
Abstract
Purpose of Review
Fractures are a prominent form of traumatic injury and shall continue to be for the foreseeable future. While the inflammatory response and the cells of the bone marrow microenvironment play significant roles in fracture healing, the nervous system is also an important player in regulating bone healing.
Recent Findings
Considerable evidence demonstrates a role for nervous system regulation of fracture healing in a setting of traumatic injury to the brain. Although many of the impacts of the nervous system on fracture healing are positive, pain mediated by the nervous system can have detrimental effects on mobilization and quality of life.
Summary
Understanding the role the nervous system plays in fracture healing is vital to understanding fracture healing as a whole and improving quality of life post-injury. This review article is part of a series of multiple manuscripts designed to determine the utility of using artificial intelligence for writing scientific reviews.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
This is one of many articles evaluating the utility of using AI to write scientific review articles on musculoskeletal topics [1]. The first draft of this review was written entirely by humans. Refer to this edition’s Comment paper for more information [2]. Since human beings first walked the Earth, fractures have been among the most prevalent forms of traumatic injury. As it currently stands, fractures are the most common large-organ consequences of trauma [3]. Despite the plethora of research invested into developing preventative measures to reduce the incidence of trauma, fractured bones will persist. Furthermore, for a multitude of reasons, many demographics (e.g., aging population, type 2 diabetics, smokers, Alzheimer’s disease) are at increased risk of fracture complications, poor healing outcomes, and nonunion, which can cause enhanced and persistent pain at the fracture site [4]. As such, optimization of the healing process needs to be considered. Improving surgical techniques to expedite fracture healing, reduce infection risk, and reduce pain is one approach to optimize the healing process; however, increasing our understanding of fracture healing may also allow for the development of better pharmacological therapeutics for fracture healing and pain.
There are three fundamental stages to fracture healing: the reactive phase, the reparative phase, and the remodeling phase [5]. When examining the first two phases, the factors typically considered the most are angiogenic mediators, proinflammatory cytokines, and osteoprogenitor cells from the periosteum and bone marrow that undergo differentiation to produce new bone. Osteoprogenitor cells undergo endochondral ossification through differentiation into chondrocytes to establish a provisional cartilaginous matrix, which eventually develops into new trabecular bone that undergoes further remodeling to become cortical bone [6]. Beyond their role as critical components of fracture healing, these processes serve within homeostatic conditions to maintain a healthy bone microenvironment [7]. In the final phase, bone remodeling is mediated by osteoblasts (OBs), osteoclasts (OCs), and other cells directing ossification. Surprisingly, there is little discussion regarding the role of neural regulation in fracture healing at any of these stages; however, tangible evidence for a role of both the peripheral (PNS) and central nervous systems (CNS) to regulate fracture healing is evident. Furthermore, the nervous system is essential for the sensing and response to fracture pain. The objective of this review is to provide insight into the neural regulation of fracture healing and how the nervous system plays a role in mediating fracture pain.
The Effects of the Peripheral Nervous System on Fracture Healing
The PNS regulates many physiological attributes of the body’s non-nervous tissues and organs by relaying visceral and somatic information to the CNS. This function can be extended to apply to bone and fracture healing via two primary nerve types: sympathetic and sensory nerve fibers [8]. In addition to relaying information from the periphery to the CNS, peripheral nerves can also modulate peripheral tissues through the antidromic release of neurotransmitters and neuropeptides [9].
Peripheral Nerve Reinnervation Following Fracture in Animal Models
Following traumatic injury, the body recruits a variety of cells and molecular factors which form the somatic inflammatory response. The recruited cells associated with fracture serve as first responders by releasing humoral cytokines which enhance the healing process. Nerve fiber recruitment is another significant component of the fracture healing process. Three days following rat tibial fracture, immunohistochemistry reveals the presence of growth-associated protein (GAP-43) in both the periosteum and the fracture hematoma [10], as an indicator of axonal growth and regeneration of free nerve endings within the organized hematoma. GAP-43 expression increases following fracture, so that at day 7 the nerve fibers are found penetrating the avascular cartilage of the callus and the hyperplastic periosteum [10]. Common to both sensory and sympathetic nerve populations is the dependence on neurotrophins to stimulate and maintain ingrowth of associated nerve fibers into bone tissue. The ingrowth of neuronal tissue prior to angiogenesis supports the concept of neurite outgrowth as a prerequisite for neovascularization, although this topic is still being discussed [11].
Mixed Nerve Denervation in Animal Models
To investigate the fundamental role of peripheral innervation in fracture healing, sciatic nerve resection models have been used to effectively deplete the fracture site of nerve input [12,13,14,15,16,17,18,19]. Gross examination of tibial fractures with sciatic denervation has shown faster callus formation and union [13, 15, 16, 19]; however, denervated calluses have reduced overall RNA concentration during endochondral ossification, suggesting that there is compromised ability to generate bony matrix. Indeed, overall mineralization and woven bone density are reduced at the callus in denervated animals, having been replaced by bone marrow [15, 16]. Sciatic nerve resection as a means to investigate the role of the PNS on fracture healing is suboptimal: the efficacy of resection to induce complete denervation is questionable, as perivascular nerve growth and release of neuropeptides have been observed at the fracture site despite complete sciatic resection [15]. Furthermore, the sciatic nerve comprised of a mixture of efferent, afferent, and autonomic fibers; thus, these studies will not decipher the roles of specific nerve types. In fact, because the sciatic nerve carries motor fibers that innervate the entirety of the posterior thigh and leg, many of the findings may potentially be attributed to reduced loading and mobility, as opposed to actual reduced sensory or autonomic innervation of bone [20]. Indeed, tibial fractures without sciatic resection may cause bone loss in ipsilateral femurs, resulting in mechanically weaker bones [21]. Nonetheless, while these points should certainly be considered, the sciatic nerve denervation models still provide some insight into the general effects of peripheral nerves during fracture healing.
Comparing fracture healing in animals with intact versus resected sciatic nerves is not the only way the functional role of the sciatic nerve has been examined. Low-intensity pulsed ultrasound (LIPUS) is reported to accelerate fracture healing. The ultrasound waves propagate mechanical energy absorbed by the fracture environment, resulting in cellular responses that improve healing [19]. To elucidate the mechanisms of LIPUS, one study examined how effects of LIPUS on fracture healing change with sciatic denervation [19]. Positive effects of LIPUS are attenuated significantly following sciatic nerve resection, suggesting that LIPUS enhances fracture healing through activation of the peripheral nerves in the fracture microenvironment [19]. Overall, sciatic nerve innervation seems to propagate fracture healing, although more selective models of peripheral nerve manipulation are needed.
