
Abstract
Production of considerable amounts of reactive oxygen species (ROS) eventually leads to oxidative stress. A key role of oxidative stress is evident in the pathologic mechanisms of endothelial dysfunction and associated cardiovascular diseases. Vascular enzymes such as NADPH oxidases, xanthine oxidase, and uncoupled endothelial nitric oxide synthase are involved in the production of ROS. The question remains whether pharmacologic approaches can effectively combat the excessive ROS production in the vasculature. Interestingly, existing registered cardiovascular drugs can directly or indirectly act as antioxidants, thereby preventing the damaging effects of ROS. Moreover, new compounds targeting NADPH oxidases have been developed. Finally, food-derived compounds appear to be effective inhibitors of oxidative stress and preserve vascular function.
Similar content being viewed by others


Introduction
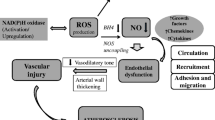
Aerobic life is inconceivable without oxygen, yet oxygen is the major factor that makes life finite. Vascular (dys)function is an illustrative example of the manifestation of oxygen’s mixed blessing on the organ level. During the past decade, a large body of evidence identified oxygen reaction products, the so-called reactive oxygen species (ROS), as important molecules in signal transmission and regulation of vascular function. However, excessive ROS production leads to oxidative stress, which has been determined to play a key role in the pathophysiology of cardiovascular diseases such as endothelial dysfunction, atherosclerosis, hypertension, and myocardial infarction. This article provides an overview of the main molecular sources of ROS in the vessel wall contributing to the development of oxidative stress and associated impaired vascular function. In addition, this review discusses the availability and potential of antioxidant treatment strategies by focusing on well-established cardiovascular drugs as well as on dietary compounds and the latest drug developments specifically designed to target ROS overproduction (Fig. 1).

Molecular sources of vascular oxidative stress and pharmacologic intervention strategies. Vascular enzymes such as NADPH oxidase 2 (1), xanthine oxidase (2), and uncoupled endothelial nitric oxide synthase (3) can promote the production of reactive oxygen species (ROS) considerably. Eventually, these processes contribute to the development of endothelial dysfunction and vascular damage. Established and novel cardiovascular drugs, as well as dietary flavonoids, can attenuate cellular ROS concentrations via direct or indirect mechanisms. ACE—angiotensin-converting enzyme; Ang—angiotensin; AT1—Ang II type 1; CaM—calmodulin; ET-1—endothelin 1; FAD—flavin adenine dinucleotide; FMN—flavin mononucleotide; L-Arg—l-arginine; L-Cit—l-citrulline; PDGF—platelet-derived growth factor; TGF-β—transforming growth factor-β
ROS in Vascular (Dys)function
ROS are generated during the reduction of oxygen and comprise two groups of molecules: 1) free radicals with short biological half-lives, such as superoxide (O •−2 ), hydroxyl (OH•), and nitric oxide (•NO), and 2) nonradicals, such as singlet oxygen (1O2), hydrogen peroxide (H2O2), hypochlorous acid (HOCl), peroxynitrite (ONOO−), and lipid hydroperoxides (LOOH), which are more stable and have longer half-lives than free radicals. Cellular ROS formation is a physiologic process in the vasculature and occurs in endothelial and smooth muscle cells as well as fibroblasts. Although ROS are small, structurally simple molecules with high and random reactivity, they appear to be subtle physiologic modulators of biochemical processes involved in intracellular signaling [1]. Signal transmission via ROS takes place at free sulfhydryl groups of cysteine residues, which are present in many enzymes, including kinases (eg, Src, Akt, protein kinase C [PKC], mitogen-activated protein kinases [MAPKs], and Rho kinases), phosphatases (eg, protein tyrosine phosphatases, SH2 domain-containing