
Abstract
Sepsis is a systemic inflammatory response syndrome occurring secondary to infection and labeled severe when end organ dysfunction or tissue hypoperfusion transpires. Sepsis-associated mortality remains high among critically ill patients, with chronic disease and immunosuppression being the most common risk factors. Studies demonstrate that early recognition and treatment are vital to decreasing mortality. Some of the least understood effects of sepsis are the associated neurologic complications. The peripheral nervous system (PNS) has gained most consideration and thought, largely due to dependence on mechanical ventilation. Central nervous system (CNS) complications related to sepsis have only more recently gained attention but continue to go unnoticed. Aside from the clinical examination, electroencephalography (EEG) is a sensitive tool for prognostication or uncovering non-convulsive seizures in encephalopathic patients. Further studies are needed to further define the urgency of a prevention and treatment plan for the deleterious effects of sepsis on the PNS and CNS.
Similar content being viewed by others

Introduction
The neurology of sepsis encompasses a wide array of clinical syndromes including septic encephalopathy, seizures, cerebrovascular events, and neuromuscular disorders that increase mortality and length of intensive care unit (ICU) stay in the critically ill population.
Sepsis, also known as endotoxemia, is a systemic inflammatory response to a suspected or known infection manifested clinically by two or more of the following: temperature >38 or <36 °C, heart rate >90 beats/min, respiratory rate >20 breaths/min or PaCO2 <32 mm Hg, and leukocyte count >12,000/mm3, <4000/mm3, or >10 % bands. Severe sepsis occurs when it results in end organ dysfunction or tissue hypoperfusion which can manifest as but not limited to a lactic acidosis or oliguria. Sepsis0induced hypotension, defined as systolic blood pressure (SBP) <90 mm Hg, mean arterial pressure (MAP) <70 mm Hg, or SBP reduction of more than 40 mm Hg or two standard deviations below normal for patient’s age, despite adequate fluid resuscitation is defined as septic shock [1, 2••].
Epidemiology
Sepsis represents the 11th leading cause of death in the USA and 7 % of all childhood deaths [3]. Severe sepsis accounts for 2 % of all hospital admissions and 10 % of all ICU admissions in the USA with a worldwide incidence up to 19 million per year, accounting for 10–50 % of deaths among ICU patients [1, 4]. Incidence remains highest among infants and the elderly, African-Americans, and males [4]. The most common risk factors for severe sepsis include history of chronic disease such as cancer or diabetes mellitus and use of immunosuppressive agents [4]. Genetic factors likely play a role as well, although no strong evidence currently supports this. Advanced age (>65 years), bacteremia, ICU admission, and community-acquired pneumonia also increase the risk for sepsis [5•]. Mortality rates vary internationally, but regardless of geography, the mortality risk arises early and persists for years, reducing the mean life span by 4 to 5 years [6]. Premorbid health status, acute organ dysfunction, and development of acute respiratory distress syndrome (ARDS) are recognized as independent predictors of long-term mortality [6]. Those who survive suffer from physical, emotional, and cognitive burdens.
Infectious sources are community- and/or health-care-acquired; pneumonia remains the leading cause of severe sepsis, followed by intra-abdominal and urinary tract infections [4]. Besides infected wounds, other sources include pelvic and gastrointestinal tract infections, and contaminated central venous catheters. A third of positive blood cultures are isolated, with Gram-negative organisms, including Escherichia coli, Klebsiella species, and Pseudomonas aeruginosa, remaining the leading culprit of severe sepsis, followed by Gram-positive organisms and then fungi. The most common Gram-positive isolates are Staphylococcus aureus and Streptococcus pneumonia [4, 6].
Early clinical detection and treatment are key to reducing sepsis-associated mortality. As outlined by the Surviving Sepsis Campaign Guidelines and more recently by the Process and ARISE trials, early recognition of sepsis using systemic inflammatory response syndrome (SIRS) criteria, early administration of antibiotics, and early volume resuscitation decrease mortality when applied within 3 h. Lack of need for invasive monitoring and strict transfusion thresholds were confirmed with recent studies when no significant differences in 60-day hospital mortality were shown between protocol-based early goal directed therapy (EGDT) group and standard therapy [2••, 7••, 8••].
Clinical Manifestations
Clinical manifestations are dependent on several factors including site of infection, causative organism, premorbid health status, and acute organ dysfunction [4]. The cardiovascular and respiratory systems are most commonly affected in the setting of severe sepsis. Acute kidney and liver injury, hyperglycemia, coagulopathy, thrombocytopenia, and adrenal and thyroid dysfunction may also occur during severe sepsis. Central nervous system (CNS) dysfunction can manifest as hyperactive or hypoactive delirium, seizures, and cerebrovascular events in the setting of severe sepsis. Even after surviving sepsis to hospital discharge, patients may continue to suffer from neurocognitive decline.
Pathophysiology
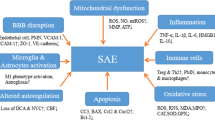
During severe sepsis, inflammatory cells including phagocytic cells of the innate immune system and lymphocytes of the adaptive immune system become dysfunctional. Pro-inflammatory and anti-inflammatory mediators released to eliminate a localized infection result in a systemic host response which leads to organ injury and secondary infection. Pro-inflammatory mediators comprised of cytokines, chemokines, prostaglandins, and nitric oxide are thought to be responsible for tissue damage as they attempt to eradicate the causative pathogen while anti-inflammatory processes which limit local and systemic injury lead to secondary infections. The systemic response in each patient differs based on the causative pathogen’s virulence and microbial load as well as host factors including age, genetics, coexisting chronic illnesses, and immunosuppression [9].