Sensory Nerve Effects on Healing
In addition to indiscriminately modulating peripheral nerves by resecting or globally stimulating the nerves, it is possible to study specific nerve fiber types via activation by fiber-type specific electrical stimulation or through pharmacological or genetic inactivation of specific nerve fibers. Sensory nerves have been isolated as one of the two primary fiber types that innervate bone, so special examination of their function in fracture healing is critical. Some studies have examined bone formation with sensory electrical stimulation of the dorsal root ganglia (DRG), using rat models of non-fracture and fracture bone growth assessment [22, 23••]. In an attempt to elicit bony bridging between L4 and L5 transverse processes, investigators electrically stimulated L4 to L6 DRG (lower lumbar region), in the absence of bone decortications or bone grafting. Novel bony bridging was observed in all rats examined at L4/L5 transverse processes and most rats at L5/L6 transverse processes, with no fusion or bridging in rats without electrical stimulation [22]. In osteoporotic rats with a closed femoral fracture, electrical stimulation of L3 and L4 DRGs resulted in fractures that healed with greater bone mineral density (BMD) and mechanical stability [23••], supporting the notion that sensory fiber activation is osteogenic. A similar role of sensory neurons was established when examining fracture healing in the presence of sensory neuron inactivation, elicited by local capsaicin injections. Sensory denervation resulted in acute reductions of collagen I fiber upregulation 3 days post-fracture, and collagen II expression 1 week post-fracture was also impaired [24]. Collagens I and II are both necessary components of fracture healing and bone homeostasis, playing roles in endochondral ossification and forming much of the organic bone matrix [25]. Just as the sciatic nerve resections resulted in larger callus formation in several instances, but with reduced ossification and generally weaker bone, sensory nerve denervation resulted in a similar trend where biomechanical testing demonstrated that denervated fractures bore ~ 21% less force to failure compared to innervated fractures [24]. This pro-healing role for sensory neurons is not altogether surprising, as many neuropeptides critical for fracture healing are primarily secreted by sensory neurons, including calcitonin gene-related peptide (CGRP) and substance P (SP), and their release is upregulated almost immediately following fracture in humans [26].
Autonomic Regulation of Fracture Healing
Autonomic nerve fibers also play a role in fracture healing. The primary focus of autonomic research has largely been to examine a role for the sympathetic neurons, despite the expression of receptors for neurotransmitters released by both sympathetic and parasympathetic nerve fibers within the cells of the bone marrow microenvironment [27]. Systemic ablation of sympathetic fibers, through peripheral 6-hydroxydopamine (6-OHDA) injections, has been used to induce sympathectomies [28]. Loss of sympathetic innervation has been shown to reduce trabecular bone volume fraction (BV/TV) and mechanical strength of both fractured and unfractured bones [28, 29]. There are also differences in callus maturation timelines with sympathectomy. Dividing callus maturation into three phases—mesenchymal, cartilaginous, and bony—studies found that sympathectomy delayed callus maturation at multiple timepoints after fracture [30, 31]. In addition, fractured and undamaged bones from animals with sympathectomy have weaker bone biomechanics, as they are significantly less resistant to torque and have reduced stiffness. BV/TV, connectivity density, trabecular bone thickness, and separation are all adversely affected with sympathectomy in fractured and unfractured bone [31]. The adverse effects of sympathectomy can be restored with local repletion of vasoactive intestinal peptide or with systemic injections of a β3 adrenergic agonist [29], suggesting that multiple neurotransmitters could be responsible for the positive effects of sympathetic nerves on bone healing and quality. Changes in immune cells in the fracture callus were also changed with sympathectomy, as CD4 + and CD8 + cells were significantly reduced both early on (5 days) and later (3 weeks) in the fracture healing process, suggesting that an interaction between sympathetic nerves and the immune system could also underlie the effects of sympathetic nerve loss on fracture healing. Because of the possible interactions between systemic sympathetic nerve ablation and the immune system, the effects of limiting sympathetic nerve denervation to the lower trunk or fracture site on fracture healing were examined. Surgical procedures to remove parts of the sympathetic trunk are methods that have been used in several instances and can be performed at different segmental levels (cervical, lumbar, periarterial). In lumbosacral ganglionectomies performed in the early twentieth century on patients, the subsequent loss of sympathetic innervation resulted in increased blood flow to the lower extremities [32, 33]. Furthermore, this form of sympathectomy resulted in increased bone growth in paralyzed patients with poliomyelitis in the lower extremity. In animal models, cervical sympathetic trunk resection elicited an increase in BMD, BV/TV, and trabecular bone 1–2 weeks after mandibular fracture in a model of distraction osteogenesis (DO) [34, 35]. It was subsequently determined that sympathectomy diminished the levels of norepinephrine (NE) and its corresponding receptor, β3-adrenergic receptor (adrb3), on mesenchymal stem cells (MSCs) at the site of distraction. Subsequent in vitro studies established that osteoanabolic factors in MSCs, including alkaline phosphatase (ALP), runt-related transcription factor 2 (RUNX2), and osteocalcin (OCN), were reduced upon exposure to NE, and that these effects were antagonized by deletion of the β3-adrenergic receptor [35]. NE is not the only neurotransmitter being used by sympathetic nerves to guide bone homeostasis and healing. Some sympathetic nerves are postnatally induced by interleukin-6 (IL-6) to switch to a cholinergic-releasing phenotype [36]. When this subset of ACh-releasing nerves is ablated, there is a decrease in bone mass. Furthermore, increases in bone mass through exercise appear to be mediated through a concurrent increase in the number of cholinergic sympathetics innervating the bone [36].
Altogether, peripheral nerve denervation studies have demonstrated that ablation has various effects on fracture healing, depending upon the breadth of denervation. Systemic sensory and sympathetic nerve and focal cholinergic sympathetic nerve loss generally diminish bone mineralization, whereas focal ablation of noradrenergic sympathetic neurons promotes healing and mineralization. Although some mechanistic factors were discussed above, the next section will expand on how the peripheral nerves affect bone homeostasis and fracture healing.
Molecular Factors in Peripheral Regulation
As evidenced by the studies described above, many nerve ablation models have conflicting results and interpretations of data. However, understanding neurogenic effects within the microenvironment of the fracture site can expand our understanding of fracture healing and provide opportunities for pharmacological therapeutics that accelerate bone repair. Several neuropeptides have already been recognized as osteoanabolic [37], and examinations of CGRP, SP, vasoactive intestinal peptide (VIP), and neuropeptide Y (NPY) are already popular targets. However, recent studies describing a role for these neuropeptides in fracture healing will be described further. Refer to Table 1 for a summary of factors in the PNS involved in fracture healing.