phosphatases, MAPK phosphatase), phospholipases (eg, phospholipase A2), and small GTPases (eg, Rac1, RhoA), as well as transcription factors (see later) and ion channels (eg, Ca2+ and K+ channels). The ROS-induced oxidation of sulfhydryl groups leads to intra- and intermolecular disulfide bonds. This posttranslationally alters the conformation of proteins, eventually resulting in altered enzyme activity or DNA binding. Among the complex protein networks transmitting information from the cell surface to the nucleus, several transcription factors (eg, nuclear factor-κB [NF-κB]/Rel family, activator protein 1, hypoxia-inducible factors 1 and 2, p53, c-myc, metal-responsive transcription factor) were first discovered to become activated or inhibited by exogenous and/or ligand-induced oxidants. In the meantime, it is known that the signal propagation upon receptor binding of growth factors such as platelet-derived growth factor (PDGF), epidermal growth factor, nerve-derived growth factor, and endothelin (ET)-1, as well as cytokines such as tumor necrosis factor (TNF)-α and interleukin (IL)-1β, is mediated via the generation of O •−2 and H2O2. Notably, the activation of G protein-coupled receptors such as β-adrenergic receptors and angiotensin (Ang) II type 1 (AT1) receptors by particular agonists results in the production of ROS and/or •NO, which act as signaling molecules in the receptor-mediated regulation of gene expression and cell growth [2•]. To prevent potentially damaging actions and maintain cellular redox balance, enzymatic antioxidant defense systems, including catalase, superoxide dismutase, glutathione peroxidase, and thioredoxin, detoxify and remove ROS. However, when the balance between ROS production and scavenging becomes disturbed, oxidative stress occurs. In this state of excessive ROS production, the ROS-stimulated signaling process switches to a ROS-mediated decrease in bioavailable •NO and damage to lipids, proteins, and DNA. As a consequence, uncontrolled cellular proliferation, apoptosis, increased cellular permeability, and cell death may occur, which in turn leads to endothelial dysfunction, inflammation, and vascular remodeling. All these events are crucial contributors to cardiovascular disease pathologies.
Sources of ROS in the Vasculature
When prolonged ROS accumulation emerges in a dysregulated vascular redox environment, next to mitochondrial respiration, three enzymes have been identified as major sources of ROS: NAD(P)H oxidases (NOX), xanthine oxidase (XO), and uncoupled endothelial nitric oxide synthase (eNOS).
NAD(P)H Oxidases
Initially, NADPH oxidases were thought to be enzymes present only in phagocytes of the innate immune response, where they are responsible for generating large amounts of O •−2 to kill invading pathogens (“oxidative burst”). Upon activation, O2 is reduced to O •−2 by the transfer of one electron from the reducing equivalent NADH or NADPH. However, these enzymes that exist only to produce free radicals appeared unexpectedly in nonphagocytic cells, and it became evident in the past 10–15 years that NADPH oxidases exist in various cell types. In the course of these discoveries, a new terminology was introduced designating nonphagocytic NADPH oxidases as the NOX family. Our current knowledge of the physiologic and pathophysiologic roles of this enzyme family in general and in the cardiovascular system in particular is compiled in several excellent reviews [3••, 4, 5••, 6, 7].
In brief, NOX family NADPH oxidases comprise seven isoforms: NOX1, NOX2 (formerly gp91phox), NOX3, NOX4, NOX5, and dual oxidases 1 and 2 (DUOX1 and DUOX2). All isoforms are transmembrane proteins and consist of multiple subunits located in both the cytosol and the cellular membrane. When the cytosolic subunits p40phox, p47phox, p67phox, the small GTPase Rac, and NOXO1 and NOXA1 are assembled with the two membrane-bound units, gp91phox (NOX2) and p22phox, the protein is fully functional and transports electrons across the membrane to reduce O2 to O •−2 .
