
Abstract
Diagnosing a myelodysplastic syndrome (MDS) can be challenging. Somatic mutations are common in MDS and might have diagnostic utility in patients with idiopathic cytopenias of undetermined significance (ICUS). However, using mutations to diagnose MDS is complicated by several issues: (1) no gene is mutated in most cases, (2) no mutated gene is highly specific for MDS, (3) clonal hematopoiesis is common in older individuals without disease, and (4) we lack outcome data for ICUS patients with clonal cytopenias of undetermined significance (CCUS). Despite these caveats, genetic sequencing can inform the diagnosis of MDS. CCUS patients more closely resemble patients with MDS than age matched controls with somatic mutations. Genetic testing can identify alternative diagnoses in cytopenic patients and help risk stratify those with proven MDS. While we cannot include somatic mutations in the diagnostic definition of MDS now, testing to recognize CCUS will help characterize outcomes in these diagnostically challenging patients.
Similar content being viewed by others


Introduction
For several of reasons, myelodysplastic syndromes (MDS) are often challenging to diagnose. MDS are heterogeneous disorders that share key clinical features, including ineffective clonal hematopoiesis, morphologic dysplasia, peripheral blood cytopenias, and a variable risk of transformation to acute myeloid leukemia (AML) [1]. However, clinical presentations can vary dramatically from patient to patient. Establishing the diagnosis of MDS requires the quantification of morphologic features such as dysplasia and bone marrow blast proportion, both of which are subject to interobserver variability even among expert hematopathologists [2, 3]. Diagnostic features of MDS are also frequently encountered in related disorders ranging from aplastic anemia (AA) and myeloproliferative neoplasms (MPN) to AML with myelodysplasia. Making an accurate diagnosis has important clinical consequences [4–6]. A more objective mechanism for diagnosing MDS, particularly in confounding cases, would be of great clinical benefit [6–8].
Hematologists have long incorporated molecular genetic information into the diagnostic evaluation of patients with myeloid diseases. In chronic myelogenous leukemia (CML), measurement of the leukocyte alkaline phosphatase has been replaced by detection of the BCR-ABL fusion transcript—the defining criterion for this disorder [9, 10]. Several subtypes of AML are defined by the presence of specific chromosomal translocations regardless of bone marrow blast proportion [11]. And, in the10 years since the discovery of JAK2 mutations in MPNs, these lesions can be diagnostic of polycythemia vera (PV), essential thrombocythemia (ET), or primary myelofibrosis (PMF) in the appropriate clinical contexts [12]. We have learned as much about the molecular genetic basis of MDS in the last decade [13•]. There are well over 40 somatically mutated genes seen recurrently in MDS, one or more of which can be identified in over 90 % of patients [14••, 15••, 16••]. We understand how mutations are associated with many disease features like ring sideroblasts, cytopenias, monocytosis, and chromosomal abnormalities. The independent prognostic value of many recurrently mutated genes has been validated in numerous studies, and mutations that act as biomarkers of response to specific therapies have been described [17, 18, 19•, 20•]. Somatic mutations are not only indicative of clonal hematopoiesis, a defining feature of MDS, they identify the molecular drivers responsible for its pathogenesis. These facts, and our experience with diagnostic molecular tests in related myeloid disorders, suggest that mutations could readily be incorporated into the diagnostic criteria for MDS [21, 22].
Unfortunately, diagnostic utility of somatic mutations in MDS is complicated by several issues. These include concerns about poor specificity, the range of genetic variability present in MDS, and a poor understanding about the implications of mutations in patients who do not meet current diagnostic criteria. This article will review current diagnostic methods for MDS and will explore the role of molecular genetic testing in this context, focusing on its challenges and how these may be overcome.
Section I—Current Diagnostic Criteria for MDS
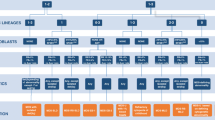
Making a diagnosis of MDS appears fairly straightforward. A patient must have at least one clinically meaningful cytopenia and a bone marrow examination with one or more “decisive” criteria for MDS (Table 1) [23]. However, meeting these criteria is not sufficient until competing explanations for these findings are excluded [11]. The list of benign conditions that can mimic MDS or confound its diagnosis is long (Fig. 1). Deficiencies of vitamin B12 or folate can cause cytopenias, megaloblastic changes, and macrocytosis. Iron deficiency can cause anemia and abnormal red cell morphology. Copper deficiency, often seen in patients with gastric bypass or chronic zinc ingestion, can result in anemia with ring sideroblasts, a defining feature of some MDS subtypes. Viral infections can cause bone marrow suppression with dysplastic features as can autoimmune conditions. These include Felty syndrome, idiopathic thrombocytopenia purpura, and systemic lupus erythematosus among others. Medications taken to treat autoimmune conditions, like methotrexate or azathioprine, can also cause cytopenias and dysplasia. Chronic alcohol abuse can cause morphologic dysplasia and cytopenias through a variety of mechanisms such as liver damage, splenomegaly, and direct bone marrow suppression. Then, there are several rare inherited conditions that can mimic MDS, such as congenital dyserythropoietic or sideroblastic anemias, which should be considered, particularly in cases with a family history of anemia. Finally, several clonal conditions that share clinical features with MDS, including AML, MPN, and AA, must also be excluded in order to make the diagnosis (Fig. 1).