The systemic inflammatory response begins with the interaction of the outer membrane component of pathogens, such as lipopolysaccharide (LPS), lipid A, or endotoxin for Gram-negative organisms, with Toll-like receptors expressed by host cells. This leads to the production of pro-inflammatory cytokines, TNF-α, and IL-1B which in turn trigger a release of prostaglandins, leukotrienes, platelet activating factor, and phospholipase A2. These mediators cause capillary leakage by damaging the endothelial lining; septic shock ensues due to vasodilation secondary to activated macrophages and neutrophils releasing nitric oxide [4].
During the inflammatory cascade, impaired fibrinolysis, activation of pro-coagulant pathways with excess fibrin deposition mediated by tissue factor, and impairment of anticoagulant pathways due to reduced activity of activated protein C, antithrombin, and tissue factor inhibitor result in microvascular thrombosis causing tissue hypoperfusion. Loss of endothelial barrier integrity also leads to tissue hypoperfusion, resulting in end organ damage. Arterial hypotension, reduced red blood cell deformability, and mitochondrial damage due to oxidative stress also contribute to impaired tissue oxygenation also leading to organ failure [4, 9].
The neurotoxic effects of these pro-inflammatory mediators have been well documented, producing not only an acute septic encephalopathy but also long-term neurocognitive sequelae [10].
Sepsis-Associated Encephalopathy
Definition
Sepsis-associated encephalopathy (SAE) also known as sepsis-associated delirium or acute brain dysfunction is a diffuse cerebral dysfunction as a consequence of the systemic inflammatory response to an infection as opposed to septic encephalopathy which refers to a distinct cerebral infection. A lumbar puncture is necessary to make this distinction and rule out underlying meningitis or encephalitis. SAE is a life-threatening yet reversible deterioration of mental status due to underlying sepsis and/or other contributing factors including sedation, antimicrobial treatment, associated comorbidities, preexisting psychiatric, or neurologic disease and remains an independent predictor of death and long-term cognitive impairment [11, 12, 13•].
Incidence and Risk Factors
The prevalence of SAE remains controversial. SAE is frequently encountered in critically ill patients in ICUs, and in up to 70 % of patients with severe systemic infection [14••]. In one single-center study, SAE was found in 41 of 232 septic patients (17.7 %) [15]. Its prevalence varies depending on how it is defined and assessed; as neurointensivists implement EEG testing, the prevalence increases significantly [16]. Patients with intestinal and biliary tract infections caused by S. aureus, Enterococcus faecium, Acinetobacter species, and P. aeruginosa, and patients with neurodegenerative disorders such as Parkinson’s disease, Alzheimer’s disease, cerebral palsy, or traumatic brain injury have an increased risk of developing SAE [15] (Table 1).
Clinical Features and Ancillary Tests
The first manifestation of SAE is an acute change in mental status, ranging from inattention, disorientation, agitation, somnolence, stupor, and coma. The severity of encephalopathy correlates with mortality. Other clinical features of SAE include roving eye movements, asterixis, tremor, multifocal myoclonus, restlessness, tachypnea, seizures, paratonic rigidity, extensor plantar responses, and flexor or extensor posturing [5•].
The EEG can show progressive slowing in correlation with the level of consciousness and may demonstrate theta, delta, or triphasic waves, burst suppression, and periodic epileptiform discharges (PLEDS) or electrographic seizures. The mortality increases with severity of EEG abnormalities: 36 % mortality with evidence of delta wave, 50 % mortality with triphasic waves, and 67 % mortality with burst suppression. The incidence of subclinical seizures in the scenario of sepsis is less than 10 % in patients under continuous EEG monitoring [17, 18••].
Neuroimaging studies are usually normal, but some patients can show multiple ischemic strokes in different vascular territories from septic emboli, multifocal subcortical white matter lesions (diffuse leukoencephalopathy), and posterior reversible encephalopathy syndrome (PRES) [19, 20]. A lumbar puncture (LP) is indicated to rule out meningitis or acute encephalitis; the cerebrospinal fluid (CSF) will usually demonstrate a normal or mildly elevated protein level with normal cell count, normal glucose, negative Gram stain, and cultures. Patients with SAE may have alteration of latency or amplitude in the somatosensory-evoked potentials, and some biomarkers such as neuron-specific enolase and S-100 β-protein could be elevated, but they have no correlation with severity of the clinical presentation; S-100 β-protein, specifically, has recently shown utility in diagnosing SAE and predicting the outcome of sepsis, but further studies are needed to evaluate the role of S-100 B-protein in diagnosing and prognosticating SAE [16, 21, 22]. S100-B levels of 0.131 μg/L were diagnostic for SAE with a reported 67.2 % specificity and 85.4 % sensitivity (area under the curve was 0.824; 95 % confidence interval 0.750–0.898). Levels of 0.197 μg/L were predictive of hospital mortality with 76.4 % specificity and 64.4 % sensitivity (area under the curve was 0.730; 95 % confidence interval 0.599–0.806) [22]. Although S-100 B-protein remains a promising biomarker, studies have yielded conflicting results regarding its use in monitoring patients with SAE prompting the need for additional studies.