The transmembranous enzyme complex is located in the membrane so that the NADPH binding domain lies in the cytoplasm, whereas the O •−2 -generating part is situated in either the extracellular or the intraorganellar space. Interestingly, the membrane-bound catalytic subunits gp91phox and p22phox, which together form the heterodimeric flavocytochrome b558, already contain the complete catalytic machinery for the electron transfer from NADPH to O2, and a basal, albeit low, production of superoxide radicals has been observed even in the absence of NOX stimulators. The cytosolic subunits have regulatory functions related to organization (p47phox and NOXO1), activation (p67phox and NOXA1), and, specifically in the case of DUOX, maturation (DUOXA1 and DUOXA2). Previous insights into the NOX family revealed obvious structural and functional differences among NOX isoforms as well as distinct regulatory mechanisms and distribution in cells and organs [3••]. In vascular endothelial cells, NOX 4, in addition to NOX2 and NOX5, is abundant, whereas in smooth muscle cells, both NOX1 and NOX4 predominate. In adventitial fibroblasts, NOX 4 prevails and in macrophages, NOX2. The O •−2 radicals generated by these cells in the vascular wall readily react with •NO, considerably lowering its vasorelaxant effect.
The activity of NADPH oxidases is stimulated particularly by Ang II but also by other vasoactive hormones (eg, ET-1), growth factors (eg, PDGF, transforming growth factor-β), and mechanical stimuli (shear stress and stretch).
Although it remains to be elucidated how the different NOX isoforms are precisely regulated and interact in vascular cells, their ROS production unmistakably contributes to cardiovascular pathologies as endothelial dysfunction, atherosclerosis, and hypertension.
Xanthine Oxidase
XO and xanthine dehydrogenase (XDH) are both xanthine oxidoreductases (XORs), a group of enzymes known as molybdenum iron-sulfur flavin hydroxylases. Historically, they were considered two distinct enzymes, although it could be demonstrated that both enzymes are encoded by the same gene and are interconverted either reversibly or irreversibly in vivo [8]. XORs catalyze the oxidation of hypoxanthine to xanthine and xanthine to uric acid. For these reactions, XDH prefers NAD+ and XO uses O2 as an electron acceptor, thereby producing NADH and O •−2 and H2O2, respectively. The protein structure of the active XOR consists of a homodimer in which each subunit harbors a molybdopterin moiety, two Fe2–S2 centers, and flavin adenine dinucleotide (FAD) as cofactors. Oxidation of a substrate takes place at the molybdenum center, from which the accepted electrons are transported rapidly via the Fe2–S2 centers to FAD, which in turn reduces the oxidant substrate NAD+ or O2 [9, 10]. Notably, reoxidation of a fully reduced XO occurs stepwise and comprises the transfer of six electrons to O2, yielding two molecules of H2O2 and two molecules of O •−2 . Next to hypoxanthine and xanthine, XOR also can hydrolyze a wide array of aldehydes and N-heterocyclic substrates, thereby contributing to the activation and/or detoxification of endogenous or xenobiotic compounds. Moreover, it exhibits nitrate and nitrite reductase activity, which generates •NO, particularly in hypoxic conditions in which eNOS is not able to produce •NO.
XORs are distributed widely in the human body, with high levels in the gut and liver. Their expression and activity are regulated on both the transcriptional and posttranslational level. For example, proinflammatory stimuli (eg, lipopolysaccharide, TNF-α, IL-1, and IL-6) and hypoxia can significantly upregulate the transcription of XO. Furthermore, a variety of patients with cardiovascular disease (e.g., coronary artery disease, heart failure, and hypertension) reveal significantly elevated XO levels. Interestingly, there is a circulating form of XO that can bind to glycosaminoglycans on the surface of endothelial cells and may acquire increased stability and oxidant-producing capacity upon binding [11]. In patients with chronic heart failure, the activity of this endothelial-bound XO increases by more than 200% [12]. Consequently, high amounts of O •−2 are produced that may quickly react with •NO released from endothelial cells to form cytotoxic ONOO−. Evidence suggests that the formation of ONOO− and the activation of downstream signaling pathways finally result in endothelial cell injury [11, 13]. In those pathophysiologic conditions, the use of XO inhibitors may be a reasonable targeted antioxidant approach.
