated in clinical trials of neuronopathic sphingolipidoses. In the future, neural stem cell therapy and gene therapy may become an option for these disorders.





Similar content being viewed by others

References
Achord, D. T., Brot, F. E., Bell, C. E., & Sly, W. S. (1978). Human beta-glucuronidase: In vivo clearance and in vitro uptake by a glycoprotein recognition system on reticuloendothelial cells. Cell, 15, 269–278.
Aerts, J. M., Groener, J. E., Kuiper, S., Donker-Koopman, W. E., Strijland, A., Ottenhoff, R., et al. (2008). Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proceedings of the National Academy of Sciences of the United States of America, 105, 2812–2817.
Ahmad, I., Hunter, R. E., Flax, J. D., Snyder, E. Y., & Erickson, R. P. (2007). Neural stem cell implantation extends life in Niemann-Pick C1 mice. Journal of Applied Genetics, 48, 269–272.
Andersson, U., Smith, D., Jeyakumar, M., Butters, T. D., Borja, M. C., Dwek, R. A., et al. (2004). Improved outcome of N-butyldeoxygalactonojirimycin-mediated substrate reduction therapy in a mouse model of Sandhoff disease. Neurobiology of Diseases, 16, 506–515.
Asheuer, M., Pflumio, F., Benhamida, S., Dubart-Kupperschmitt, A., Fouquet, F., Imai, Y., et al. (2004). Human CD34+ cells differentiate into microglia and express recombinant therapeutic protein. Proceedings of the National Academy of Sciences of the United States of America, 101, 3557–3562.
Austin, J. H. (1965). Metachromatic leukodystrophy. In C. C. Carter (Ed.), Medical aspects of mental retardation (p. 768). Springfield, IL: Charles C Thomas.
Baudry, M., Yao, Y., Simmons, D., Liu, J., & Bi, X. (2003). Postnatal development of inflammation in a murine model of Niemann-Pick type C disease: Immunohistochemical observations of microglia and astroglia. Experimental Neurology, 184, 887–903.
Beutler, E., & Grabowski, G. A. (2001). Gaucher disease. In C. R. Scriver, C. R. Beaudet, W. S. Sly, & D. Valle (Eds.), The metabolic, molecular basis of inherited diseases (8th ed., pp. 3635–3668). New York, NY: McGraw-Hill.
Biswas, S., Biesiada, H., Williams, T. D., & LeVine, S. M. (2003). Substrate reduction intervention by l-cycloserine in twitcher mice (globoid cell leukodystrophy) on a B6;CAST/Ei background. Neuroscience Letters, 347, 33–36.
Blanz, J., Stroobants, S., Lullmann-Rauch, R., Morelle, W., Ludemann, M., D’Hooge, R., et al. (2008). Reversal of peripheral and central neural storage and ataxia after recombinant enzyme replacement therapy in alpha-mannosidosis mice. Human Molecular Genetics, 17, 3437–3445.
Boelens, J. J. (2006). Trends in haematopoietic cell transplantation for inborn errors of metabolism. Journal of Inherited Metabolic Disease, 29, 413–420.
Bradová, V., Smíd, F., Ulrich-Bott, B., Roggendorf, W., Paton, B. C., & Harzer, K. (1993). Prosaposin deficiency: further characterization of the sphingolipid activator protein-deficient sibs. Multiple glycolipid elevations (including lactosylceramidosis), partial enzyme deficiencies and ultrastructure of the skin in this generalized sphingolipid storage disease. Human Genetics, 92, 143–152.
Brady, R. O. (2006). Enzyme replacement for lysosomal diseases. Annual Review of Medicine, 57, 283–296.
Brady, R. O., Pentchev, P. G., Gal, A. E., Hibbert, S. R., & Dekaban, A. S. (1974). Replacement therapy for inherited enzyme deficiency. Use of purified glucocerebrosidase in Gaucher’s disease. New England Journal of Medicine, 291, 989–993.
Broekman, M. L., Baek, R. C., Comer, L. A., Fernandez, J. L., Seyfried, T. N., & Sena-Esteves, M. (2007). Complete correction of enzymatic deficiency and neurochemistry in the GM1-gangliosidosis mouse brain by neonatal adeno-associated virus-mediated gene delivery. Molecular Therapy, 15, 30–37.
Buser, A. M., Schmid, D., Kern, F., Erne, B., Lazzati, T., & Schaeren-Wiemers, N. (2009). The myelin protein MAL affects peripheral nerve myelination: A new player influencing p75 neurotrophin receptor expression. European Journal of Neuroscience, 29, 2276–2290.
Butters, T. D., Dwek, R. A., & Platt, F. M. (2003). Therapeutic applications of imino sugars in lysosomal storage disorders. Current Topics in Medicinal Chemistry, 3, 561–574.
Cachon-Gonzalez, M. B., Wang, S. Z., Lynch, A., Ziegler, R., Cheng, S. H., & Cox, T. M. (2006). Effective gene therapy in an authentic model of Tay-Sachs-related diseases. Proceedings of the National Academy of Sciences of the United States of America, 103, 10373–10378.
Canuel, M., Korkidakis, A., Konnyu, K., & Morales, C. R. (2008). Sortilin mediates the lysosomal targeting of cathepsins D and H. Biochemical and Biophysical Research Communications, 373, 292–297.
Capablo, J. L., Saenz de Cabezon, A., Fraile, J., Alfonso, P., Pocovi, M., & Giraldo, P. (2008). Neurological evaluation of patients with Gaucher disease diagnosed as type 1. Journal of Neurology, Neurosurgery and Psychiatry, 79, 219–222.
Chen, C. S., Patterson, M. C., Wheatley, C. L., O’Brien, J. F., & Pagano, R. E. (1999). Broad screening test for sphingolipid-storage diseases. Lancet, 354, 901–905.
Cho, K. H., Kim, M. W., & Kim, S. U. (1997). Tissue culture model of Krabbe’s disease: Psychosine cytotoxicity in rat oligodendrocyte culture. Developmental Neuroscience, 19, 321–327.
Choudhury, A., Dominguez, M., Puri, V., Sharma, D. K., Narita, K., Wheatley, C. L., et al. (2002). Rab proteins mediate Golgi transport of caveola-internalized glycosphingolipids and correct lipid trafficking in Niemann-Pick C cells. Journal of Clinical Investigation, 109, 1541–1550.
Choudhury, A., Sharma, D. K., Marks, D. L., & Pagano, R. E. (2004). Elevated endosomal cholesterol levels in Niemann-Pick cells inhibit rab4 and perturb membrane recycling. Molecular Biology of the Cell, 15, 4500–4511.
Ciron, C., Cressant, A., Roux, F., Raoul, S., Cherel, Y., Hantraye, P., et al. (2009). Human alpha-iduronidase gene transfer mediated by adeno-associated virus types 1, 2, and 5 in the brain of nonhuman primates: Vector diffusion and biodistribution. Human Gene Therapy, 20, 350–360.
Clarke, J. T., Skomorowski, M. A., & Chang, P. L. (1989). Marked clinical difference between two sibs affected with juvenile metachromatic leukodystrophy. American Journal of Medical Genetics, 33, 10–13.