Differential Diagnosis
The differential diagnosis of SAE include acute CNS infections such as bacterial, viral, fungal, or parasitic meningitis, epidural or subdural empyema, brain abscess, immune-mediated encephalitis, alcohol or drug intoxication/withdrawal, non-convulsive status epilepticus, Wernicke encephalopathy, PRES, serotonin syndrome, neuroleptic malignant syndrome (NMS), and malignant catatonia [5•].
Treatment and Prognosis
There is no specific treatment for SAE. Management should be focused on treating the underlying infectious process complemented with general measures. Minimizing or discontinuing analgesics or sedatives is done as soon the patient can tolerate. Identifying and eliminating reversible factors such as hypoxemia, hypercapnia, hypotension, hyperthermia or hypothermia, hepatic or renal dysfunction, and metabolic or electrolytic disturbances is also necessary. Scant evidence in animal models suggests that inhibition of inducible nitric oxide synthase (iNOS) could prevent lipopolysaccharide (LPS)-induced neuronal apoptosis. There are no clinical data, however, demonstrating good outcome in human models. Insulin therapy may provide some protection against SAE as hyperglycemia is associated with increased oxidative stress and apoptosis. Currently, the main goal of treatment remains control of sepsis [21, 23–26].
Critical Illness Polyneuropathy and Myopathy
The systemic inflammatory response elicited by critical illness can affect the peripheral nerves, skeletal muscle, or both. Critical illness polyneuropathy (CIP) and critical illness myopathy (CIM) are the most commonly acquired neuromuscular conditions in the setting of severe sepsis, acute respiratory distress syndrome, prolonged mechanical ventilation, and exposure to neuro/myotoxic agents, reaching a risk of 25–45 % in ICU patients and up to 100 % in patients with multiorgan failure [27••]. Despite the decrease in sepsis-related mortality, critical-illness-associated weakness continues to climb. Sepsis has been identified as an independent risk factor for neuromuscular disorders with Gram-negative bacteremia being an independent risk factor [27••, 28]. Hyperglycemia is also considered an independent risk factor for CIP or CIM [25]. Many patients have electrophysiologic and morphologic features of both, with previous use of corticosteroids or neuromuscular blocking agents posing higher risk of developing neuromuscular dysfunction. CIP and the CIM delay weaning from mechanical ventilation and prolong the onset to mobilization, thus extending the intensive care unit course, placing the patient at risk for infection and thromboembolism, and in effect raising ICU costs [28–30].
CIP
Although the pathophysiology of CIP in unknown, it is hypothesized that the humoral and cellular responses which induce release of pro-inflammatory mediators produce microcirculatory dysfunction resulting in not only end organ failure but also distal sensory and motor axonal degeneration, considered a classic electrophysiologic pattern of CIP. Sepsis, however, may not be the only factor playing a role in axonal damage. CIP has been associated with other disorders which may or may not coexist with septic patients: prolonged mechanical ventilation, renal replacement therapy, hyperglycemia, catecholamine administration, vitamin deficiencies, metabolic derangements, severe burns, immunologic disorders, Guillain–Barré syndrome (GBS), certain antibiotics (e.g., gentamicin), neuromuscular blocking agents, changes in osmolality, toxin-producing bacteria, female gender, and old age [31–34].
CIP or CIM or both are often preceded by SAE and, therefore, sensory testing is unreliable. They should be suspected in patients with weakness and difficulty weaning from the mechanical ventilator due to phrenic nerve involvement and diaphragmatic and intercostal muscle weakness [35]. Physical examination findings of CIP include weaning failure, symmetric atrophy, flaccid quadriparesis with lower extremity distal muscles most severely affected, sparing of cranial nerves and facial muscles, and reduced or absent reflexes; approximately one third of patients have retained reflexes [36]. Responsive patients will have distal loss of pain, temperature, and vibration sense. Nerve conduction studies (NCSs) of upper and lower extremities and the phrenic nerve, when respiratory neuromuscular failure is considered, and repetitive nerve stimulation should be completed, demonstrating reduced compound motor action potential (CMAP) and sensory nerve action potential (SNAP) amplitudes; conduction velocity is normal or near normal. Needle electromyography (EMG) demonstrates reduced recruitment of motor unit potentials and abnormal spontaneous activity, at times accompanied by fibrillation potentials and positive sharp waves [28, 29]. Abnormalities can be detected as early as 48 h into the critical illness period [37].
Nerve biopsy shows a non-inflammatory distal axonal degeneration of motor and sensory nerves; normal histology is associated with rapidly reversible neuropathies. Chromatolysis of anterior horn cells and degeneration of dorsal root ganglion cells have also been described as a secondary occurrence. Muscle may show denervation atrophy with necrosis, vacuoles, and increased glycogen deposition, suggesting a coexisting CIM [36].
Entrapment neuropathy is also common in the ICU setting. Loss of subcutaneous fat predisposes the superficial peripheral nerves to compression, particularly the peroneal nerve at the fibular head, and the ulnar nerve at the elbow. This can often be avoided with proper limb positioning, early mobilization, and minimizing sedation [38, 39].