Calcitonin Gene-Related Peptide
CGRP is a polypeptide primarily known for its nociceptive signaling as a neurotransmitter and as a vasodilatory, proangiogenic molecule [38]. Its main receptors are the calcitonin gene-related peptide receptor (CLR) and receptor activity modifying protein 1 (RAMP1). In addition to its known principal functions, CGRP can potentially bind to OBs, as OBs express CLR. This results in increased osteogenic gene expression of factors such as osteoprotegrin (OPG) [38]. The expression of CLR on OBs in conjunction with proangiogenic properties of CGRP makes it a prime candidate for improved bone healing. Cell culture studies support this notion as well. In cell culture studies, transfected M2 macrophages were manipulated to overexpress CGRP in vitro. At 1 and 3 days following transfection, CGRP impaired mRNA expression of pro-osteogenic genes bone morphogenetic proteins-2 (BMP-2) and -6 (BMP-6), wnt10, and oncostatin M within the M2 macrophages, but then pro-osteogenic gene expression was increased at 5 and 7 days post-transfection [48]. The investigators then co-cultured transfected M2 macrophages with MC3T3 osteoblastic precursor cells and assessed osteoblastic differentiation and osteogenic gene expression. Expression of osteoblastic differentiation mRNA factors ALP, RUNX2, osterix, and osteopontin in MC3T3 cells when co-cultured with the CGRP-overexpressing M2 macrophages decreased and then increased in a time-dependent fashion that mirrored osteogenic gene expression in the M2 macrophages. All these gene expression effects were negated by the use of veterporfin, a yes-associated protein-1 (Yap-1) inhibitor, suggesting that the osteogenic effects of CGRP are mediated by Yap-1. Another study found that pharmacological inhibition of CGRP signaling results in reduced phosphorylated Ras/extracellular signal-regulated kinase (pERK) in the fracture microenvironment [49]. Activated ERK has been shown to promote OB differentiation; thus, altered pERK activity could be another mechanism by which CGRP promotes fracture healing. Further, CGRP increases OB production of cyclic adenosine monophosphate (cAMP), increases IGF-1 production, and inhibits the proinflammatory cytokine tumor necrosis factor-α (TNF-α), a promoter of osteoclastogenesis [50, 51].
In animal models, while CGRP-deficient mice have shown no changes in fracture healing in one instance [30], other studies show impaired callus formation, maturity, and OB activity in fracture healing [49, 52, 53•]. Research to specifically alter CGRP signaling at the site of fracture or in defined cell populations has helped to understand some of the discrepancies within knockout animals. A prominent role for CGRP in altering proliferation and activation of osteoblasts has been suggested. One recently defined role for CGRP is to drive the proliferation of periosteal progenitor cells following fracture. In these experiments, CGRPexpressing nerve fibers were shown to innervate the periosteum and expression of CGRP receptor components on periosteal progenitor cells was identified. To interrogate the role of the CGRP, investigators depleted the CGRP receptor on the periosteal progenitor cells, resulting in a loss of callus size and extent of cartilage in the callus [53•]. Surprisingly, however, there was little effect of these manipulations on biomechanical properties of the fractured bones. Another study looked at the effects of CGRP-impregnated fibrin sealant to determine whether it would affect patellar regeneration following partial patellectomy. In these experiments, CGRP sealant increased bone area and BMD compared to control, whereas inclusion of the CGRP antagonist in the fibrin sealant trended towards decreasing bone area and BMD compared to control at 8 and 16 weeks post-operatively in rabbits [54]. Moreover, patella-patellar tendon complexes possessed greater load to failure and stiffness than controls and antagonists. As mentioned previously, acute increases in CGRP release via electrical stimulation of the DRG improve fracture healing in osteoporotic fractures in rats [23••], an effect that was reversed by administration of a CGRP antagonist. Overall, these increases in CGRP with fracture and the pro-osteogenic signaling mechanisms of the neuropeptide suggest that it promotes fracture healing.
Substance P
The primary receptor of SP is the neurokinin-1-tachykinin receptor (NK-1R). Antagonism of this receptor reduced the expression of osteocalcin and collagens I and II [40]. Moreover, biomechanical testing demonstrated that NK-1R inhibition reduced the loading capacity of the femurs both acutely (6 weeks post-operatively) and chronically (3 months post-operatively). Impairment of SP might produce these results due to its activation of the wnt signaling pathway, as fracture experiments in type 1 diabetes (T1D) rats demonstrate evidence of compromised wnt signaling. SP treatment of these rats restored wnt signaling, increased OPG, reduced RANKL expression, and improved fracture healing [39]. Thus, at least in T1D rats, SP induces osteogenesis.
Vasoactive Intestinal Peptide
Although the neuropeptide VIP is typically thought of in the context of the intestinal tract as a promoter of digestion, VIP receptor types 1 (VPAC1) and 2 (VPAC2) are expressed on OCs, prevent OC differentiation, and have been examined as a potential therapeutic in inflammatory bone disease [41, 42]. Furthermore, it has recently been the focus of examination in the context of fracture healing. Not surprisingly, VIP expression is significantly reduced with sympathectomy [28]. Exogenous VIP treatment to animals symphathectomized by 6-OHDA treatment partially restored bone volume losses and biomechanical deficits induced by the sympathectomy, suggesting a pro-osteogenic role for VIP in fracture healing [28].
Neuropeptide Y
A role for NPY, which is expressed primarily in noradrenergic sympathetic neurons, in fracture healing has also been examined. Increased expression of NPY can lead to osteogenesis of MSCs by binding to Y1 receptors, the primary NPY receptor, and upregulating RUNX2 expression [43]. Deletion of the Y1 receptor results in delayed healing; while wild-type mice experience partial to complete bridging 6 weeks after fracture, Y1-deficient mice did not experience any cases of complete bridging 6 weeks post-surgery [55]. An examination of NPY-secreting nerve fiber distribution in angulated fractures found that NPY fibers penetrate the periosteum and fibrous callus, especially on the concave side of an angular fracture, and innervate the tissue on that side [56]. Because the concave side of an angulated fracture is associated with bone formation, it is thought that NPY innervation is involved, in part, in this process during fracture healing. As observed with CGRP inhibition, NPY inhibitors reduce phosphorylated ERK in fractures [49]. Altogether, NPY release appears to be pro-osteogenic, and loss of NPY signaling may be responsible for the loss of fracture healing observed with sympathectomy in animal fracture models.