Uncoupled Endothelial Nitric Oxide Synthase
eNOS is a homodimer consisting of two monomers that each contain a reductase domain capable of binding NADPH, FAD, and flavin mononucleotide (FMN) and an oxygenase domain that carries a prosthetic heme group. These heme groups are necessary for the dimerization of both monomers to form the active NOS dimer and for the electron transfer from the flavin to the heme of the opposite monomer. To become a fully operational enzyme complex, the oxygenase domain binds (6R)-5,6,7,8-tetrahydrobiopterin (BH4), molecular oxygen, and the substrate l-arginine. Finally, a zinc ion binds at the interfaces of the NOS dimer, which has mainly a structural function [14]. eNOS catalyzes an electron transfer within the reductase domain from NADPH via the flavins FAD and FMN to the heme in the oxygenase domain. Calcium-induced binding of calmodulin to the reductase domain increases this electron transfer. At the heme of the oxygenase domain, O2 is reduced and l-arginine is oxidized in two steps via N-hydroxy-l-arginine to l-citrulline and •NO. eNOS activity is regulated by the intracellular calcium concentration and phosphorylation in the reductase and calmodulin-binding domain. Depending on the stimulus, several kinases can phosphorylate eNOS, including Akt/protein kinase B, protein kinase A, 5′-adenosine monophosphate-activated protein kinase, and calmodulin-dependent kinase II.
If the electron flow becomes disturbed within eNOS (in vitro this may occur because of the absence of the cofactor BH4 or the substrate l-arginine), O •−2 will be generated from the oxygenase by dissociation of the ferrous-dioxygen complex. This state of eNOS uncoupling has been observed not only experimentally, but also in humans with essential hypertension [15] or hypercholesterolemia-associated endothelial dysfunction [16] and in chronic smokers [17]. Several molecular triggers have been identified that likely contribute to eNOS uncoupling in vivo. During normal catalytic function of eNOS, BH4 acts as an electron donor. Its oxidized form can be recycled to BH4 by the enzyme itself. However, in an environment of increased O •−2 production—as a result, for example, of elevated NOX and/or XO activity—O •−2 can react with eNOS formed •NO to the highly reactive ONOO−. ONOO− has been shown to oxidize BH4 to biologically inactive products that cannot be recycled further. Eventually, this results in an uncoupling of eNOS. This phenomenon also is observed when l-arginine levels fall below those required to saturate eNOS. Because the K m of eNOS for l-arginine lies between 1 and 10 µM and intracellular l-arginine concentrations are up to 10 times higher than plasma concentrations (approximately 80–120 µM), it appears unlikely that l-arginine might become a limiting factor in vivo [18]. However, there is evidence from hypertensive humans that augmented activity and/or expression of the l-arginine catabolizing enzyme arginase may be responsible for locally diminished l-arginine concentrations [19]. Finally, there is indication that the endogenous eNOS inhibitor asymmetric dimethyl-l-arginine can uncouple eNOS, because it competitively displaces l-arginine from its eNOS binding site [20].
Pharmacologic Interventions in Vascular Oxidative Stress
Registered Cardiovascular Drugs with Direct Antioxidant Activity
The current awareness that oxidative stress plays a pivotal role in the pathophysiologic processes of vascular dysfunction resulted in several treatment strategies to alter ROS levels by decreasing production and/or increasing radical scavenging. In that respect, it is interesting to note that part of the pharmacologic activity of long-established cardiovascular drugs might be a result of their antioxidant properties. For example, it has been shown that antiarrhythmic drugs can inhibit lipid peroxidation [21]. Also, several β-blockers dose-dependently inhibit membrane lipid peroxidation [22]. The β-blocker carvedilol contains a β- and α1-adrenoceptor-blocking pharmacophore as well as a specific carbazole moiety to enhance its antioxidant activity. Also, the metabolites of carvedilol are active antioxidants [23, 24].
By comparing the various classes of Ca2+ antagonists, studies showed that the dihydropyridines (eg, nifedipine, nisoldipine) were particularly effective in protecting against H2O2-induced decreases in contractile function of rat hearts [25] and low-density lipoprotein oxidation [26].