CIM
If needle EMG demonstrates small motor unit potentials (MUPs), a concomitant myopathy needs to be considered, especially if SNAPs are normal. EMG findings most suggestive of a myopathy occur 2–5 days after symptom onset and include reduced CMAP amplitudes, prolonged CMAP duration, and preserved SNAPs. NCS will show low-amplitude CMAPs with normal conduction velocities and distal latencies. Depending on the time course of the illness, CMAP duration may or may not be prolonged. SNAP amplitudes are preserved unless CIP, prior diabetic neuropathy, or limb edema is present. Three critical illness myopathies have been identified: non-necrotizing cachectic myopathy, thick filament myopathy, and acute necrotizing myopathy. Cachectic myopathy also known as disuse atrophy is typically a diagnosis of exclusion with muscle biopsy showing type 2 fiber atrophy. Thick filament myopathy is often seen in patients requiring ventilator support, high-dose steroids, or neuromuscular blocking agents. Focal or diffuse absence of thick myosin filaments is seen on muscle biopsy. Acute necrotizing myopathy is caused by various etiologies, resulting in myoglobinuria and elevated creatinine kinase (CK) level with biopsy revealing muscle fiber necrosis. Elevated CK without other findings is not diagnostic due to low sensitivity [40••]. Muscle biopsy remains the gold standard for confirming muscle involvement [41, 42••]. To minimize morbidity associated with open muscle biopsy, obtaining core needle samples for quantifying myosin/actin ratios via gel electrophoresis has also been proposed for diagnosis of CIM [37].
The predominant clinical features of a myopathy are weaning failure, flaccid quadriparesis-proximal greater than distal limb involvement, and weakness of the diaphragm and neck flexors often with sparing of facial muscles. Although rare, facial muscles can be involved with occurrence of opthalmoplegia [27••]. Muscle stretch reflexes may be normal, reduced, or absent [28, 29].
CIM has been reported with the use of corticosteroids including methylprednisolone, hydrocortisone, prednisone, betamethasone, and dexamethasone; the minimal dose and duration of treatment of steroids resulting in CIM have been difficult to approximate due to confounding factors limiting studies (different routes of administration, dosage, drug formulations, and treatment schedules) [36]. The same holds true of the neuromuscular blockers; the most common paralytics associated with CIM include pancuronium and vecuronium but other neuromuscular blockers have also been implicated [36].
CIP and CIM may coexist in critically ill patients, but CIM appears to be the most common cause of weakness. Clinically and electrophysiologically, it may be difficult to differentiate between the two. The term critical polyneuromyopathy has therefore emerged. Suffering of both entities has been shown to be associated with persistent disability; outcome for CIM has better prognosis compared to CIP in recent data when assessing patients at 1-year follow-up [43, 44].
Differential diagnosis includes Guillain–Barré syndrome (GBS), metabolic or toxic neuropathies, or neuropathies secondary to nutritional deficiencies [27••].
Treatment of CIP and CIM is aimed at treating the underlying critical illness while also preventing decubitus ulcers, deep vein thrombosis (DVTs), and implementing early mobilization with physiotherapy. Passive and active exercises are recommended as soon as possible. Avoiding and treating infection, hyperglycemia or hypoglycemia, hypotension, hypoxemia, electrolyte imbalance, nutritional deficiencies, and renal failure all play a crucial role in the management of CIP and CIM [28, 29]. Studies have shown reduction of CIP and CIM incidence with intensive insulin control which also increases risk of hypoglycemia, increasing mortality among adults [45•, 46, 47•]. Insulin carries anti-inflammatory effects, endothelial protection, and neuroprotective effects in animals [48]. Electrical muscle stimulation has also been utilized, but efficacy remains unclear with scarce data and need for randomized trials. Aggressive treatment of sepsis and multiorgan failure is considered the most effective means in reducing CIP and CIM incidence [27••]. Despite treatment, patients continue to suffer from decreased exercise capacity months to years after diagnosis [27••]. Some recover after 4 to 12 weeks but symptoms can be prolonged lasting more than 4 months [36]. (Table 2)
Imaging
Imaging patterns have been shown to indicate an underlying link between cerebral microvascular dysfunction and sepsis. Impairment of not only microvascular circulation but also cerebral autoregulation and neurovascular and neurometabolic coupling leads to a decrease of cerebral perfusion during sepsis [49]. In order to maintain the integrity of cerebral blood flow (CBF), it is prudent to maintain an adequate MAP goal and pH/paCO2 level.
Head CT and brain MRI are often normal in the setting of severe sepsis or septic shock. However, ischemic lesions, hemorrhages, infectious/inflammatory (mycotic) aneurysms, microabscesses, vasogenic edema, and multifocal necrotizing leukoencephalopathies have all been neuroimaging patterns associated with sepsis. CNS imaging can help distinguish between structural lesions, alterations of perfusion and oxygenation due to impaired autoregulation, and treatment-related effects.
Initial assessment after a neurologic examination is a non-contrast head CT. Findings related to sepsis may include ischemic strokes secondary to septic emboli or cardioembolic sources, including paroxysmal atrial fibrillation; hemorrhagic strokes may result from anticoagulation, DIC, or coagulopathy, or subarachnoid hemorrhage (SAH) due to mycotic aneurysm rupture. MRI of the brain particularly diffusion-weighted restriction sequence has a much higher sensitivity for detection of cytotoxic cerebral edema, ischemic strokes (watershed or embolic), and brain abscesses [18••]. Acute strokes, within 7 to 14 days of onset of sepsis, have been associated with worse prognosis in these patients [20, 50, 51]. Other CNS findings during sepsis include PRES particularly in patients with predisposing factors such as underlying autoimmune disorder or those receiving chemotherapy [49].