Neurotrophins
Neuropeptides are not the only factors involved in the healing process. Brain-derived neurotrophic factor (BDNF) is a neurotrophin that has been shown to accelerate fracture healing [57]. BDNF treatment enhances the proliferation of MLO-Y4 osteocyte-like cells in vitro and the differentiation of MSC into OBs [58]. BDNF works through binding to its receptor, tropomyosin-related kinase B receptor (TrkB), resulting in activation of the Akt signaling pathway and subsequent inhibition of asparagine endopeptidase (AEP). Genetic knockout of AEP results in increased trabecular bone density and can partially reverse the loss of bone density induced by ovariectomy [44]. Moreover, the use of a TrkB receptor agonist, R13, produced equivalent results and increased OPG expression in the bone. This specific study also found that 7,8-DHF, a BDNF agonist, inhibits RANKL and promotes OPG as well. Interestingly, another study found essentially the opposite results when examining 7,8-DHF in fracture healing. In their study, 7,8-DHF treatment reduced callus sizes and the mechanical stability of healed bones, and had no effects on pro-OB RNA expression [59]; however, these studies were performed in male mice. Clearly, more work needs to be done to fully understand the effects of BDNF on fracture healing and whether there is a sex-specific effect.
Another neurotrophin, nerve growth factor (NGF), can potentially play a significant role in healing. Its receptor, tyrosine kinase receptor type 1 (TrkA), is widely expressed in osteoprogenitor cells and OBs, and NGF itself has been shown to be produced by OBs [20, 45]. NGF has been shown to upregulate vascular endothelial growth factor (VEGF) and BMP expression in bone [46, 47]. Expectedly, it is thus deemed an accelerator and promoter of fracture healing [37, 60•]. Injections of β-NGF post-fracture in mice promote endochondral ossification and result in union up to 2 weeks faster compared to mice with no treatment [6]; thus, NGF improves overall outcomes and at a faster rate than in the absence of exogenous NGF treatment.
The Effects of Central Nervous System Injury on Fracture Healing
Superficially, it may appear that the CNS involvement in fracture healing is sparse, but there are significant interactions between the CNS and bone. Direct pathways linking the CNS to bone healing have flourished in the past two decades, with much research centered on the use of traumatic injury models.
Traumatic Brain Injury
The literature regarding the effects of traumatic brain injury (TBI) on fracture healing is conflicting. Systemic inflammation caused by traumatic injury to the head often results in increased proinflammatory cytokine circulation, which can stimulate bone resorption and reduce bone formation [61, 62]. Central influences of these injury-related responses include diminished release of pituitary hormones such as growth hormone (GH) secretion, which can alter aspects of osteogenesis [62,63,64]. From a neurological perspective, hyperadrenergic activity leading to autonomic dysfunction after acute brain injury is not uncommon following TBI [65]. An increased sympathetic state effectively suppresses osteoblastogenesis and promotes osteoclastogenesis through direct targeting of β adrenergic receptors found on OBs, resulting in bone loss [66].
Clinical Findings
Despite the potentially negative effects of TBI on fracture healing, many in the orthopedic community perceive nervous system trauma to be an accelerant of fracture healing. Numerous reports have been published describing faster fracture healing outcomes in patients, due, in part to robust callus formation, in conjunction with the coincidence of TBI and fracture [67,68,69,70,71]. A multitude of subsequent clinical and animal studies have been conducted to further validate or reject these findings. Some of these studies have found that patients with head injury exhibit a time to fracture union and healing that is significantly reduced when compared to those with no head injury [67, 72, 73, 74•]. This phenomenon has been reproduced in fracture injury of femurs, tibiae, and humeri [67, 72, 73]. Moreover, the callus-to-femoral diaphysis ratio is greater in head injury patients as assessed by radiographs [72, 73, 74•, 75]. Patients with TBI concomitant to fracture can have greater callus volume than fracture-only counterparts [76]. When these phenomena were investigated on a cellular level, it has been shown that serum of patients with TBI can result in higher proliferation rates of human fetal osteoblast (hFOB) cells and expression of OB mRNA differentiation markers [73]. Further in vitro analysis of hFOB1.19 and primary OB response to TBI patient serum collected up to 1-week post-injury finds significant increases in their proliferative capacity [75]. Contradictory findings have also been observed when examining the correlation between severity of TBI and healing in patients; whereas a strong positive correlation between the extent of TBI and callus ratio exists in one study [74•], another study found no correlation [77]. Histologic examination of the “callus,” at 3 weeks post-injury, shows mature woven bone in the periarticular periphery indicative of potential heterotopic ossification as opposed to normal fracture healing responses [67].
Preclinical Findings
Discrepancies regarding the relationship between TBI and fracture healing also exist in rodent models. In rats, callus diameter is significantly reduced 3 weeks post-injury as opposed to normal fractures, but the opposite results are observed in mice [78,79,80]. Despite this indicating improved outcomes in mice, one study found mice subjected to repeated mild TBI had significant reductions in mineralized bone formation at the callus site, as well as reduced bone connectivity density [81]. Nonetheless, there is a majority consensus of improved healing. Assays of callus mineral density and increased torsional strength show modest increases in mice 4 weeks post-operatively in a severe TBI plus fracture model [80]. Callus stiffness, an indicator of healing, in head-injury rats is greater than bones in non-TBI rats, again supporting that TBI hastens the fracture healing process [78]. Moreover, C3H10T1/2 cell line proliferation is increased in vitro when exposed to serum derived from TBI rats, indicating increased MSC proliferation [78], although this serum had no growth effects on NIH3T3 fibroblast cell line or OB lineage cells.
Underlying Mechanisms
Just as the effects of CNS injury on bone healing are complex, the underlying molecular basis behind these effects is poorly defined. It is evident there are multiple mechanisms that regulate healthy bone, and it is likely that neurological, hormonal, and humoral factors all play a role in directing healing outcomes. When delving into the subject from a neurological perspective, some key neuropeptides and neurotransmitters which have been described previously to promote fracture healing come into play. Refer to Table 2 for a summary of molecular factors involved with CNS injury and where they are located. One factor is NPY, which has been found to be elevated in TBI patient groups with fracture [43]. Following TBI, there is an increase of NPY in the cerebrospinal fluid (CSF). This increase extends to the serum due to leakage of CSF that often results because of CNS injury.
Another factor is CGRP, which plays a role in both the PNS and CNS. Following TBI with fracture, there is an increase of CGRP concentrations in the DRG [82]. Most studies report an increase in CGRP in the serum following CNS trauma [51, 83, 84], yet this is not entirely consistent [82]. Similarly, NGF and epidermal growth factor (EGF) are neurotrophins [85] that are elevated in serum following TBI and fracture, suggesting that they might mediate TBI-induced improvements in fracture healing [84, 86].