Colle, M. A., Piguet, F., Bertrand, L., Raoul, S., Bieche, I., Dubreil, L., et al. (2010). Efficient intracerebral delivery of AAV5 vector encoding human ARSA in non-human primate. Human Molecular Genetics, 19, 147–158.
Consiglio, A., Quattrini, A., Martino, S., Bensadoun, J. C., Dolcetta, D., Trojani, A., et al. (2001). In vivo gene therapy of metachromatic leukodystrophy by lentiviral vectors: Correction of neuropathology and protection against learning impairments in affected mice. Nature Medicine, 7, 310–316.
Cosma, M. P., Pepe, S., Annunziata, I., Newbold, R. F., Grompe, M., Parenti, G., et al. (2003). The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases. Cell, 113, 445–456.
Cosma, M. P., Pepe, S., Parenti, G., Settembre, C., Annunziata, I., Wade-Martins, R., et al. (2004). Molecular and functional analysis of SUMF1 mutations in multiple sulfatase deficiency. Human Mutation, 23, 576–581.
Cutler, R. G., Kelly, J., Storie, K., Pedersen, W. A., Tammara, A., Hatanpaa, K., et al. (2004). Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America, 101, 2070–2075.
Desnick, R., Ioannou, Y. A., & Eng, C. M. (2001). α-Galactosidase A deficiency: Fabry disease. In C. R. Scriver, A. L. Beaudet, W. S. Sly, & D. Valle (Eds.), The metabolic, molecular basis of inherited diseases (8th ed., pp. 3733–3774). New York, NY: McGraw-Hill.
Dierks, T., Schlotawa, L., Frese, M. A., Radhakrishnan, K., von Figura, K., & Schmidt, B. (2009). Molecular basis of multiple sulfatase deficiency, mucolipidosis II/III and Niemann-Pick C1 disease—Lysosomal storage disorders caused by defects of non-lysosomal proteins. Biochimica et Biophysica Acta, 1793, 710–725.
Dierks, T., Schmidt, B., Borissenko, L. V., Peng, J., Preusser, A., Mariappan, M., et al. (2003). Multiple sulfatase deficiency is caused by mutations in the gene encoding the human C(alpha)-formylglycine generating enzyme. Cell, 113, 435–444.
Duchen, L. W., Eicher, E. M., Jacobs, J. M., Scaravilli, F., & Teixeira, F. (1980). Hereditary leucodystrophy in the mouse: The new mutant twitcher. Brain, 103, 695–710.
Eckhardt, M., Hedayati, K. K., Pitsch, J., Lullmann-Rauch, R., Beck, H., Fewou, S. N., et al. (2007). Sulfatide storage in neurons causes hyperexcitability and axonal degeneration in a mouse model of metachromatic leukodystrophy. Journal of Neuroscience, 27, 9009–9021.
Elliot-Smith, E., Speak, A. O., Lloyd-Evans, E., Smith, D. A., van der Spoel, A. C., Jeyakumar, M., et al. (2008). Beneficial effects of substrate reduction therapy in a mouse model of GM1 gangliosidosis. Molecular Genetics and Metabolism, 94, 204–211.
Elstein, D., Hollak, C., Aerts, J. M., van Weely, S., Maas, M., Cox, T. M., et al. (2004). Sustained therapeutic effects of oral miglustat (Zavesca, N-butyldeoxynojirimycin, OGT 918) in type I Gaucher disease. Journal of Inherited Metabolic Disease, 27, 757–766.
Enquist, I. B., Lo Bianco, C., Ooka, A., Nilsson, E., Månsson, J. E., Ehinger, M., et al. (2007). Murine models of acute neuronopathic Gaucher disease. Proceedings of the National Academy of Sciences of the United States of America, 104, 17483–17488.
Escolar, M. L., Poe, M. D., Provenzale, J. M., Richards, K. C., Allison, J., Wood, S., et al. (2005). Transplantation of umbilical-cord blood in babies with infantile Krabbe’s disease. New England Journal of Medicine, 352, 2069–2081.
Eskelinen, E. L., & Saftig, P. (2009). Autophagy: A lysosomal degradation pathway with a central role in health and disease. Biochimica et Biophysica Acta, 1793, 664–673.
Fan, J. Q. (2008). A counterintuitive approach to treat enzyme deficiencies: Use of enzyme inhibitors for restoring mutant enzyme activity. Biological Chemistry, 389, 1–11.
Fan, J. Q., & Ishii, S. (2003). Cell-based screening of active-site specific chaperone for the treatment of Fabry disease. Methods in Enzymology, 363, 412–420.
Fan, J. Q., Ishii, S., Asano, N., & Suzuki, Y. (1999). Accelerated transport and maturation of lysosomal alpha-galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nature Medicine, 5, 112–115.
Farfel-Becker, T., Vitner, E., Dekel, H., Leshem, N., Enquist, I. B., Karlsson, S., et al. (2009). No evidence for activation of the unfolded protein response in neuronopathic models of Gaucher disease. Human Molecular Genetics, 18, 1482–1488.
Ficicioglu, C. (2008). Review of miglustat for clinical management in Gaucher disease type 1. Therapeutics and Clinical Risk Management, 4, 425–431.
Fletcher, J. M. (2006). Screening for lysosomal storage disorders—a clinical perspective. Journal of Inherited Metabolic Disease, 29, 405–408.
Frank, M., van der Haar, M. E., Schaeren-Wiemers, N., & Schwab, M. E. (1998). rMAL is a glycosphingolipid-associated protein of myelin and apical membranes of epithelial cells in kidney and stomach. Journal of Neuroscience, 18, 4901–4913.
Fredman, P. (1998). Sphingolipids and cell signalling. Journal of Inherited Metabolic Disease, 21, 472–480.
Fressinaud, C., & Vallat, J. M. (1994). Basic fibroblast growth factor improves recovery after chemically induced breakdown of myelin-like membranes in pure oligodendrocyte cultures. Journal of Neuroscience Research, 38, 202–213.
Fukuda, T., Ewan, L., Bauer, M., Mattaliano, R. J., Zaal, K., Ralston, E., et al. (2006). Dysfunction of endocytic and autophagic pathways in a lysosomal storage disease. Annals of Neurology, 59, 700–708.
Fuller, M., Rozaklis, T., Lovejoy, M., Zarrinkalam, K., Hopwood, J. J., & Meikle, P. J. (2008). Glucosylceramide accumulation is not confined to the lysosome in fibroblasts from patients with Gaucher disease. Molecular Genetics and Metabolism, 93, 437–443.
Gieselmann, V. (2008). Metachromatic leukodystrophy: Genetics, pathogenesis and therapeutic options. Acta Paediatrica. Supplement, 97, 15–21.
Gillard, B. K., Clement, R. G., & Marcus, D. M. (1998). Variations among cell lines in the synthesis of sphingolipids in de novo and recycling pathways. Glycobiology, 8, 885–890.