Survivors of sepsis have been studied to assess chronic MRI brain changes. Using diffusion tensor imaging and voxel-based morphometry, a greater degree of atrophy and white matter changes have been associated with prolonged duration of previous delirium and worse cognitive outcome at 1-year follow-up [52–54]. Other studies have shown left hippocampal atrophy in patients with SAE; this may be explained by asymmetrical distribution of noradrenaline, a protective, anti-inflammatory neurotransmitter, that is more prevalent within the right hemisphere compared to the left, explaining why the left hippocampus is more vulnerable to inflammation secondary to sepsis [53].
EEG
SAE occurs early in the setting of severe sepsis or septic shock, and its severity is associated with worse outcome. Up to 20 % of critically ill patients develop seizures, 90 % of which are non-convulsive [55]. Continuous electroencephalography (cEEG) monitoring is a useful tool in detecting underlying non-convulsive status epilepticus and other EEG abnormalities such as triphasic waves, burst suppression, PLEDs, or generalized periodic epileptiform discharges (GPEDs) and can be advantageous in prognostication. It can also be useful in monitoring level of sedation (Fig. 1).

A 44-year-old man with urosepsis. EEG demonstrates mild slowing of the background and periodic lateralized epileptiform discharges (PLEDs)
Most studies looking at EEG patterns in the setting of sepsis consist of case series or case control studies, with the majority being limited studies due to small sample size, retrospective designs, selection bias, effect of sedating medications, and heterogeneous populations. Evidence therefore is sparse regarding utility of EEG monitoring in the critical care setting, and recommendations are yet to be defined [56••]. European Society of Intensive Care Medicine (ESICM) recommendations on the use of cEEG monitoring in critically ill patients without acute primary brain injury state EEG monitoring should be pursued in patients with unexplained impaired mental status or other neurologic deficits, particularly in patients with severe sepsis, renal failure, and/or hepatic failure graded as a weak recommendation, low quality of evidence [56••].
An alpha rhythm or slowing of the alpha rhythm with theta activity is observed among patients with absent or mild to moderate encephalopathy such as delirium. Stupor or coma often correlate with delta activity, triphasic waves, or burst suppression pattern suggesting severe deep brain impairment [55]. The presence of slowing with primarily theta-delta activity and lack of reactivity have been associated with more severe encephalopathy and worse outcome [57]. Acute ischemic injury has been shown to correlate with focal attenuation or suppression in prior prospective studies [50]. Triphasic waves are more common in patients with hepatic failure or sepsis with or without multiorgan failure [58]. GPEDs were present in septic patients in a large case control study which was associated with a prolonged ICU stay; other studies showed that periodic discharges lasting longer than 24 h were independent predictors of poor outcome [59]. Electrographic seizures have also been shown to be independent predictors of poor outcome and sepsis an independent risk factor for developing electrographic seizures in two different case series [18••, 57].
A retrospective study of MICU patients, 60 % with severe sepsis, at Columbia University Medical Center were monitored with cEEG. Twenty-two percent of patients had either electrographic seizures or GPEDs; 10 % had electrographic seizures, 67 % of which were non-convulsive, and 17 % with GPEDs. Electrographic seizures and PEDs were more predominant in patients with sepsis, 32 versus 9 %, and associated with a significant increase in death or severe disability at hospital discharge after controlling for other determinants.
A recently published prospective single-center observational study evaluated cEEG abnormalities in patients with severe sepsis and impact on outcome [60]. Their findings showed non-convulsive seizures, and periodic discharges were common among encephalopathic patients with severe sepsis but not present in patients with a higher illness severity score. This could be due to inability of severe brain injury to generate seizures or discharges or because sicker patients require intubation and sedation which would treat any electrographic abnormalities. Lack of EEG reactivity was more common among patients on continuous sedation and did impact outcome with higher mortality at discharge and one year. The study was underpowered to comment on predictors of functional and cognitive outcome [60].
Despite these studies, there is insufficient evidence to conclude the significance of periodic discharges and seizures on outcome in septic patients especially due to poor standardized EEG terminology and pattern identification in previous years. Nonetheless, it is suggested that prolonged EEG up to 24 to 48 h is essential to capture non-convulsive seizures, although large prospective studies are needed to further evaluate sepsis-associated encephalopathy and EEG patterns.
If status epilepticus is detected on cEEG, it is essential to rapidly control any seizure activity. A loading dose of an intravenously administered antiepileptic drug is administered or often a combination of drugs is required. In patients with refractory or super-refractory status epilepticus, the use of anesthetic drugs, propofol, or large doses of midazolam is necessitated. Use of anesthetic drugs can add to a myriad of systemic dysfunction suffered by septic patients. Propofol infusion syndrome which can result in cardiovascular collapse is more common in patients treated with a dose greater than 5 mg/kg/h or an infusion ongoing for 3 or more days with the risk factors of young age, critical illness such as sepsis, high fat, and low carbohydrate intake, and use of vasopressors or steroids increasing the threat [61]. Treatment of propofol infusion syndrome often entails not only stopping the drug but also renal replacement therapy and even extracorporeal membrane oxygenation (ECMO) at times. Use of lorazepam infusions is discouraged due to risk of propylene glycol toxicity [62•]. Side effects of the inhaled halogenated anesthetics including isoflurane and desflurane are associated with hypotension, infection, and paralytic ileus. Case reports have also described hemorrhagic leukoencephalitis and bowel infarction in association with the inhaled anesthetics [63•].