Neural Connections to Fracture Pain
Much of the discussion thus far has examined the effects of neural function within the bone microenvironment. With some exceptions, the nervous system works to accelerate fracture healing. However, sensory nerve activation also mediates fracture pain. While bone repair should be the primary goal for the treatment of fracture patients, doing so in a way that can also diminish the pain associated with trauma and bone fracture is critical. Indeed, alleviating acute pain generally has positive effects on bone healing [87]. Between 30 and 50% of fracture patients develop chronic pain following bone fracture [88, 89]; thus, understanding the changes in the nervous system that are induced by fracture is critical to address both acute and chronic fracture pain.
In the periosteum, there is a diffuse presence of sensory Aδ and C fibers, which are subtypes that are predominantly nociceptive in nature. These fibers, upon fracture, become damaged and send nociceptive signals to the brain [90]. Increases in NGF and other inflammatory mediators in the bone microenvironment result in enhanced peripheral sensitization [91]. Examination of interventions that may attenuate pain by decreasing inflammation and nociceptive signaling while maintaining healing has been highlighted in recent animal studies. Administration of anti-NGF therapy in mice has been shown to reduce pain behaviors up to 70% while maintaining bone healing outcomes [92, 93]. However, anti-NGF therapy has also been labeled as having a multitude of adverse effects, making it a suboptimal candidate for widespread therapy. Interestingly, one recent finding has shown that exogenous delivery of adenosine can mitigate the nociceptive effects of NGF and reduce pain while simultaneously improving healing outcomes in mice [94]. In addition to NGF, caspase-6 has been identified as a potential prime regulator of fracture pain, and its inhibition in mice has also shown reduced pain behaviors following fracture [95]. These recent findings may provide a potential therapeutic mechanism for improved fracture healing outcomes with reduced pain in the future.
During the healing process, nerve fibers begin to sprout into the healing bone, as early as 3 days post-fracture [10]. Normal healing dictates that this is subsequently followed by synaptic pruning to avoid impingement and overstimulation. However, ectopic nerve sprouting may result in impaired pruning and constant stimulation. Aberrant signaling by peripheral sensory neurons, either via peripheral sensitization or via continuous activation of impinged axons, can drive the development of central sensitization, whereby structural, functional, and chemical changes in the CNS amplify peripheral input to enhance the perception of pain. One functional change observed with fracture in the nervous system is an increase in glial cell proliferation. One to 4 weeks following fracture, there is increased expression of astrocyte and microglial gene markers in the peripheral and central somatosensory systems [96]. In the spinal cord, increased microglia were found to be associated with allodynia, reduced weight bearing, and heat hypersensitivity, as treatment with a microglial inhibitor reduced nociceptive responses. Moreover, systemic inhibition of SP through the injection of a neurokinin 1 (NK1) receptor antagonist reduced microglia and astrocyte activation and subsequent increases in nociceptive hypersensitivity in a mouse fracture model [96], suggesting a role for afferent neuropeptide signaling in driving activation of microglia and astrocytes.
Nonunion is another significant driving force of pain development [97, 98]. Nonunions in mice have been shown to present a significantly increased pain response, accompanied by CGRP and GAP-43-positive increased nerve density in the bone marrow [97, 99]. Indeed, it is suggested that ongoing pain in nonunion may be a result of improper pruning and the development of neuroma-like nerve endings within the bone [99]. Furthermore, in nonunion studies in rodents, there is an increased expression of proinflammatory cytokines TNF-α and interleukin-1β (IL-1β) in the serum [100], potentially leading to increased pain at the fracture site.
Not all chronic pain develops due to nonunion. Complex regional pain syndrome (CRPS) is a pathologic condition in which the body responds abnormally to tissue injury, resulting in chronic pain [101, 102••]. While NGF overexpression plays a role in developing this condition, humoral response of increased proinflammatory cytokines is among the chief underlying causes of CRPS [98]. The release of these cytokines, however, is still neuronally mediated, with CGRP and SP increases likely being the driving force for this cytokine storm [101, 103]. Chemically sympathectomized mice have a significantly higher paw withdrawal threshold and increased weight-bearing 3 and 7 weeks post-fracture than those with fracture only [102••], suggesting that sympathetic nerves drive some of the pain in CRPS. The opposite was true for parasympathetic nerves, as administration of nicotine, as a parasympathetic agonist, inhibited pain behaviors in the CRPS animal model. Both sympathectomy and nicotine significantly decreased IL-1β release [102••], suggesting that the mechanism of action is via modulation of the immune response to injury. Although the nervous system’s presence at the fracture site is crucial for proper healing, it can also propagate the development of pain.
Conclusion
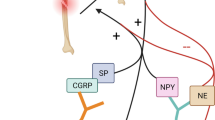
The nervous system is important in the fracture healing process. In the PNS, increased stimulation of the fracture with sensory nerve fibers proves critical in creating a more mechanically stable, healed bone. On the other hand, although a diffuse sympathetic response is necessary, focal sympathetic innervation of the fracture appears to have dual effects: adrenergic sympathetic nerves slow healing, while cholinergic sympathetic nerves accelerate healing. Many of the effects of the PNS are mediated through secreted molecular factors, which unfortunately also activate nociceptors in accordance with evolutionary protective measures to guard against injury. Thus, improved healing outcomes with neural regulation largely coexist with a caveat of increased pain. Traumatic injuries to the brain result in altered peripheral neurotrophin and neuropeptide release and some poorly understood changes in fracture healing outcomes and timeline. These effects, as well as the effects of the PNS on fracture healing, can be observed in Fig. 1. Given the uncertainty of the roles of the PNS and CNS in fracture healing, there is continuing need for increased research into neural regulation of fracture healing. Understanding how the PNS and CNS regulate fracture healing and how they contribute to the development of acute and chronic pain is critical for future exploration of therapeutic interventions that could target the pain pathways while minimizing negative effects on the fracture healing process.

Nervous system regulation of fracture healing. The effects of TBI on fracture healing are contested; however, evidence suggests that mild brain injury impairs healing, whereas severe brain injury promotes healing. Peripheral nerves innervating the fracture site secrete a variety of factors that promote healing. Created with BioRender.com
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Kacena MA, Plotkin LI, Fehrenbacher JC. The use of artificial intelligence in writing scientific review articles. Curr Osteoporos Rep. 2024. https://doi.org/10.1007/s11914-023-00852-0.