Registered Cardiovascular Drugs with Indirect Antioxidant Activity
The antioxidant activity of angiotensin-converting enzyme (ACE) inhibitors, especially the sulfhydryl group containing captopril, has been discussed extensively [27]. It also was suggested that enalapril and captopril might indirectly enhance the tissue antioxidant defense mechanisms [28]. The in vivo relevance of the direct systemic antiradical effects, however, have been strongly disputed [29]. Indeed, the general antioxidant (radical scavenging) action of drugs is not likely to be very effective clinically. The concentrations needed are probably too high to reach. Thus, the putative antioxidant action of ACE inhibitors might be displayed in a more confined tissue area. Ang II potently stimulates NADPH oxidases in various cells of the vasculature, promoting the generation of O •−2 [30]. ACE inhibition thus limits the formation of O •−2 [31]. A similar indirect antioxidant effect may be expected from the Ang II receptor blockers, the “sartans.”
Similarly, part of the pleiotropic effect of statins has been explained by an indirect antioxidant mechanism, in this case by their inhibitory effect on the coupling of the AT1 receptor to the NADPH oxidase [32].
If source-directed inhibition of ROS generation improves vascular function, XO inhibition also should have a beneficial effect. Indeed, a recent review describes encouraging results for the XO inhibitor allopurinol [33••].
Newly Designed Antioxidant Drugs
Specific medical-chemical approaches have been used to design antioxidant drugs for the vascular system. In the late 1980s, the so-called 21-aminosteroids were designed to protect against acute and delayed vascular consequences after a subarachnoid hemorrhage [34].
Based on the antioxidant chromanol moiety of vitamin E (α-tocopherol), quaternary ammonium analogues of α-tocopherol were developed and have been proven to successfully decrease the heart infarct size in animals [35]. Many other antioxidant hybrid molecules have been designed, and this line of research continues. Losartan-antioxidant hybrids [36•], antiarrhythmic-α-tocopherol hybrids [37], and troglitazone, an α-tocopherol-thiazolidinedione hybrid that is a peroxisome proliferator-activated receptor-γ ligand with antioxidant activity are examples in this regard. The clinical application of the latter was discontinued because of hepatotoxicity [38].
New peptide inhibitors have been developed to specifically target NADPH oxidases [5••]. The 18-amino acid peptide gp91ds-tat interferes with NADPH oxidase and subunit assembly and attenuates vascular O •−2 production. Nine of its amino acids mimic the region of p22phox that interacts with p47phox.
PR39 is a naturally occurring 39-amino acid proline- and arginine-rich peptide originally isolated from pig intestine. PR39 binds to the Src homology domain 3 (SH3) p47phox and, like gp91ds-tat, prevents the association between p47phox and NOX proteins.
A specific NADPH oxidase inhibitor, VAS2870, was found recently and is awaiting further characterization.
The frequently used NADPH inhibitor apocynin is a methoxy-catechol extracted from the Picroria kurroa plant and oxidized by intracellular peroxidases into active metabolites. Moreover, nitration and nitrosylation of the NADPH oxidase lead to inhibition of the enzyme. In addition, PKC plays an important role in the activation of NADPH oxidase through phosphorylation of p47phox. In this way, PKC inhibitors also decrease O •−2 production by the NADPH oxidase.
As mentioned, with a shortage of BH4, NOS uncouples and produces O •−2 rather than •NO. However, supplementation with BH4 restores the •NO/O •−2 balance.
Polyphenolic Antioxidant Modulators
The cardiotoxicity of doxorubicin, a generally used agent for heart failure, can be prevented with antioxidants. The flavonoid 7-monohydroxyethylrutoside effectively protects against doxorubicin-induced toxicity [39]. Interestingly, this flavonoid effectively protects against the neutrophil infiltration of ischemic heart tissue in mice after reperfusion [40].
Recently, the antioxidant and anti-inflammatory action of flavonoids was explored in more detail. It has been suggested that in the inhibition of the proinflammatory transcription factor NF-κB by the flavonoids, attenuating effects on poly(ADP-ribose) polymerase are involved [41].