Giri, S., Jatana, M., Rattan, R., Won, J. S., Singh, I., & Singh, A. K. (2002). Galactosylsphingosine (psychosine)-induced expression of cytokine-mediated inducible nitric oxide synthases via AP-1 and C/EBP: Implications for Krabbe disease. The FASEB Journal, 16, 661–672.
Giri, S., Khan, M., Rattan, R., Singh, I., & Singh, A. K. (2006). Krabbe disease: Psychosine-mediated activation of phospholipase A2 in oligodendrocyte cell death. Journal of Lipid Research, 47, 1478–1492.
Givogri, M. I., Galbiati, F., Fasano, S., Amadio, S., Perani, L., Superchi, D., et al. (2006). Oligodendroglial progenitor cell therapy limits central neurological deficits in mice with metachromatic leukodystrophy. Journal of Neuroscience, 26, 3109–3119.
Goker-Alpan, O., Schiffmann, R., LaMarca, M. E., Nussbaum, R. L., McInerney-Leo, A., & Sidransky, E. (2004). Parkinsonism among Gaucher disease carriers. Journal of Medical Genetics, 41, 937–940.
Grabowski, G. A., Kolodny, E. H., Weinreb, N. J., Rosenbloom, B. E., Prakash-Cheng, A., et al. (2006). Gaucher disease: Phenotypic and genetic variation. Chapter 146.1. In C. R. Scriver, W. S. Sly, A. Beaudet, D. Valle, & B. Childs (Eds.), The metabolic, molecular bases of inherited diseases. New York, NY: McGraw-Hill.
Gravel, R. A., Kaback, M. M., Proia, R., Sandhoff, K., Suzuki, K., & Suzuki, K. (2001). The GM2 gangliosidosis. In C. R. Scriver, A. L. Beaudet, W. S. Sly, & D. Valle (Eds.), The metabolic, molecular basis of inherited diseases (8th ed., pp. 3827–3876). New York, NY: McGraw-Hill.
Green, D. R., & Kroemer, G. (2004). The pathophysiology of mitochondrial cell death. Science, 305, 626–629.
Grubb, J. H., Vogler, C., Levy, B., Galvin, N., Tan, Y., & Sly, W. S. (2008). Chemically modified beta-glucuronidase crosses blood-brain barrier and clears neuronal storage in murine mucopolysaccharidosis VII. Proceedings of the National Academy of Sciences of the United States of America, 105, 2616–2621.
Hahn, C. N., del Pilar Martin, M., Schroder, M., Vanier, M. T., Hara, Y., Suzuki, M., et al. (1997). Generalized CNS disease and massive GM1-ganglioside accumulation in mice defective in lysosomal acid beta-galactosidase. Human Molecular Genetics, 6, 205–211.
Han, X., Holtzman, D. M., McKeel, D. W., Kelley, J., Jr, & Morris, J. C. (2002). Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer’s disease: Potential role in disease pathogenesis. Journal of Neurochemistry, 82, 809–818.
Hannun, Y. A., & Bell, R. M. (1987). Lysosphingolipids inhibit protein kinase C: Implications for the sphingolipidoses. Science, 235, 670–674.
Hara, T., Nakamura, K., Matsui, M., Yamamoto, A., Nakahara, Y., Suzuki-Migishima, R., et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature, 441, 885–889.
Harzer, K., Paton, B. C., Poulos, A., Kustermann-Kuhn, B., Roggendorf, W., Grisar, T., et al. (1989). Sphingolipid activator protein deficiency in a 16-week-old atypical Gaucher disease patient and his fetal sibling: biochemical signs of combined sphingolipidoses. European Journal of Pediatrics, 149, 31–39.
Haskins, M. (2009). Gene therapy for lysosomal storage diseases (LSDs) in large animal models. ILAR Journal, 50, 112–121.
Hein, L. K., Meikle, P. J., Hopwood, J. J., & Fuller, M. (2007). Secondary sphingolipid accumulation in a macrophage model of Gaucher disease. Molecular Genetics and Metabolism, 92, 336–345.
Heitner, R., Elstein, D., Aerts, J., Weely, S., & Zimran, A. (2002). Low-dose N-butyldeoxynojirimycin (OGT 918) for type I Gaucher disease. Blood Cells, Molecules, and Diseases, 28, 127–133.
Holleran, W. M., Ginns, E. I., Menon, G. K., Grundmann, J. U., Fartasch, M., McKinney, C. E., et al. (1994). Consequences of beta-glucocerebrosidase deficiency in epidermis. Ultrastructure and permeability barrier alterations in Gaucher disease. Journal of Clinical Investigation, 93, 1756–1764.
Holleran, W. M., Takagi, Y., Menon, G. K., Legler, G., Feingold, K. R., & Elias, P. M. (1993). Processing of epidermal glucosylceramides is required for optimal mammalian cutaneous permeability barrier function. Journal of Clinical Investigation, 91, 1656–1664.
Im, D. S., Heise, C. E., Nguyen, T., O’Dowd, B. F., & Lynch, K. R. (2001). Identification of a molecular target of psychosine and its role in globoid cell formation. Journal of Cell Biology, 153, 429–434.
Jatana, M., Giri, S., & Singh, A. K. (2002). Apoptotic positive cells in Krabbe brain and induction of apoptosis in rat C6 glial cells by psychosine. Neuroscience Letters, 330, 183–187.
Jeyakumar, M., Lee, J. P., Sibson, N. R., Lowe, J. P., Stuckey, D. J., Tester, K., et al. (2009). Neural stem cell transplantation benefits a monogenic neurometabolic disorder during the symptomatic phase of disease. Stem Cells, 27, 2362–2370.
Jeyakumar, M., Smith, D. A., Williams, I. M., Borja, M. C., Neville, D. C., Butters, T. D., et al. (2004). NSAIDs increase survival in the Sandhoff disease mouse: Synergy with N-butyldeoxynojirimycin. Annals of Neurology, 56, 642–649.
Jeyakumar, M., Thomas, R., Elliot-Smith, E., Smith, D. A., van der Spoel, A. C., d’Azzo, A., et al. (2003). Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain, 126, 974–987.
Jonas, S., van Loo, B., Hyvonen, M., & Hollfelder, F. (2008). A new member of the alkaline phosphatase superfamily with a formylglycine nucleophile: Structural and kinetic characterisation of a phosphonate monoester hydrolase/phosphodiesterase from Rhizobium leguminosarum. Journal of Molecular Biology, 384, 120–136.
Kanazawa, T., Nakamura, S., Momoi, M., Yamaji, T., Takematsu, H., Yano, H., et al. (2000). Inhibition of cytokinesis by a lipid metabolite, psychosine. Journal of Cell Biology, 149, 943–950.
Kaptzan, T., West, S. A., Holicky, E. L., Wheatley, C. L., Marks, D. L., Wang, T., et al. (2009). Development of a Rab9 transgenic mouse and its ability to increase the lifespan of a murine model of Niemann-Pick type C disease. American Journal of Pathology, 174, 14–20.
Kasperzyk, J. L., d’Azzo, A., Platt, F. M., Alroy, J., & Seyfried, T. N. (2005). Substrate reduction reduces gangliosides in postnatal cerebrum-brainstem and cerebellum in GM1 gangliosidosis mice. Journal of Lipid Research, 46, 744–751.