Nazzal MK, Morris AJ, Parker RS, et al. Using AI to write a review article examining the role of the nervous system on skeletal homeostasis and fracture healing. Curr Osteoporos Rep. 2024. https://doi.org/10.1007/s11914-023-00854-y.
Einhorn TA, Gerstenfeld LC. Fracture healing: mechanisms and interventions. Nat Rev Rheumatol. 2015;11(1):45–54.
Ekegren CL, et al. Incidence, costs and predictors of non-union, delayed union and mal-union following long bone fracture. Int J Environ Res Public Health. 2018;15(12):2845.
Davis KM, et al. Muscle-bone interactions during fracture healing. J Musculoskelet Neuronal Interact. 2015;15(1):1–9.
Rivera KO, et al. Local injections of beta-NGF accelerates endochondral fracture repair by promoting cartilage to bone conversion. Sci Rep. 2020;10(1):22241.
Karnik, S.J., et al. Megakaryocyte secreted factors regulate bone marrow niche cells during skeletal homeostasis, aging, and disease. Calcif Tissue Int. 2023.
Brazill JM, et al. Nerves in bone: evolving concepts in pain and anabolism. J Bone Miner Res. 2019;34(8):1393–406.
Chiu IM, von Hehn CA, Woolf CJ. Neurogenic inflammation and the peripheral nervous system in host defense and immunopathology. Nat Neurosci. 2012;15(8):1063–7. https://doi.org/10.1038/nn.3144.
Li J, et al. Bone reinnervation after fracture: a study in the rat. J Bone Miner Res. 2001;16(8):1505–10.
Li Z, et al. Fracture repair requires TrkA signaling by skeletal sensory nerves. J Clin Invest. 2019;129(12):5137–50.
Nordsletten L, et al. The neuronal regulation of fracture healing. Effects of sciatic nerve resection in rat tibia. Acta Orthop Scand. 1994;65(3):299–304.
Madsen JE, et al. Fracture healing and callus innervation after peripheral nerve resection in rats. Clin Orthop Relat Res. 1998;351:230–40.
Madsen JE, et al. Neural involvement in post-traumatic osteopenia: an experimental study in the rat. Bone. 1996;18(5):411–6.
Hukkanen M, et al. Effect of sciatic nerve section on neural ingrowth into the rat tibial fracture callus. Clin Orthop Relat Res. 1995;311:247–57.
Aro H. Effect of nerve injury on fracture healing. Callus formation studied in the rat. Acta Orthop Scand. 1985;56(3):233–7.
Aro H, Eerola E, Aho AJ. Development of nonunions in the rat fibula after removal of periosteal neural mechanoreceptors. Clin Orthop Relat Res. 1985;199:292–9.
Jiang SD, Jiang LS, Dai LY. Spinal cord injury causes more damage to bone mass, bone structure, biomechanical properties and bone metabolism than sciatic neurectomy in young rats. Osteoporos Int. 2006;17(10):1552–61.
Lam WL, et al. The role of the sensory nerve response in ultrasound accelerated fracture repair. J Bone Joint Surg Br. 2012;94(10):1433–8.
Tomlinson RE, et al. NGF-TrkA signaling in sensory nerves is required for skeletal adaptation to mechanical loads in mice. Proc Natl Acad Sci U S A. 2017;114(18):E3632–41.
Kirkeby OJ, et al. Fracture weakens ipsilateral long bones: mechanical and metabolic changes after femoral or tibial injury in rats. J Orthop Trauma. 1993;7(4):343–7.
Lau YC, et al. Dorsal root ganglion electrical stimulation promoted intertransverse process spinal fusion without decortications and bone grafting: a proof-of-concept study. Spine J. 2014;14(10):2472–8.
•• Mi J, et al. Implantable electrical stimulation at dorsal root ganglions accelerates osteoporotic fracture healing via calcitonin gene-related peptide. Adv Sci (Weinh). 2022;9(1): e2103005. Discusses role of dorsal root ganglia and sensory nerve fibers on bone mineral density in fracture setting. Identifies CGRP as an important neuropeptide for this DRG mechanism.
Apel PJ, et al. Effect of selective sensory denervation on fracture-healing: an experimental study of rats. J Bone Joint Surg Am. 2009;91(12):2886–95.
Nair A, et al. Characterization of collagen response to bone fracture healing using polarization-SHG. Sci Rep. 2022;12(1):18453.
Onuoha GN. Circulating sensory peptide levels within 24 h of human bone fracture. Peptides. 2001;22(7):1107–10.
Elefteriou F. Impact of the autonomic nervous system on the skeleton. Physiol Rev. 2018;98(3):1083–112.
Shi L, et al. Vasoactive intestinal peptide promotes fracture healing in sympathectomized mice. Calcif Tissue Int. 2021;109(1):55–65.
Wagner, J.M., et al. Role of autonomous neuropathy in diabetic bone regeneration. Cells. 2022;11(4).
Niedermair, T., et al. Impact of the sensory and sympathetic nervous system on fracture healing in ovariectomized mice. Int J Mol Sci. 2020;21(2).
Niedermair T, et al. Absence of substance P and the sympathetic nervous system impact on bone structure and chondrocyte differentiation in an adult model of endochondral ossification. Matrix Biol. 2014;38:22–35.
Ito H, Asami G. Lumbosacral sympathetic ganglionectomy its value as a therapeutic measure for thromboangiitis obliterans (with a sidelight upon alleged sympathetic innervation of the tonus of the skeletal muscles). Am J Surg. 1932;15(1):26–38.
Harris R, McDonald J. The effect of lumbar sympathectomy upon the growth of legs paralyzed by anterior poliomyelitis. JBJS. 1936;18(1):35–45.
Wang T, et al. Effects of sympathetic innervation loss on mandibular distraction osteogenesis. J Craniofac Surg. 2012;23(5):1524–8.
Du Z, et al. Sympathetic denervation-induced MSC mobilization in distraction osteogenesis associates with inhibition of MSC migration and osteogenesis by norepinephrine/adrb3. PLoS ONE. 2014;9(8): e105976.
Gadomski S, et al. A cholinergic neuroskeletal interface promotes bone formation during postnatal growth and exercise. Cell Stem Cell. 2022;29(4):528-544 e9.
Sun S, et al. No pain, no gain? The effects of pain-promoting neuropeptides and neurotrophins on fracture healing. Bone. 2020;131: 115109.
Wang Q, et al. Research progress in calcitonin gene-related peptide and bone repair. Biomolecules. 2023;13(5):838.
Wang X, Su N. Neurokinin-1-tachykinin receptor agonist promotes diabetic fracture healing in rats with type 1 diabetes via modulation of Wnt/beta-catenin signalling axis. Saudi J Biol Sci. 2021;28(4):2139–45.