Because flavonoids are a substantial part of fruits and vegetables, it is tempting to suggest that these polyphenols contribute to their cardiovascular effects. In fact, a high intake of fruits and vegetables has been associated with a decrease in blood pressure in humans [42]. The endothelial-protective effects of flavonoids may be related to other activities of these compounds, such as inhibition of ET-1 synthesis [43] or inhibition of arginase [44], increasing the availability of l-arginine, which forms the rate-limiting factor for cellular •NO production. It has further been proposed that the prevailing mode of action of flavonoids is inhibition of NADPH oxidase, thus lowering the O •−2 generation that leads to elevation of •NO levels in the cell. The long-term effect of flavonoids is probably mediated by alterations in gene expression and protein synthesis, such as eNOS [45].
It will be interesting to investigate whether the heretofore unsolved mechanism of the antihypertensive effect of a tomato extract, which is observed even when the extract is administered on top of regular pharmacologic treatment in humans, also differs with regard to short-term and long-term administration. Paste from genetically modified tomatoes enriched in quercetin inhibited the development of hypertension in spontaneously hypertensive rats [46].
Polyphenols and their metabolites probably preserve the •NO/O •−2 balance by inhibition of NADPH oxidase combined with O •−2 scavenging. From that view, polyphenols differ from the antioxidant vitamins ascorbic acid and α-tocopherol, which increase the efficacy of eNOS by ensuring the level of the cofactor BH4 [47].
Conclusions
Several already-registered cardiovascular-effective compounds possess prominent antioxidant activity, which may explain some of their therapeutic activity. Changes in cellular redox status with antioxidant vitamins also may improve •NO availability, either by supporting the eNOS cofactor BH4 level or by decreasing the O •−2 -mediated abolition of •NO. More promising is the inhibition of the vascular O •−2 -generating NADPH oxidase. It appears that here, too, established, successful drugs possess this inhibitory effect. Moreover, vasoactive food-derived flavonoids exhibit this effect in addition to their well-known antioxidant action.
Our increasing knowledge of the molecular sources of oxidative stress and its role in vascular function will enable the development of new vasoactive pharmacologic and dietary modulators of the pathogenesis of cardiovascular disease.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Poli G, Leonarduzzi G, Biasi F, Chiarpotto E: Oxidative stress and cell signalling. Curr Med Chem 2004, 11:1163–1182.
• Ushio-Fukai M: Vascular signaling through G protein-coupled receptors: new concepts. Curr Opin Nephrol Hypertens 2009, 18:153–159, This is a comprehensive review of the role of ROS in G protein-coupled receptor signaling.
•• Bedard K, Krause K: The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007, 87:245–313, This excellent review provides detailed information about the role of NOX in health and disease.
Cave AC, Brewer AC, Narayanapanicker A, et al.: NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal 2006, 8:691–728.
•• Guzik TJ, Harrison DG: Vascular NADPH oxidases as drug targets for novel antioxidant strategies. Drug Disc Today 2006, 11:524–533, This very interesting article discusses new approaches for inhibiting NADPH oxidases. The authors make it clear that besides direct ROS scavenging, an indirect antioxidant action also is a strategy for modulating vascular oxidative stress.
Nauseef WM: Biological role for the NOX family NADPH oxidases. J Biol Chem 2008, 283:16961–16965.
Ushio-Fukai M: Compartmentalization of redox signaling through NADPH-oxidase-derived ROS. Antioxid Redox Signal 2009, 11:1289–1299.
Nishino T, Okamoto K, Eger BT, et al.: Mammalian xanthine oxidoreductase—mechanisms of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J 2008, 275:3278–3289.
Berry CE, Hare JM: Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications. J Physiol 2004, 555:589–606.
Harrison R: Structure and function of xanthine oxidoreductase: where are we now? Free Rad Biol Med 2002, 33:774–797.
Radi R, Rubbo H, Bush K, Freeman BA: Xanthine oxidase binding to glucosaminoglycans: kinetics and superoxide dismutase interactions of immobilized xanthine oxidase-heparin complexes. Arch Biochem Biophys 1997, 339:125–135.