Kasperzyk, J. L., El-Abbadi, M. M., Hauser, E. C., D’Azzo, A., Platt, F. M., & Seyfried, T. N. (2004). N-Butyldeoxygalactonojirimycin reduces neonatal brain ganglioside content in a mouse model of GM1 gangliosidosis. Journal of Neurochemistry, 89, 645–653.
Kennedy, D. W., & Abkowitz, J. L. (1997). Kinetics of central nervous system microglial and macrophage engraftment: Analysis using a transgenic bone marrow transplantation model. Blood, 90, 986–993.
Klein, D., Schmandt, T., Muth-Kohne, E., Perez-Bouza, A., Segschneider, M., Gieselmann, V., et al. (2006). Embryonic stem cell-based reduction of central nervous system sulfatide storage in an animal model of metachromatic leukodystrophy. Gene Therapy, 13, 1686–1695.
Klein, D., Yaghootfam, A., Matzner, U., Koch, B., Braulke, T., & Gieselmann, V. (2009). Mannose 6-phosphate receptor-dependent endocytosis of lysosomal enzymes is increased in sulfatide-storing kidney cells. Biological Chemistry, 390, 41–48.
Ko, D. C., Milenkovic, L., Beier, S. M., Manuel, H., Buchanan, J., & Scott, M. P. (2005). Cell-autonomous death of cerebellar purkinje neurons with autophagy in Niemann-Pick type C disease. PLoS Genetics, 1, 81–95.
Kobayashi, T., Beuchat, M. H., Lindsay, M., Frias, S., Palmiter, R. D., Sakuraba, H., et al. (1999). Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nature Cell Biology, 1, 113–118.
Kobayashi, T., Goto, I., Okada, S., Orii, T., Ohno, K., & Nakano, T. (1992). Accumulation of lysosphingolipids in tissues from patients with GM1 and GM2 gangliosidoses. Journal of Neurochemistry, 59, 1452–1458.
Koike, M., Shibata, M., Waguri, S., Yoshimura, K., Tanida, I., Kominami, E., et al. (2005). Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease). American Journal of Pathology, 167, 1713–1728.
Kollmann, K., Pohl, S., Marschner, K., Encarnação, M., Sakwa, I., Tiede, S., et al. (2010). Mannose phosphorylation in health and disease. European Journal of Cell Biology, 89, 117–123.
Kolter, T., & Sandhoff, K. (1999). Sphingolipids-their metabolic pathways and the pathobiochemistry of neurodegenerative diseases. Angewandte Chemie International Edition, 38, 1532–1568.
Korkotian, E., Schwarz, A., Pelled, D., Schwarzmann, G., Segal, M., & Futerman, A. H. (1999). Elevation of intracellular glucosylceramide levels results in an increase in endoplasmic reticulum density and in functional calcium stores in cultured neurons. The Journal of Biological Chemistry, 274, 21673–21678.
Kornfeld, S. (1986). Trafficking of lysosomal enzymes in normal and disease states. Journal of Clinical Investigation, 77, 1–6.
Kretz, K. A., Carson, G. S., Morimoto, S., Kishimoto, Y., Fluharty, A. L., & O’Brien, J. S. (1990). Characterization of a mutation in a family with saposin B deficiency: A glycosylation site defect. Proceedings of the National Academy of Sciences of the United States of America, 87, 2541–2544.
Krivit, W. (2004). Allogeneic stem cell transplantation for the treatment of lysosomal and peroxisomal metabolic diseases. Springer Seminars in Immunopathology, 26, 119–132.
Krivit, W., Peters, C., & Shapiro, E. G. (1999). Bone marrow transplantation as effective treatment of central nervous system disease in globoid cell leukodystrophy, metachromatic leukodystrophy, adrenoleukodystrophy, mannosidosis, fucosidosis, aspartylglucosaminuria, Hurler, Maroteaux-Lamy, and Sly syndromes, and Gaucher disease type III. Current Opinion in Neurology, 12, 167–176.
Kwon, H. J., Abi-Mosleh, L., Wang, M. L., Deisenhofer, J., Goldstein, J. L., Brown, M. S., et al. (2009). Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell, 137, 1213–1224.
Lamark, T., Kirkin, V., Dikic, I., & Johansen, T. (2009). NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle, 8, 1986–1990.
Lebrand, C., Corti, M., Goodson, H., Cosson, P., Cavalli, V., Mayran, N., et al. (2002). Late endosome motility depends on lipids via the small GTPase Rab7. EMBO Journal, 21, 1289–1300.
Lee, W. C., Courtenay, A., Troendle, F. J., Stallings-Mann, M. L., Dickey, C. A., DeLucia, M. W., et al. (2005). Enzyme replacement therapy results in substantial improvements in early clinical phenotype in a mouse model of globoid cell leukodystrophy. The FASEB Journal, 19, 1549–1551.
Lee, J. P., Jeyakumar, M., Gonzalez, R., Takahashi, H., Lee, P. J., Baek, R. C., et al. (2007). Stem cells act through multiple mechanisms to benefit mice with neurodegenerative metabolic disease. Nature Medicine, 13, 439–447.
Lefrancois, S., Zeng, J., Hassan, A. J., Canuel, M., & Morales, C. R. (2003). The lysosomal trafficking of sphingolipid activator proteins (SAPs) is mediated by sortilin. EMBO Journal, 22, 6430–6437.
Leventhal, A. R., Chen, W., Tall, A. R., & Tabas, I. (2001). Acid sphingomyelinase-deficient macrophages have defective cholesterol trafficking and efflux. The Journal of Biological Chemistry, 276, 44976–44983.
Li, S. C., Li, Y. T., Moriya, S., & Miyagi, T. (2001). Degradation of G(M1) and G(M2) by mammalian sialidases. Biochemical Journal, 360, 233–237.
Lieberman, R. L., Wustman, B. A., Huertas, P., Powe, A. C., Jr, Pine, C. W., Khanna, R., et al. (2007). Structure of acid beta-glucosidase with pharmacological chaperone provides insight into Gaucher disease. Nature Chemical Biology, 3, 101–107.
Liu, Y., Wada, R., Kawai, H., Sango, K., Deng, C., Tai, T., et al. (1999). A genetic model of substrate deprivation therapy for a glycosphingolipid storage disorder. Journal of Clinical Investigation, 103, 497–505.
Lloyd-Evans, E., Morgan, A. J., He, X., Smith, D. A., Elliot-Smith, E., Sillence, D. J., et al. (2008). Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nature Medicine, 14, 1247–1255.
Lloyd-Evans, E., Pelled, D., Riebeling, C., Bodennec, J., de-Morgan, A., Waller, H., et al. (2003). Glucosylceramide and glucosylsphingosine modulate calcium mobilization from brain microsomes via different mechanisms. The Journal of Biological Chemistry, 278, 23594–23599.
Lui, K., Commens, C., Choong, R., & Jaworski, R. (1988). Collodion babies with Gaucher’s disease. Archives of Disease in Childhood, 63, 854–886.