Hofman M, et al. Effect of neurokinin-1-receptor blockage on fracture healing in rats. Sci Rep. 2019;9(1):9744.
Castro-Vazquez D, et al. The neuropeptide VIP limits human osteoclastogenesis: clinical associations with bone metabolism markers in patients with early arthritis. Biomedicines. 2021;9(12):1880.
Martinez C, et al. A clinical approach for the use of VIP axis in inflammatory and autoimmune diseases. Int J Mol Sci. 2019;21(1):65.
Gu XC, et al. Neuropeptide Y accelerates post-fracture bone healing by promoting osteogenesis of mesenchymal stem cells. Neuropeptides. 2016;60:61–6.
Xiong J, et al. A TrkB agonist prodrug prevents bone loss via inhibiting asparagine endopeptidase and increasing osteoprotegerin. Nat Commun. 2022;13(1):4820.
Tomlinson RE, et al. NGF-TrkA signaling by sensory nerves coordinates the vascularization and ossification of developing endochondral bone. Cell Rep. 2016;16(10):2723–35.
Yang X, et al. Nerve growth factor promotes osteogenic differentiation of MC3T3-E1 cells via BMP-2/Smads pathway. Ann Anat. 2022;239: 151819.
Yang S, et al. Effects of exogenous nerve growth factor on the expression of BMP-9 and VEGF in the healing of rabbit mandible fracture with local nerve injury. J Orthop Surg Res. 2021;16(1):74.
Zhang Q, et al. CGRP-modulated M2 macrophages regulate osteogenesis of MC3T3-E1 via Yap1. Arch Biochem Biophys. 2021;697: 108697.
Tang P, et al. NPY and CGRP inhibitor influence on ERK pathway and macrophage aggregation during fracture healing. Cell Physiol Biochem. 2017;41(4):1457–67.
Xu J, et al. The effects of calcitonin gene-related peptide on bone homeostasis and regeneration. Curr Osteoporos Rep. 2020;18(6):621–32.
Song Y, et al. Increased levels of calcitonin gene-related peptide in serum accelerate fracture healing following traumatic brain injury. Mol Med Rep. 2012;5(2):432–8.
Appelt J, et al. The neuropeptide calcitonin gene-related peptide alpha is essential for bone healing. EBioMedicine. 2020;59: 102970.
• Wee NKY, et al. Inhibition of CGRP signaling impairs fracture healing in mice. J Orthop Res. 2023;41(6):1228–39. Discusses the role of CGRP signaling on callus formation and biomechanical properties of bone following fracture.
Chen H, et al. Calcitonin gene-related peptide influences bone-tendon interface healing through osteogenesis: investigation in a rabbit partial patellectomy model. Orthop J Sports Med. 2021;9(7):23259671211003984.
Sousa DM, et al. Neuropeptide Y modulates fracture healing through Y1 receptor signaling. J Orthop Res. 2013;31(10):1570–8.
Long H, et al. Neuropeptide Y innervation during fracture healing and remodeling. A study of angulated tibial fractures in the rat. Acta Orthop. 2010;81(5):639–46.
Zhang Z, et al. BDNF promoted osteoblast migration and fracture healing by up-regulating integrin beta1 via TrkB-mediated ERK1/2 and AKT signalling. J Cell Mol Med. 2020;24(18):10792–802.
Loy TL, et al. Effects of BDNF and PEC nanoparticles on osteocytes. Molecules. 2020;25(18):4151.
Johnstone MR, et al. The TrkB agonist, 7,8-dihydroxyflavone, impairs fracture healing in mice. J Musculoskelet Neuronal Interact. 2021;21(2):263–71.
• Sekiguchi H, et al. Expression of nerve growth factor in the callus during fracture healing in a fracture model in aged mice. Biomed Mater Eng. 2022;33(2):131–7. Examines NGF expression in fracture healing and its changes with aging. Aged mice showed elevated expression of NGF compared to young mice in fracture healing.
Hardy R, Cooper MS. Bone loss in inflammatory disorders. J Endocrinol. 2009;201(3):309–20.
Bajwa NM, Kesavan C, Mohan S. Long-term consequences of traumatic brain injury in bone metabolism. Front Neurol. 2018;9:115.
Kesavan C, et al. Growth hormone effects on bone loss-induced by mild traumatic brain injury and/or hind limb unloading. Sci Rep. 2019;9(1):18995.
Yu H, et al. Reduced bone mass accrual in mouse model of repetitive mild traumatic brain injury. J Rehabil Res Dev. 2014;51(9):1427–37.
Hinson HE, Sheth KN. Manifestations of the hyperadrenergic state after acute brain injury. Curr Opin Crit Care. 2012;18(2):139–45.
Elefteriou F. Regulation of bone remodeling by the central and peripheral nervous system. Arch Biochem Biophys. 2008;473(2):231–6.
Spencer RF. The effect of head injury on fracture healing. A quantitative assessment. J Bone Joint Surg Br. 1987;69(4):525–8.
Perkins R, Skirving AP. Callus formation and the rate of healing of femoral fractures in patients with head injuries. J Bone Joint Surg Br. 1987;69(4):521–4.
Garland DE, Dowling V. Forearm fractures in the head-injured adult. Clin Orthop Relat Res. 1983;176:190–6.
Garland DE, Rothi B, Waters RL. Femoral fractures in head-injuries adults. Clin Orthop Relat Res. 1982;166:219–25.
Newman RJ, Stone MH, Mukherjee SK. Accelerated fracture union in association with severe head injury. Injury. 1987;18(4):241–6.
Giannoudis PV, et al. Accelerated bone healing and excessive callus formation in patients with femoral fracture and head injury. Injury. 2006;37(Suppl 3):S18-24.
Cadosch D, et al. Humoral factors enhance fracture-healing and callus formation in patients with traumatic brain injury. J Bone Joint Surg Am. 2009;91(2):282–8.
• Shim DW, et al. Accelerated tibia fracture healing in traumatic brain injury in accordance with increased hematoma formation. BMC Musculoskelet Disord. 2022;23(1):1110. Discusses the role of traumatic brain injury on fracture healing rates and callus sizes.
Gautschi OP, et al. Serum-mediated osteogenic effect in traumatic brain-injured patients. ANZ J Surg. 2009;79(6):449–55.
Ravi P, et al. Correlation between traumatic brain injuries and callus formation in long bone fractures. Indian J Orthop. 2022;56(5):837–46.
Yang TY, et al. The effects of an injury to the brain on bone healing and callus formation in young adults with fractures of the femoral shaft. J Bone Joint Surg Br. 2012;94(2):227–30.