Landmesser U, Spiekermann S, Dikalov S, et al.: Vascular oxidative stress and endothelial dysfunction in patients with chronic heart failure: role of xanthine-oxidase and extracellular superoxide dismutase. Circulation 2002, 106:3073–3078.
Ungvari Z, Gupte SA, Recchia FA, et al.: Role of oxidative-nitrosative stress and downstream pathways in various forms of cardiomyopathy and heart failure. Curr Vasc Pharmacol 2005, 3:221–229.
Förstermann U: Janus-faced role of endothelial NO synthase in vascular disease: uncoupling of oxygen reduction from NO synthesis and its pharmacological reversal. Biol Chem 2006, 387:1521–1533.
Higashi Y, Sasaki S, Nakagawa K, et al.: Tetrahydrobiopterin enhances forearm vascular response to acetylcholine in both normotensive and hypertensive individuals. Am J Hypertens 2002, 15:326–332.
Stroes E, Kastelein J, Cosentino F, et al.: Tetrahydrobiopterin restores endothelial function in hypercholesterolemia. J Clin Invest 1997, 99:41–46.
Heitzer T, Brockhoff C, Mayer B, et al.: Tetrahydrobiopterin improves endothelium-dependent vasodilation in chronic smokers: evidence for a dysfunctional nitric oxide synthase. Circ Res 2000, 86:E36–E41.
Moncada S, Higgs EA: Molecular mechanisms and therapeutic strategies related to nitric oxide. FASEB J 1995, 9:1319–1330.
Holowatz LA, Kenney WL: Up-regulation of arginase activity contributes to attenuated reflex cutaneous vasodilation in hypertensive humans. J Physiol 2007, 581:863–872.
Sydow K, Munzel T: ADMA and oxidative stress. Atheroscl Suppl 2003, 4:41–51.
Rekka E, Mannhold RM, Bast A, et al.: Molecular pharmacological aspects of antiarrhythmic activity I. Class I and class III compounds and lipid peroxidation. Biochem Pharmacol 1990, 39:95–100.
Yue T-L, Cheng H-Y, Lysko PG, et al.: Carvedilol, a new vasodilator and beta adrenoceptor antagonist, is an antioxidant and free radical scavenger. J. Pharmacol Exp Ther 1992, 263:92–98.
Dulin B, Abraham WT: Pharmacology of carvedilol. Am J Cardiol 2004, 93:3B–6B.
Jonsson G, Abdelnoor M, Seljeflot I, et al.: The antioxidant effects of long-term treatment are more pronounced for carvedilol than for atenolol in post-myocardial infarction patients. J Cardiovasc Pharmacol 2007, 49:27–32.
Kauder WF, Watts JA: Antioxidant properties of dihydropyridines in isolated rat hearts. Biochem Pharmacol 1996, 51:811–819.
Nègre-Salvayre A, Salvayre R: Protection by Ca2+ channel blockers (nifedipine, diltiazem and verapamil) against the toxicity of oxidized low density lipoprotein to cultured lymphoid cells. Br J Pharmacol 1992, 107:738–744.
Bartosz M, Kedziora J, Bartosz G: Antioxidant and prooxidant properties of captopril and enalapril. Free Rad Biol Med 1997, 23:729–735.
de Cavanagh EM, Inserra F, Ferder L, et al.: Enalapril and captopril enhance glutathione-dependent antioxidant defenses in mouse tissue. Am J Physiol Regul Integr Comp Physiol 2000, 278:R572–R577.
Lapenna D, De Gioia S, Ciofani G, et al.: Captopril has no significant scavenging antioxidant activity in human plasma in vitro or in vivo. Br J Clin Pharmacol 1996, 42:451–456.
Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW: Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res 1994, 74:1141–1148.
van der Giet M, Erinola M, Zidek W, et al.: Captopril and quinapril reduce reactive oxygen species. Eur J Clin Invest 2002, 32:732–737.