Systemic manifestations of status epilepticus will also complicate a patient’s ICU course. In the setting of convulsive status epilepticus, patients can develop a metabolic acidosis secondary to muscle contraction leading to anaerobic metabolism with lactic acid formation. Aspiration pneumonia or ventilator-associated pneumonia, fever, cardiac arrhythmias, arterial hypotension secondary to the intravenous anesthetics or takotsubo cardiomyopathy, neurogenic pulmonary edema, hyperglycemia, and acute renal failure are all potential systemic manifestations of status epilepticus [62•]. Due to a prolonged immobile state, patients are high risk for DVTs, pulmonary emboli, infection (colitis, urinary tract infections, pneumonia), and pressure ulcers. Poor outcome is associated with prolonged mechanical ventilation, development of pneumonia, or cardiac dysrhythmias [62•, 64].
Conclusions
Aggressive treatment of sepsis and multiorgan failure are crucial to remedying sepsis-associated encephalopathy and critical polyneuromyopathy. Both the PNS and particularly CNS complications of sepsis are often overlooked and escape necessary investigations, largely due to other ongoing systemic derangements requiring much attention. Imaging using CT and MRI should be performed to assess for intracranial hemorrhage, ischemia, and infection. Diagnostic tools such as EEG and EMG/NCS are essential means to assess for non-convulsive seizures and critical polyneuromyopathy, respectively. Because neurocognitive decline and prolonged physical disability are risks of severe sepsis, early attention and analysis are imperative. Further studies are needed to assess which collective therapies should be utilized in the most vulnerable patients with severe sepsis to prevent cognitive and physical disability and predict which early interventions will impact outcome.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Widmann C. Long-term cerebral consequences of sepsis. The Lancet. 2014;13(6):630–6.
Dellinger RP, Carlet JM, Gerlach H, et al. Surviving sepsis campaign guidelines for management of severe sepsis and septic shock. CritCare Med. 2004;32(3):858–73. Guidelines aimed to reduce sepsis associated mortality by improving diagnosis, management, and treatment using bundles in healthcare.
King EG, Bauza GJ, Mella JR, et al. Pathophysiologic mechanisms in septic shock. Laboratory Investigation. 2014;94:4–12.
Angus DC, Van der Poll T. Severe Sepsis and Septic Shock. N Eng J Med. 2013;369(21):2063.
Hocker SE, Wijdicks EF. Neurologic complications of sepsis. Continuum (Minneap Minn). 2014;20(3 Neurology of Systemic Disease):598–613. A through overview of all facets of neurologic complications of sepsis, focusing especially on sepsis associated encephalopathy, with case studies provided to emphasize key learning points.
Mayr FB, Yende S, Angus DC. Epidemiology of severe sepsis. Virulence. 2014;5(1):4–11.
Yealy DM, Kellum JA, Huang DT, et al. A randomized trial of protocol based care for early septic shock. N Eng J Med. 2014;370(18):1683–93. Multicenter trial conducted at tertiary care centers found no mortality benefit for early goal directed therapy (EGDT) in septic shock as recommended in the 2001 EGDT trial.
Rivers E, Nguyen B, Havstad S, et al. Early Goal-Directed Therapy Collaborative Group. “Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345(19):1368–77. Single center landmark trial in the field of critical care which has since received much criticism regarding aspects of bundled care to reduce sepsis associated mortality.
Remick DG. Pathophysiology of sepsis. Am J Pathol. 2007;170(5):1435–44.
Semmler A, Hermann S, Mormann F. Sepsis causes neuroinflammation and concomitant decrease of cerebral metabolism. J Neuroinflammation. 2008;5(38):1–10.
Iacobone E, Bailly-Salin J, Polito A, et al. Sepsis-associated encephalopathy and its differential diagnosis. Crit Care Med. 2009;37(10 Suppl):S331–6.
Golzari SE, Mahmoodpoor A. Sepsis-associated encephalopathy versus sepsis-induced encephalopathy. Lancet Neurol. 2014;13(10):967–8.
Zampieri FG, Park M, Machado FS, et al. Sepsis-associated encephalopathy: not just delirium. Clinics. 2011;66(10):1825–31. An important reference to differentiate the aspects between SAE and delirium in ICU patients.
Gofton TE, Young GB. Sepsis-associated encephalopathy. Nature Reviews Neurology. 2012;8(10):557–66. One of the most extensive review about SAE in the last 5 years. The main purpose was to analyze the evidence in the pathophysiological aspects, diagnosis and management of SAE. It has been cited multiple times in similar works about SAE.
Zhang LN, Wang XT, Ai YH, et al. Epidemiological features and risk factors of sepsis-associated encephalopathy in intensive care unit patients: 2008–2011. Chin Med J (Engl). 2012;125(5):828–31.