Maegawa, G. H., van Giersbergen, P. L., Yang, S., Banwell, B., Morgan, C. P., Dingemanse, J., et al. (2009). Pharmacokinetics, safety and tolerability of miglustat in the treatment of pediatric patients with GM2 gangliosidosis. Molecular Genetics and Metabolism, 97, 284–291.
Malhotra, J. D., & Kaufman, R. J. (2007). The endoplasmic reticulum and the unfolded protein response. Seminars in Cell & Developmental Biology, 18, 716–731.
Martino, S., Marconi, P., Tancini, B., Dolcetta, D., De Angelis, M. G., Montanucci, P., et al. (2005). A direct gene transfer strategy via brain internal capsule reverses the biochemical defect in Tay-Sachs disease. Human Molecular Genetics, 14, 2113–2123.
Matsuda, J., Kido, M., Tadano-Aritomi, K., Ishizuka, I., Tominaga, K., Toida, K., et al. (2004). Mutation in saposin D domain of sphingolipid activator protein gene causes urinary system defects and cerebellar Purkinje cell degeneration with accumulation of hydroxy fatty acid-containing ceramide in mouse. Human Molecular Genetics, 13, 2709–2723.
Matsuda, J., Suzuki, O., Oshima, A., Yamamoto, Y., Noguchi, A., Takimoto, K., et al. (2003). Chemical chaperone therapy for brain pathology in G(M1)-gangliosidosis. Proceedings of the National Academy of Sciences of the United States of America, 100, 15912–15917.
Matsushima, G. K., Taniike, M., Glimcher, L. H., Grusby, M. J., Frelinger, J. A., Suzuki, K., et al. (1994). Absence of MHC class II molecules reduces CNS demyelination, microglial/macrophage infiltration, and twitching in murine globoid cell leukodystrophy. Cell, 78, 645–656.
Matzner, U., Herbst, E., Hedayati, K. K., Lullmann-Rauch, R., Wessig, C., Schroder, S., et al. (2005). Enzyme replacement improves nervous system pathology and function in a mouse model for metachromatic leukodystrophy. Human Molecular Genetics, 14, 1139–1152.
Matzner, U., Lullmann-Rauch, R., Stroobants, S., Andersson, C., Weigelt, C., Eistrup, C., et al. (2009). Enzyme replacement improves ataxic gait and central nervous system histopathology in a mouse model of metachromatic leukodystrophy. Molecular Therapy, 17, 600–606.
McInnes, B., Potier, M., Wakamatsu, N., Melancon, S. B., Klavins, M. H., Tsuji, S., et al. (1992). An unusual splicing mutation in the HEXB gene is associated with dramatically different phenotypes in patients from different racial backgrounds. Journal of Clinical Investigation, 90, 306–314.
Meikle, P. J., Hopwood, J. J., Clague, A. E., & Carey, W. F. (1999). Prevalence of lysosomal storage disorders. JAMA, 281, 249–254.
Molander-Melin, M., Pernber, Z., Franken, S., Gieselmann, V., Mansson, J. E., & Fredman, P. (2004). Accumulation of sulfatide in neuronal and glial cells of arylsulfatase A deficient mice. Journal of Neurocytology, 33, 417–427.
Moser, H. W., Linke, T., Fensom, A. H., Levade, T., & Sandhoff, K. (2001). Acid ceramidase deficiency: Farber lipogranulomatosis. In C. R. Scriver, A. L. Beaudet, W. S. Sly, & D. Valle (Eds.), The metabolic, molecular basis of inherited diseases (8th ed., pp. 3573–3585). New York, NY: McGraw-Hill.
Myerowitz, R., Lawson, D., Mizukami, H., Mi, Y., Tifft, C. J., & Proia, R. L. (2002). Molecular pathophysiology in Tay-Sachs and Sandhoff diseases as revealed by gene expression profiling. Human Molecular Genetics, 11, 1343–1350.
Narita, K., Choudhury, A., Dobrenis, K., Sharma, D. K., Holicky, E. L., Marks, D. L., et al. (2005). Protein transduction of Rab9 in Niemann-Pick C cells reduces cholesterol storage. The FASEB Journal, 19, 1558–1560.
Neuenhofer, S., Conzelmann, E., Schwarzmann, G., Egge, H., & Sandhoff, K. (1986). Occurrence of lysoganglioside lyso-GM2 (II3-Neu5Ac-gangliotriaosylsphingosine) in GM2 gangliosidosis brain. Biological Chemistry Hoppe-Seyler, 367, 241–244.
Ni, X., & Morales, C. R. (2006). The lysosomal trafficking of acid sphingomyelinase is mediated by sortilin and mannose 6-phosphate receptor. Traffic, 7, 889–902.
O’Brien, J. S., Carson, G. S., Seo, H. C., Hiraiwa, M., & Kishimoto, Y. (1994). Identification of prosaposin as a neurotrophic factor. Proceedings of the National Academy of Sciences of the United States of America, 91, 9593–9596.
O’Brien, J. S., & Kishimoto, Y. (1991). Saposin proteins: structure, function, and role in human lysosomal storage disorders. The FASEB Journal, 5, 301–308.
Opitz, J. M., Stiles, F. C., Wise, D., Race, R. R., Sanger, R., von Gemmingen, G. R., et al. (1965). The genetics of angiokeratoma corporis diffusum (Fabry’s Disease) and its linkage relations with the Xg locus. American Journal of Human Genetics, 17, 325–342.
Ozkara, H. A., & Topcu, M. (2004). Sphingolipidoses in Turkey. Brain and Development, 26, 363–366.
Pacheco, C. D., Kunkel, R., & Lieberman, A. P. (2007). Autophagy in Niemann-Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Human Molecular Genetics, 16, 1495–1503.
Pagano, R. E. (2003). Endocytic trafficking of glycosphingolipids in sphingolipid storage diseases. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 358, 885–891.
Parenti, G. (2009). Treating lysosomal storage diseases with pharmacological chaperones: From concept to clinics. EMBO Molecular Medicine, 1, 268–279.
Parenti, G., Zuppaldi, A., Gabriela Pittis, M., Rosaria Tuzzi, M., Annunziata, I., Meroni, G., et al. (2007). Pharmacological enhancement of mutated alpha-glucosidase activity in fibroblasts from patients with Pompe disease. Molecular Therapy, 15, 508–514.
Pelled, D., Lloyd-Evans, E., Riebeling, C., Jeyakumar, M., Platt, F. M., & Futerman, A. H. (2003). Inhibition of calcium uptake via the sarco/endoplasmic reticulum Ca2+-ATPase in a mouse model of Sandhoff disease and prevention by treatment with N-butyldeoxynojirimycin. The Journal of Biological Chemistry, 278, 29496–29501.
Pelled, D., Trajkovic-Bodennec, S., Lloyd-Evans, E., Sidransky, E., Schiffmann, R., & Futerman, A. H. (2005). Enhanced calcium release in the acute neuronopathic form of Gaucher disease. Neurobiology of Diseases, 18, 83–88.