Adam O, Laufs U: Antioxidative effects of statins. Arch Toxicol 2008, 82:885–892.
•• George J, Struthers AD: Role of urate, xanthine oxidase and the effects of allopurinol in vascular oxidative stress. Vasc Health Risk Manag 2009, 5:265–272, This article discusses the role of XO in vascular oxidative stress. The research on allopurinol as an inhibitor of XO and its encouraging effect on endothelial dysfunction are reported.
Hall ED: Efficacy and mechanisms of action of the cytoprotective lipid peroxidation inhibitor tirilazad mesylate in subarachnoid haemorrhage. Eur J Anaesthesiol 1996, 13:279–289.
Petty MA, Grisar JM, de Jong W: Protective effects of an alpha-tocopherol analogue against myocardial reperfusion injury in rats. Eur J Pharmacol 1992, 210:85–90.
• Garcia G, Rodriguez-Puyol M, Alajarin R, et al.: Losartan-antioxidant hybrids: novel molecules for the prevention of hypertension-induced cardiovascular damage. J Med Chem 2009, 52:7229–7227, This article provides a nice example of the medical-chemical approach to designing chimeric molecules that combine an established cardiovascular pharmacologic effect with an antioxidant action.
Koini EN, Papazafiri P, Vassilopoulos A, et al.: 5,7,8-Trimethyl-benzopyran and 5,7,8-trimethyl-1,4-benzoxazine aminoamide derivatives as novel antiarrhythmics against ischemia-reperfusion injury. J Med Chem 2009, 52:2328–2340.
Hamblin M, Chang L, Zhang J, et al.: The role of peroxisome proliferator-activated receptor gamma in blood pressure regulation. Curr Hypertens Rep 2009, 11:239–245.
Bast A, Haenen GR, Bruynzeel AM, et al.: Protection by flavonoids against anthracycline cardiotoxity: from chemistry to clinical trials. Cardiovasc Tox 2007, 7:154–159.
de Celle T, Heeringa P, Strzelecka AE, et al.: Sustained protective effects of 7-monohydroxyethylrutoside in an in vivo model of cardiac ischemia-reperfusion. Eur J Pharmacol 2004, 494:205–212.
Weseler AR, Geraets L, Moonen HJJ, et al.: Poly(ADP-ribose)polymerase-1-inhibiting flavonoids attenuate cytokine release in blood from male patients with chronic obstructive pulmonary disease or type 2 diabetes. J Nutr 2009, 139:952–957.
John JH, Ziebland S, Yudkin P, et al.: Effects of fruit and vegetable consumption on plasma antioxidant concentrations and blood pressure; a randomized controlled trial. Lancet 2002, 359:1969–1974.
Caton PW, Pothecary MR, Lees DM, et al.: Regulation of vascular endothelial function by procyanidin-rich foods and beverages. J Agric Food Chem 2010 Jan 28 (Epub ahead of print).
Schnorr O, Brossette T, Momma TY, et al.: Cocoa flavanols lower vascular arginase activity in human endothelial cells in vitro and in erythrocytes in vivo. Arch Biochem Biophys 2008, 476:211–215.
Schewe T, Steffen Y, Sies H, et al.: How do flavanols improve vascular function? A position paper. Arch Biochem Biophys 2008, 476:102–106.
de Leeuw PW, Bast A: Tomato extract for hypertension? Cardiovasc Drugs Ther 2009, 23:107–108.
Steffen Y, Gruber C, Schewe T, et al.: Mono-O-methylated flavanols and other flavonoids as inhibitors of endothelial NADPH oxidase. Arch Biochem Biophys 2008, 469:209–219.
Acknowledgment
The authors acknowledge financial support from the European Union (grant 226588, entitled Flaviola) within the Seventh Framework Programme.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Weseler, A.R., Bast, A. Oxidative Stress and Vascular Function: Implications for Pharmacologic Treatments. Curr Hypertens Rep 12, 154–161 (2010). https://doi.org/10.1007/s11906-010-0103-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11906-010-0103-9