Ebersoldt M, Sharshar T, Annane D. Sepsis-associated delirium. Intensive Care Med. 2007;33(6):941–50.
Young GB, Bolton CF, Archibald YM, et al. The electroencephalogram in sepsis-associated encephalopathy. Journal of Clinical Neurophysiology. 1992;9(1):145–52.
Oddo M, Carrera E, Claassen J, et al. Continuous electroencephalography in the medical intensive care unit. Crit Care Med. 2009;37(6):2051–6. Retrospective study of CEEG findings in MICU patients without a primary neurologic injury discovered frequent periodic epileptiform discharges and nonconvulsive seizures in patients with sepsis.
Luitse MJ, van Asch CJ, Klijn CJ. Deep coma and diffuse white matter abnormalities caused by sepsis-associated encephalopathy. The Lancet. 2013;381(9884):2222.
Sharshar T, Carlier R, Bernard F, et al. Brain lesions in septic shock: a magnetic resonance imaging study. Intensive Care Med. 2007;33(5):798–806.
Chaudhry N, Duggal AK. Sepsis associated encephalopathy. Advances in Medicine. 2014;Article ID 762320:16.
Yao B, Zhang L, Ai Y. Serum S100β is a better biomarker than neuron-specific enolase for sepsis-associated encephalopathy and determining its prognosis: a prospective and observational study. Neurochem Res. 2014;39(7):1263–9.
Kadoi Y, Goto F. Selective inducible nitric oxide inhibition can restore hemodynamics, but does not improve neurological dysfunction in experimentally-induced septic shock in rats. Anesthesia & Analgesia. 2004;99(1):212–20.
Wang H, Wu YB, Du XH. Effect of dexamethasone on nitric oxide synthase and Caspase-3 gene expressions in endotoxemia in neonate rat brain. Biomed Environ Sci. 2005;18(3):181–6.
Van den Berghe G, Schoonheydt K, Becx P, et al. Insulin therapy protects the central and peripheral nervous system of intensive care patients. Neurology. 2005;64(8):1348–53.
Sierra A, Gottfried‐Blackmore A, Milner TA, et al. Steroid hormone receptor expression and function in microglia. Glia. 2008;56(6):659–74.
Zhou C, WU L, Ni F, et al. Critical illness polyneuropathy and myopathy: a systematic review. Neural Regen Res. 2014;9(10):101–10. Detailed review of the pathophysiology, clinical symptomatology, neurophysiologic findings, and treatment of critical illness polyneuropathy and myopathy.
Friedrich O, Reid MB, Van den Berghe G. The sick and the weak: neuropathies/myopathies in the critically ill. Physiol Rev. 2015;95(3):1025–109.
Chawla J, Gruener G. Management of critical illness polyneuropathy and myopathy. Neurol Clin. 2010;28(4):961–77.
Hund E. Neurologic complications of sepsis: critical illness polyneuropathy and myopathy. J Neurol. 2001;248(11):649–53.
Witt NJ, Zochodne DW, Bolton CF, et al. Peripheral nerve function in sepsis and multiple organ failure. Chest. 1991;99(1):176–84.
Zink W, Kollmar R, Schwab S. Critical illness polyneuropathy and myopathy in the intensive care unit. Nat Rev Neurol. 2009;5(7):372–9.
Wilmshurst PT, Treacher DF, Lantos PL, et al. Critical illness poly-neuropathy following severe hyperpyrexia. QJM. 1995;88(5):351–5.
Hermans G, De Jonghe B, Bruyninckx F, et al. Interventions for preventing critical illness polyneuropathy and critical illness myopathy. Cochrane Database Syst Rev. 2009;21(1):CD006832.
Mehta S. Neuromuscular disease causing acute respiratory failure. Respiratory Care. 2006;51:1016–21.
Gorson KC. Approach to neuromuscular disorders in the intensive care unit. Neurocritical Care. 2005;3:195–212.
Schweickert WD, Hall J. ICU-acquired weakness. CHEST. 2007;131(5):1541–9.
Kress JP, Hall JB. ICU-acquired weakness and recovery from critical illness. N Engl J Med. 2014;370:1626–35.
Kalb RG. ICU-acquired weakness and recovery from critical illness. N Engl J Med. 2014;371:287.
Hermans G, De Jonghe B, Bruyninckx F, et al. Clinical review: critical illness polyneuropathy and myopathy. Crit Care. 2008;12(6):238. Systematic review of the epidemiology, pathophysiology, risk factors, diagnostic challenges, interventions, and treatment for critical illness polyneuropathy and myopathy.
Lacomis D, Giuliani MJ, Van Cott A, et al. Acute myopathy of intensive care: clinical, electromyographic, and pathologic aspects. Ann Neurol. 1996;40(4):645–54.
Khan J, Harrison TB, Rich MM, et al. Early development of critical illness myopathy and neuropathy in patients with severe sepsis. Neurology. 2006;29(12):1421–5. Prospective cohort study evaluating prevalence, time of onset, and cause of neuromuscular dysfunction in critically ill patients with severe sepsis.
Guarneri B, Bertolini G, Latronico N. Long-term outcome in patients with critical illness myopathy or neuropathy: the Italian multicentre CRIMYNE study. J Neurol Neurosurg Psychiatry. 2008;79:838–41.