Phaneuf, D., Wakamatsu, N., Huang, J. Q., Borowski, A., Peterson, A. C., Fortunato, S. R., et al. (1996). Dramatically different phenotypes in mouse models of human Tay-Sachs and Sandhoff diseases. Human Molecular Genetics, 5, 1–14.
Pineda, M., Perez-Poyato, M. S., O’Callaghan, M., Vilaseca, M. A., Pocovi, M., Domingo, R., et al. (2010). Clinical experience with miglustat therapy in pediatric patients with Niemann-Pick disease type C: A case series. Molecular Genetics and Metabolism, 99, 358–366.
Platt, F. M., Neises, G. R., Dwek, R. A., & Butters, T. D. (1994a). N-Butyldeoxynojirimycin is a novel inhibitor of glycolipid biosynthesis. The Journal of Biological Chemistry, 269, 8362–8365.
Platt, F. M., Neises, G. R., Karlsson, G. B., Dwek, R. A., & Butters, T. D. (1994b). N-Butyldeoxygalactonojirimycin inhibits glycolipid biosynthesis but does not affect N-linked oligosaccharide processing. The Journal of Biological Chemistry, 269, 27108–27114.
Platt, F. M., Neises, G. R., Reinkensmeier, G., Townsend, M. J., Perry, V. H., Proia, R. L., et al. (1997). Prevention of lysosomal storage in Tay-Sachs mice treated with N-butyldeoxynojirimycin. Science, 276, 428–431.
Puri, V., Watanabe, R., Dominguez, M., Sun, X., Wheatley, C. L., Marks, D. L., et al. (1999). Cholesterol modulates membrane traffic along the endocytic pathway in sphingolipid-storage diseases. Nature Cell Biology, 1, 386–388.
Radu, C. G., Cheng, D., Nijagal, A., Riedinger, M., McLaughlin, J., Yang, L. V., et al. (2006). Normal immune development and glucocorticoid-induced thymocyte apoptosis in mice deficient for the T-cell death-associated gene 8 receptor. Molecular and Cellular Biology, 26, 668–677.
Ramakrishnan, H., Hedayati, K. K., Lullmann-Rauch, R., Wessig, C., Fewou, S. N., Maier, H., et al. (2007). Increasing sulfatide synthesis in myelin-forming cells of arylsulfatase A-deficient mice causes demyelination and neurological symptoms reminiscent of human metachromatic leukodystrophy. Journal of Neuroscience, 27, 9482–9490.
Reczek, D., Schwake, M., Schroder, J., Hughes, H., Blanz, J., Jin, X., et al. (2007). LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell, 131, 770–783.
Rommerskirch, W., & von Figura, K. (1992). Multiple sulfatase deficiency: Catalytically inactive sulfatases are expressed from retrovirally introduced sulfatase cDNAs. Proceedings of the National Academy of Sciences of the United States of America, 89, 2561–2565.
Ron, I., & Horowitz, M. (2005). ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Human Molecular Genetics, 14, 2387–2398.
Rosengren, B., Fredman, P., Mansson, J. E., & Svennerholm, L. (1989). Lysosulfatide (galactosylsphingosine-3-O-sulfate) from metachromatic leukodystrophy and normal human brain. Journal of Neurochemistry, 52, 1035–1041.
Sango, K., Yamanaka, S., Hoffmann, A., Okuda, Y., Grinberg, A., Westphal, H., et al. (1995). Mouse models of Tay-Sachs and Sandhoff diseases differ in neurologic phenotype and ganglioside metabolism. Nature Genetics, 11, 170–176.
Sano, R., Annunziata, I., Patterson, A., Moshiach, S., Gomero, E., Opferman, J., et al. (2009). GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca(2+)-dependent mitochondrial apoptosis. Molecular Cell, 36, 500–511.
Saravanan, K., Schaeren-Wiemers, N., Klein, D., Sandhoff, R., Schwarz, A., Yaghootfam, A., et al. (2004). Specific downregulation and mistargeting of the lipid raft-associated protein MAL in a glycolipid storage disorder. Neurobiology of Diseases, 16, 396–406.
Sawkar, A. R., Adamski-Werner, S. L., Cheng, W. C., Wong, C. H., Beutler, E., Zimmer, K. P., et al. (2005). Gaucher disease-associated glucocerebrosidases show mutation-dependent chemical chaperoning profiles. Chemistry & Biology, 12, 1235–1244.
Sawkar, A. R., Cheng, W. C., Beutler, E., Wong, C. H., Balch, W. E., & Kelly, J. W. (2002). Chemical chaperones increase the cellular activity of N370S beta-glucosidase: A therapeutic strategy for Gaucher disease. Proceedings of the National Academy of Sciences of the United States of America, 99, 15428–15433.
Schaeren-Wiemers, N., Bonnet, A., Erb, M., Erne, B., Bartsch, U., Kern, F., et al. (2004). The raft-associated protein MAL is required for maintenance of proper axon–glia interactions in the central nervous system. Journal of Cell Biology, 166, 731–742.
Schiffmann, R., Fitzgibbon, E. J., Harris, C., DeVile, C., Davies, E. H., Abel, L., et al. (2008). Randomized, controlled trial of miglustat in Gaucher’s disease type 3. Annals of Neurology, 64, 514–522.
Schmidt, B., Selmer, T., Ingendoh, A., & von Figura, K. (1995). A novel amino acid modification in sulfatases that is defective in multiple sulfatase deficiency. Cell, 82, 271–278.
Schnabel, D., Schröder, M., Fürst, W., Klein, A., Hurwitz, R., Zenk, T., et al. (1992). Simultaneous deficiency of sphingolipid activator proteins 1 and 2 is caused by a mutation in the initiation codon of their common gene. The Journal of Biological Chemistry, 267, 3312–3315.
Schott, I., Hartmann, D., Gieselmann, V., & Lullmann-Rauch, R. (2001). Sulfatide storage in visceral organs of arylsulfatase A-deficient mice. Virchows Archiv, 439, 90–96.
Schueler, U. H., Kolter, T., Kaneski, C. R., Blusztajn, J. K., Herkenham, M., Sandhoff, K., et al. (2003). Toxicity of glucosylsphingosine (glucopsychosine) to cultured neuronal cells: A model system for assessing neuronal damage in Gaucher disease type 2 and 3. Neurobiology of Diseases, 14, 595–601.
Settembre, C., Annunziata, I., Spampanato, C., Zarcone, D., Cobellis, G., Nusco, E., et al. (2007). Systemic inflammation and neurodegeneration in a mouse model of multiple sulfatase deficiency. Proceedings of the National Academy of Sciences of the United States of America, 104, 4506–4511.
Settembre, C., Fraldi, A., Jahreiss, L., Spampanato, C., Venturi, C., Medina, D., et al. (2008). A block of autophagy in lysosomal storage disorders. Human Molecular Genetics, 17, 119–129.
Sevin, C., Benraiss, A., Van Dam, D., Bonnin, D., Nagels, G., Verot, L., et al. (2006). Intracerebral adeno-associated virus-mediated gene transfer in rapidly progressive forms of metachromatic leukodystrophy. Human Molecular Genetics, 15, 53–64.