Boes M, et al. Osteogenic effects of traumatic brain injury on experimental fracture-healing. J Bone Joint Surg Am. 2006;88(4):738–43.
Tsitsilonis S, et al. The effect of traumatic brain injury on bone healing: an experimental study in a novel in vivo animal model. Injury. 2015;46(4):661–5.
Locher RJ, et al. Traumatic brain injury and bone healing: radiographic and biomechanical analyses of bone formation and stability in a combined murine trauma model. J Musculoskelet Neuronal Interact. 2015;15(4):309–15.
Kesavan C, Rundle C, Mohan S. Repeated mild traumatic brain injury impairs fracture healing in male mice. BMC Res Notes. 2022;15(1):25.
Zhang D, et al. The influence of brain injury or peripheral nerve injury on calcitonin gene-related peptide concentration variation and fractures healing process. Artif Cells Blood Substit Immobil Biotechnol. 2009;37(2):85–91.
Song Y, et al. The role of the hippocampus and the function of calcitonin gene-related peptide in the mechanism of traumatic brain injury accelerating fracture-healing. Eur Rev Med Pharmacol Sci. 2017;21(7):1522–31.
Xu YQ, et al. Expressions and significance of calcitonin gene-related peptide and nerve growth factor in rabbit model of traumatic brain injury complicated with tibial fracture: preliminary results. Eur Rev Med Pharmacol Sci. 2019;23(12):5040–50.
Yamada M, Ikeuchi T, Hatanaka H. The neurotrophic action and signalling of epidermal growth factor. Prog Neurobiol. 1997;51(1):19–37.
Zhuang YF, Li J. Serum EGF and NGF levels of patients with brain injury and limb fracture. Asian Pac J Trop Med. 2013;6(5):383–6.
McVeigh LG, et al. Assessment, quantification, and management of fracture pain: from animals to the clinic. Curr Osteoporos Rep. 2020;18(5):460–70.
Aulenkamp JL, et al. Chronic pain following fracture-related surgery: posttraumatic rather than postsurgical origin promotes chronification-a prospective observational study with 1-year follow-up. Anesth Analg. 2022;134(5):974–86.
Mehta SP, et al. Baseline pain intensity is a predictor of chronic pain in individuals with distal radius fracture. J Orthop Sports Phys Ther. 2015;45(2):119–27.
Mitchell SAT, Majuta LA, Mantyh PW. New insights in understanding and treating bone fracture pain. Curr Osteoporos Rep. 2018;16(4):325–32.
Magnusdottir R, et al. Fracture-induced pain-like behaviours in a femoral fracture mouse model. Osteoporos Int. 2021;32(11):2347–59.
Rapp AE, et al. Analgesia via blockade of NGF/TrkA signaling does not influence fracture healing in mice. J Orthop Res. 2015;33(8):1235–41.
Majuta LA, et al. Orthopedic surgery and bone fracture pain are both significantly attenuated by sustained blockade of nerve growth factor. Pain. 2015;156(1):157–65.
Shih YV, et al. Multi-functional small molecule alleviates fracture pain and promotes bone healing. Adv Sci (Weinh). 2023;10(36):e2303567.
Cui W, et al. Spinal caspase-6 regulates AMPA receptor trafficking and dendritic spine plasticity through netrin-1 in postoperative pain after orthopedic surgery for tibial fracture in mice. Pain. 2021;162(1):124–34.
Li WW, et al. Substance P spinal signaling induces glial activation and nociceptive sensitization after fracture. Neuroscience. 2015;310:73–90.
Kasai Y, et al. Increased calcitonin gene-related peptide expression in DRG and nerve fibers proliferation caused by nonunion fracture in rats. J Pain Res. 2021;14:3565–71.
Avin KG, et al. Single-cell RNAseq provides insight into altered immune cell populations in human fracture nonunions. J Orthop Res. 2023;41(5):1060–9.
Chartier SR, et al. Exuberant sprouting of sensory and sympathetic nerve fibers in nonhealed bone fractures and the generation and maintenance of chronic skeletal pain. Pain. 2014;155(11):2323–36.
Cheng A, et al. Impaired bone healing following treatment of established nonunion correlates with serum cytokine expression. J Orthop Res. 2019;37(2):299–307.
Alves CJ, et al. Fracture pain-traveling unknown pathways. Bone. 2016;85:107–14.
•• Sahbaie P, et al. Autonomic regulation of nociceptive and immunologic changes in a mouse model of complex regional pain syndrome. J Pain. 2022;23(3):472–86. Discusses the role of the autonomic nervous system on the development of complex regional pain syndrome and the release of proinflammatory interleukins.
Shi X, et al. Facilitated spinal neuropeptide signaling and upregulated inflammatory mediator expression contribute to postfracture nociceptive sensitization. Pain. 2015;156(10):1852–63.
Funding
Funding for these studies was provided in part by the NIH AG060621 (MAK), AR065971 (MKN), and HL110854 (AJM). This work was also supported in part by Indiana University School of Medicine, the Indiana Clinical and Translational Sciences Institute (funded in part by NIH UM1TR004402), the Indiana Center for Musculoskeletal Health, the Department of Pharmacology and Toxicology, and the Department of Orthopaedic Surgery. This material is also the result of work supported with resources and the use of facilities at the Richard L. Roudebush VA Medical Center, Indianapolis, IN: VA Merit #BX006399 (MAK) and #RX003552 (MAK). NIH NS102415 (FAW), Congressionally-directed Medical Research Program, W81XWH-18-1-0433 (FAW), and Indiana Traumatic Spinal Cord & Brain Injury Research Grant Program (FAW).
Author information
Authors and Affiliations
Contributions
JCF, RMN, FAW, and MAK conceived the review. MKN, RSP, and AJM performed the literature search and data analysis. MKN, RSP, and AJM, with input from all co-authors, wrote the draft of the review. All authors revised the manuscript and approved the final content of the manuscript.
Corresponding authors
Ethics declarations
Conflict of Interest
Dr. Kacena is Editor-in-Chief for Current Osteoporosis Reports. Drs. Fehrenbacher & Natoli are Section Editors for Current Osteoporosis Reports.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Disclaimer
The presented contents are solely the responsibility of the authors and do not necessarily represent the official views of any of the aforementioned agencies.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nazzal, M.K., Morris, A.J., Parker, R.S. et al. Do Not Lose Your Nerve, Be Callus: Insights Into Neural Regulation of Fracture Healing. Curr Osteoporos Rep 22, 182–192 (2024). https://doi.org/10.1007/s11914-023-00850-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-023-00850-2