Lacomis D, Petrella JT, Giuliani MJ, et al. Causes of neuromuscular weakness in the intensive care unit: a study of ninety-two patients. Muscle Nerve. 1998;21:610–7.
Morris PE, Goad A, Thompson C, et al. Early intensive care unit mobility therapy in the treatment of acute respiratory failure. Crit Care Med. 2008;36(8):2238–43. Prospective cohort study which demonstrated an early mobility protocol in the critically ill decreased ICU and hospital length of stay.
Hermans G, Wilmer A, Meersseman W, et al. Impact of intensive insulin therapy on neuromuscular complications and ventilator dependency in the medical intensive care unit. Am J Resp Crit Care Med. 2007;175(5):480–9.
NICE-SUGAR Study Investigators, Finfeer S, Chittock DR, et al. Intensive versus conventional glucose control in critically ill patients. N Eng J Med. 2009;360(13):1283–97. Large randomized trial demonstrating decreased mortality in critically ill patients with blood sugar target of 180 mg or less.
Ishii DN, Lupien SB. Insulin-like growth factors protect against diabetic neuropathy: effects on sensory nerve regeneration in rats. J Neurosci Res. 1995;40(1):138–44.
Taccone FS, Scolletta S, Franchi F, et al. Brain perfusion in sepsis. Curr Vasc Pharmacol. 2013;11:170–86.
Polito A, Eischwald F, Maho AL, et al. Pattern of brain injury in the acute setting of human septic shock. Crit Care. 2013;17:R204. Prevalence of brain injury in the setting of septic shock was studied using MRI; ischemic strokes and leukoencephalopathy were most prevalent in septic shock.
Bartynski WS, Boardman JF, Zeigler ZR, et al. Posterior reversible encephalopathy syndrome in infection, sepsis, and shock. AJNR Am J Neuroradiol. 2006;27:2179–90.
Gunther ML, Morandi A, Krauskopf E, et al. The association between brain volumes, delirium duration, and cognitive outcomes in intensive care unit survivors: the VISIONS cohort magnetic resonance imaging study. Crit Care Med. 2012;40:2022–32.
Semmler A, Widmann CN, Okulla T, et al. Persistent cognitive impairment, hippocampal atrophy and EEG changes in sepsis survivors. J Neurol Neurosurg Psychiatry. 2013;84:62–9.
Morandi A, Rogers BP, Gunther ML, et al. The relationship between delirium duration, white matter integrity, and cognitive impairment in intensive care unit survivors as determined by diffusion tensor imaging: the VISIONS prospective cohort magnetic resonance imaging study. Crit Care Med. 2012;40:2182–9.
Hosokawa N, Gaspard N, Su F, et al. Clinical neurophysiological assessment of sepsis-associated brain dysfunction: a systematic review. Crit Care. 2014;18(6):674.
Claassen J, Taccone FS, Horn P, et al. Recommendations on the use of EEG monitoring in critically ill patients: consensus statement from the neurointensive care section of the ESICM. Intensive Care Med. 2013;39:1337–51. Systematic review of 42 studies recommending the use of CEEG monitoring to rule out nonconvulsive status epilepticus in comatose patients who are brain injured, post cardiac arrest, suspected refractory status epilepticus, or without brain injury yet suffer persistent altered consciousness.
Kurtz P, Gaspard N, Wahl AS, et al. Continuous electroencephalography in a surgical intensive care unit. Intensive Care Med. 2014;40:228–34.
Sutter R, Stevens RD, Kaplan PW. Clinical and imaging correlates of EEG patterns in hospitalized patients with encephalopathy. J Neurol. 2013;260:1087–98.
Foreman B, Claassen J, Abou Khaled K, et al. Generalized periodic discharges in the critically ill: a case control study of 200 patients. Neurology. 2012;79:1951–60.
Gilmore EJ, Gaspard N, Choi HA, et al. Acute brain failure in severe sepsis: a prospective study in the medical intensive care unit utilizing continuous EEG monitoring. Intensive Care Medicine. 2015;41(4):686–94.
Diedrich DA, Brown DR. Analytic reviews: propofol infusion syndrome in the ICU. J Intensive Care Med. 2011;26:59–72.
Wijdicks EF. The multifaceted care of status epilepticus. Epilepsia. 2013;54(6):61–3. The systemic complications of status epilepticus- infectious, hemodynamic, thromboembolic, and cognitive- are discussed with associated complex treatment plans.
Hocker S. Systemic complications of status epilepticus – an update. Eplepsy Behav. 2015;49:83–7. An update on the neurocardiogenic, pulmonary, and infectious complications of status epilepticus as well as the adverse effects of the pharmacologic treatment to treat status epilepticus, prolonged immobility, and other comorbidities associated with critical illness are discussed.
Hocker SE, Britton JW, Mandrekar JN, et al. Predictors of outcome in refractory status epilepticus. JAMA Neurol. 2013;70:72–7.
Acknowledgments
A special thank you to Dr. John Brust for taking the time to review this article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Rochelle Sweis, Jorge Ortiz, and José Biller declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Neurology of Systemic Diseases
Rights and permissions
About this article

Cite this article
Sweis, R., Ortiz, J. & Biller, J. Neurology of Sepsis. Curr Neurol Neurosci Rep 16, 21 (2016). https://doi.org/10.1007/s11910-016-0623-z
Published:
DOI: https://doi.org/10.1007/s11910-016-0623-z