Sevin, C., Verot, L., Benraiss, A., Van Dam, D., Bonnin, D., Nagels, G., et al. (2007). Partial cure of established disease in an animal model of metachromatic leukodystrophy after intracerebral adeno-associated virus-mediated gene transfer. Gene Therapy, 14, 405–414.
Shapiro, B. E., Pastores, G. M., Gianutsos, J., Luzy, C., & Kolodny, E. H. (2009). Miglustat in late-onset Tay-Sachs disease: A 12-month, randomized, controlled clinical study with 24 months of extended treatment. Genetics in Medicine, 11, 425–433.
Sharma, D. K., Brown, J. C., Choudhury, A., Peterson, T. E., Holicky, E., Marks, D. L., et al. (2004). Selective stimulation of caveolar endocytosis by glycosphingolipids and cholesterol. Molecular Biology of the Cell, 15, 3114–3122.
Sidman, R. L., Li, J., Stewart, G. R., Clarke, J., Yang, W., Snyder, E. Y., et al. (2007). Injection of mouse and human neural stem cells into neonatal Niemann-Pick A model mice. Brain Research, 1140, 195–204.
Sikora, J., Harzer, K., & Elleder, M. (2007). Neurolysosomal pathology in human prosaposin deficiency suggests essential neurotrophic function of prosaposin. Acta Neuropathologica, 113, 163–175.
Sillence, D. J., Puri, V., Marks, D. L., Butters, T. D., Dwek, R. A., Pagano, R. E., et al. (2002). Glucosylceramide modulates membrane traffic along the endocytic pathway. Journal of Lipid Research, 43, 1837–1845.
Sinha, S., & Levine, B. (2008). The autophagy effector Beclin 1: A novel BH3-only protein. Oncogene, 27(Suppl 1), S137–S148.
Smith, D., Wallom, K. L., Williams, I. M., Jeyakumar, M., & Platt, F. M. (2009). Beneficial effects of anti-inflammatory therapy in a mouse model of Niemann-Pick disease type C1. Neurobiology of Diseases, 36, 242–251.
Snyder, E. Y., Taylor, R. M., & Wolfe, J. H. (1995). Neural progenitor cell engraftment corrects lysosomal storage throughout the MPS VII mouse brain. Nature, 374, 367–370.
Sorkin, A., & von Zastrow, M. (2009). Endocytosis and signalling: Intertwining molecular networks. Nature Reviews. Molecular Cell Biology, 10, 609–622.
Spiegel, R., Bach, G., Sury, V., Mengistu, G., Meidan, B., Shalev, S., et al. (2005). A mutation in the saposin A coding region of the prosaposin gene in an infant presenting as Krabbe disease: First report of saposin A deficiency in humans. Molecular Genetics and Metabolism, 84, 160–166.
Stenmark, H. (2009). Rab GTPases as coordinators of vesicle traffic. Nature Reviews. Molecular Cell Biology, 10, 513–525.
Strazza, M., Luddi, A., Carbone, M., Rafi, M. A., Costantino-Ceccarini, E., & Wenger, D. A. (2009). Significant correction of pathology in brains of twitcher mice following injection of genetically modified mouse neural progenitor cells. Molecular Genetics and Metabolism, 97, 27–34.
Sueyoshi, N., Maehara, T., & Ito, M. (2001). Apoptosis of Neuro2a cells induced by lysosphingolipids with naturally occurring stereochemical configurations. Journal of Lipid Research, 42, 1197–1202.
Sugiyama, E., Uemura, K., Hara, A., & Taketomi, T. (1990). Effects of various lysosphingolipids on cell growth, morphology and lipid composition in three neuroblastoma cell lines. Biochemical and Biophysical Research Communications, 169, 673–679.
Sun, Y., Liou, B., Ran, H., Skelton, M. R., Williams, M. T., Vorhees, C. V., et al. (2010). Neuronopathic Gaucher disease in the mouse: Viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits. Human Molecular Genetics, 19, 1088–1097.
Sun, Y., Witte, D. P., Zamzow, M., Ran, H., Quinn, B., Matsuda, J., et al. (2007). Combined saposin C and D deficiencies in mice lead to a neuronopathic phenotype, glucosylceramide and alpha-hydroxy ceramide accumulation, and altered prosaposin trafficking. Human Molecular Genetics, 16, 957–971.
Suzuki, Y., Oshima, A., & Nanba, E. (2001). Beta-galactosidase deficiency (beta-galactosidosis): GM1 gangliosidosis and Morquio B disease. In C. R. Scriver, A. L. Beaudet, W. S. Sly, & D. Valle (Eds.), The metabolic, molecular basis of inherited diseases (8th ed., pp. 3775–3809). New York, NY: McGraw-Hill.
Suzuki, M., Sugimoto, Y., Ohsaki, Y., Ueno, M., Kato, S., Kitamura, Y., et al. (2007). Endosomal accumulation of Toll-like receptor 4 causes constitutive secretion of cytokines and activation of signal transducers and activators of transcription in Niemann-Pick disease type C (NPC) fibroblasts: A potential basis for glial cell activation in the NPC brain. Journal of Neuroscience, 27, 1879–1891.
Svennerholm, L., Vanier, M. T., & Mansson, J. E. (1980). Krabbe disease: A galactosylsphingosine (psychosine) lipidosis. Journal of Lipid Research, 21, 53–64.
Takamura, A., Higaki, K., Kajimaki, K., Otsuka, S., Ninomiya, H., Matsuda, J., et al. (2008). Enhanced autophagy and mitochondrial aberrations in murine G(M1)-gangliosidosis. Biochemical and Biophysical Research Communications, 367, 616–622.
Tanaka, K., & Webster, H. D. (1993). Effects of psychosine (galactosylsphingosine) on the survival and the fine structure of cultured Schwann cells. Journal of Neuropathology and Experimental Neurology, 52, 490–498.
te Vruchte, D., Lloyd-Evans, E., Veldman, R. J., Neville, D. C., Dwek, R. A., Platt, F. M., et al. (2004). Accumulation of glycosphingolipids in Niemann-Pick C disease disrupts endosomal transport. The Journal of Biological Chemistry, 279, 26167–26175.
Tessitore, A., del, P. M. M., Sano, R., Ma, Y., Mann, L., Ingrassia, A., et al. (2004). GM1-ganglioside-mediated activation of the unfolded protein response causes neuronal death in a neurodegenerative gangliosidosis. Molecular Cell, 15, 753–766.
Toda, K., Kobayashi, T., Goto, I., Kurokawa, T., & Ogomori, K. (1989). Accumulation of lysosulfatide (sulfogalactosylsphingosine) in tissues of a boy with metachromatic leukodystrophy. Biochemical and Biophysical Research Communications, 159, 605–611.
Toda, K., Kobayashi, T., Goto, I., Ohno, K., Eto, Y., Inui, K., et al. (1990). Lysosulfatide (sulfogalactosylsphingosine) accumulation in tissues from patients with metachromatic leukodystrophy. Journal of Neurochemistry, 55, 1585–1591.
Tybulewicz, V. L., Tremblay, M. L., LaMarca, M. E., Willemsen, R., Stubblefield, B. K., Winfield, S., et al. (1992). Animal model of Gaucher’s disease from targeted disruption of the mouse glucocerebrosidase gene. Nature, 357, 407–410.
Urayama, A., Grubb, J. H., Banks, W. A., & Sly, W. S. (2007). Epinephrine enhances lysosomal enzyme delivery across the blood brain barrier by up-regulation of the mannose 6-phosphate receptor. Proceedings of the National Academy of Sciences of the United States of America, 104, 12873–12878.
Urayama, A., Grubb, J. H., Sly, W. S., & Banks, W. A. (2004). Developmentally regulated mannose 6-phosphate receptor-mediated transport of a lysosomal enzyme across the blood-brain barrier. Proceedings of the National Academy of Sciences of the United States of America, 101, 12658–12663.
van der Voorn, J. P., Kamphorst, W., van der Knaap, M. S., & Powers, J. M. (2004). The leukoencephalopathy of infantile GM1 gangliosidosis: Oligodendrocytic loss and axonal dysfunction. Acta Neuropathologica, 107, 539–545.
Vanier, M. T. (1999). Lipid changes in Niemann-Pick disease type C brain: Personal experience and review of the literature. Neurochemical Research, 24, 481–489.
Vergarajauregui, S., Connelly, P. S., Daniels, M. P., & Puertollano, R. (2008). Autophagic dysfunction in mucolipidosis type IV patients. Human Molecular Genetics, 17, 2723–2737.
Vogler, C., Levy, B., Grubb, J. H., Galvin, N., Tan, Y., Kakkis, E., et al. (2005). Overcoming the blood-brain barrier with high-dose enzyme replacement therapy in murine mucopolysaccharidosis VII. Proceedings of the National Academy of Sciences of the United States of America, 102, 14777–14782.
von Figura, K., Gieselmann, V., & Jaeken, J. (2001). Metachromatic leukodystrophy. In C. R. Scriver, A. L. Beaudet, W. S. Sly, & D. Valle (Eds.), The metabolic, molecular basis of inherited diseases (8th ed., pp. 3695–3724). New York, NY: McGraw-Hill.
Wada, R., Tifft, C. J., & Proia, R. L. (2000). Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proceedings of the National Academy of Sciences of the United States of America, 97, 10954–10959.
Walkley, S. U., & Vanier, M. T. (2009). Secondary lipid accumulation in lysosomal disease. Biochimica et Biophysica Acta, 1793, 726–736.
Wei, H., Kim, S. J., Zhang, Z., Tsai, P. C., Wisniewski, K. E., & Mukherjee, A. B. (2008). ER and oxidative stresses are common mediators of apoptosis in both neurodegenerative and non-neurodegenerative lysosomal storage disorders and are alleviated by chemical chaperones. Human Molecular Genetics, 17, 469–477.
Weinreb, N. J., Charrow, J., Andersson, H. C., Kaplan, P., Kolodny, E. H., Mistry, P., et al. (2002). Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: A report from the Gaucher Registry. American Journal of Medicine, 113, 112–119.
Wenger, D. A., Rafi, M. A., Luzi, P., Datto, J., & Costantino-Ceccarini, E. (2000). Krabbe disease: Genetic aspects and progress toward therapy. Molecular Genetics and Metabolism, 70, 1–9.
Wenger, D. A., Suzuki, K., Suzuki, Y., & Suzuki, K. (2001). Galactosylceramide lipidosis. Globoid cell leukodystrophy (Krabbe disease). In C. R. Scriver, A. L. Beaudet, W. S. Sly, & D. Valle (Eds.), The metabolic, molecular basis of inherited diseases (8th ed., pp. 3669–3694). New York, NY: McGraw-Hill.
White, A. B., Givogri, M. I., Lopez-Rosas, A., Cao, H., van Breemen, R., Thinakaran, G., et al. (2009). Psychosine accumulates in membrane microdomains in the brain of krabbe patients, disrupting the raft architecture. Journal of Neuroscience, 29, 6068–6077.
Whitfield, P. D., Nelson, P., Sharp, P. C., Bindloss, C. A., Dean, C., Ravenscroft, E. M., et al. (2002). Correlation among genotype, phenotype, and biochemical markers in Gaucher disease: Implications for the prediction of disease severity. Molecular Genetics and Metabolism, 75, 46–55.
Wittke, D., Hartmann, D., Gieselmann, V., & Lullmann-Rauch, R. (2004). Lysosomal sulfatide storage in the brain of arylsulfatase A-deficient mice: Cellular alterations and topographic distribution. Acta Neuropathologica, 108, 261–271.
Wraith, J. E., & Imrie, J. (2009). New therapies in the management of Niemann-Pick type C disease: Clinical utility of miglustat. Therapeutics and Clinical Risk Management, 5, 877–887.
Wraith, J. E., Vecchio, D., Jacklin, E., Abel, L., Chadha-Boreham, H., Luzy, C., et al. (2010). Miglustat in adult and juvenile patients with Niemann-Pick disease type C: Long-term data from a clinical trial. Molecular Genetics and Metabolism, 99, 351–357.
Wu, Y. P., & Proia, R. L. (2004). Deletion of macrophage-inflammatory protein 1 alpha retards neurodegeneration in Sandhoff disease mice. Proceedings of the National Academy of Sciences of the United States of America, 101, 8425–8430.
Yamada, H., Martin, P., & Suzuki, K. (1996). Impairment of protein kinase C activity in twitcher Schwann cells in vitro. Brain Research, 718, 138–144.
Yue, Z., Friedman, L., Komatsu, M., & Tanaka, K. (2009). The cellular pathways of neuronal autophagy and their implication in neurodegenerative diseases. Biochimica et Biophysica Acta, 1793, 1496–1507.
Zaka, M., Rafi, M. A., Rao, H. Z., Luzi, P., & Wenger, D. A. (2005). Insulin-like growth factor-1 provides protection against psychosine-induced apoptosis in cultured mouse oligodendrocyte progenitor cells using primarily the PI3K/Akt pathway. Molecular and Cellular Neurosciences, 30, 398–407.
Zaka, M., & Wenger, D. A. (2004). Psychosine-induced apoptosis in a mouse oligodendrocyte progenitor cell line is mediated by caspase activation. Neuroscience Letters, 358, 205–209.
Zeger, M., Popken, G., Zhang, J., Xuan, S., Lu, Q. R., Schwab, M. H., et al. (2007). Insulin-like growth factor type 1 receptor signaling in the cells of oligodendrocyte lineage is required for normal in vivo oligodendrocyte development and myelination. Glia, 55, 400–411.
Zhao, H., & Grabowski, G. A. (2002). Gaucher disease: Perspectives on a prototype lysosomal disease. Cellular and Molecular Life Sciences, 59, 694–707.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Eckhardt, M. Pathology and Current Treatment of Neurodegenerative Sphingolipidoses. Neuromol Med 12, 362–382 (2010). https://doi.org/10.1007/s12017-010-8133-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12017-010-8133-7