Diagnostic overlap between MDS and other clonal disorders and benign conditions that can mimic MDS. HIV human immunodeficiency virus, EBV Epstein-Barr virus, Hep hepatitis, LGL large granular lymphocyte leukemia
The WHO guidelines recognize the challenge of diagnosing patients with unexplained cytopenias, no increased blast proportion, and a normal karyotype [11]. They recommend that before making the diagnosis of MDS, patients with unilineage dysplasia as their sole diagnostic finding demonstrate 6 months of persistent cytopenia. Recovery of counts in that time would suggest a cause other than MDS. The presence of an acquired chromosomal abnormality can be helpful as it indicates clonal hematopoiesis. This does not exclude benign causes of cytopenias but might make it more likely that MDS or another clonal disorder is present. The WHO guidelines include a list of chromosomal abnormalities that can serve as presumptive evidence of the diagnosis, even in the absence of dysplasia or increased blast proportion (Table 1). These include several of the most frequent cytogenetic abnormalities seen in MDS such as del(5q), del(7q), loss of chromosome 7, or a complex karyotype (defined as the presence of three or more concurrent abnormalities). It is important to note that several recurrent chromosomal abnormalities are not included in this list. Lesions such as del(20q) and trisomy 8 are found in MDS but are present in many other myeloid disorders and hence lack the necessary specificity required to serve as diagnostic evidence of MDS. Loss of chromosome Y is also not considered a diagnostic clonal marker because it can frequently be seen in older men in the absence of hematologic disease. In practice, less than 50 % of patients with MDS will have an abnormal karyotype, and for lower risk patients with no increasing in blasts, this fraction is closer to 25 % [24, 25]. Cytopenic patients who do not reach the arbitrary cutoffs of 10 % dysplasia any lineage or 5 % bone marrow blast proportion will rarely carry a disease-defining chromosomal abnormality. In contrast, somatic mutations indicative of clonal hematopoiesis can be found in over 80 % of MDS patients with lower risk disease [26]. This begs the question of whether certain somatic mutations can substitute for chromosomal abnormalities as presumptive evidence of MDS in the absence of disease-defining morphologic criteria.
For reasons that are addressed below, the short answer to this question at the moment is no. Somatic mutations should not replace our current diagnostic standards for MDS as they have for several myeloproliferative disorders.
Section II—Challenges to the Diagnostic Utility of Somatic Mutation in MDS
To understand why somatic mutations are not reliably diagnostic of MDS, it will be helpful to review the features of good diagnostic biomarkers. The first is frequency. A mutation present in the majority of cases of given disease is extremely useful for its diagnosis. Even if the mutation occurs in other disorders, the clinical context can help exclude these alternative diagnoses as unlikely. If a diagnosis is suspected on clinical grounds but its highly frequent, associated mutation is absent, the diagnosis must be considered much less likely or ruled out altogether. This describes JAK2 mutations in the diagnosis of PV. Even though JAK2 mutations can be found in a variety of myeloid disorders, including MDS, a mutation should be present in a patient clinically suspected of having PV. If no JAK2 mutation can be identified after appropriate testing, an alternative diagnosis such as secondary or congenital polycythemia must be given greater weight as the diagnosis of PV is essentially excluded.
The second feature that can support the utility of a diagnostic biomarker is specificity. A mutation uniquely associated with a disease can be considered presumptive evidence of its diagnosis. This is true even if the majority of the patients with the disorder lack this particular mutation. Consider MPL mutations which are present in 5 % of ET. While these mutations are not present in the majority of ET patients, they are extremely rare in other conditions. Clinical context can exclude confounding conditions such as MF and refractory anemia with ring sideroblasts (RARS) and thrombocytosis (RARS-T). Therefore, a typical MPL mutation in a patient with isolated thrombocytosis strongly supports the diagnosis of ET. Several other MPN-associated gene mutations, such as those in CALR for PMF and ET, KIT in mastocytosis, and the FIPL1-PDGFRA rearrangement in the hypereosinophilic syndrome, can also be considered specific enough for diagnostic use in the appropriate clinical contexts. It is also helpful if the molecular lesion in question is the founding abnormality that gives rise to the disease. This implies a degree of specificity but also ensures that the lesion will be present in the majority of clonal cells making easier to detect. The BCR-ABL fusion gene in CML is the classic example of causative, disease-defining mutation that meets both the frequency and the specificity criteria described above, making it an excellent diagnostic marker.
The examples above are MPNs where acquired mutations are routinely incorporated into diagnostic criteria [11]. There are several reasons why this is not yet the case for MDS. To begin with, the clinical context for MDS is much more challenging to interpret than it is for MPNs. There are a limited number of molecular mechanisms that cause myeloproliferation and alternative diagnoses for patients with elevated cell counts are few and relatively rare. In contrast, cytopenias and myelodysplasia, the defining features of MDS, can be caused by a broad array of molecular abnormalities and many potential alternative causes of these findings are frequent. There is no single gene that is mutated in the majority of cases of MDS. Mutations of TET2 and SF3B1 are each present in only a quarter to a third of patients [15••, 16••, 26]. A handful of other genes (ASXL1, SRSF2, DNMT3A, and RUNX1) are mutated in 10–20 %, but the majority of recurrently mutated genes identified in MDS are present in fewer than 5 % of cases. Therefore, no mutated gene meets the frequency criteria that would allow a diagnosis of MDS to be excluded if a particular mutation was not identified.
Mutations could still be diagnostically useful if they were highly specific for MDS. Unfortunately, this is not the case. Like the del(20q) and trisomy 8 chromosomal abnormalities, many genes mutated in MDS are observed in other myeloid disorders where they have very different clinical implications. For example, ASXL1 mutations are more common in chronic myelomonocytic leukemia (CMML) than in MDS but can also occur in MPN, AA, and AML [27–29]. Mutations of TET2 and DNMT3A are found in these myeloid disorders but can occur in lymphoid neoplasms as well [30]. Mutations of SF3B1 are the most specific as they are strongly associated with the presence of ring sideroblasts, a readily identifiable morphologic feature used to make the diagnosis of MDS [31•]. However, SF3B1 mutations can also occur in MPN/MDS overlap syndromes like CMML and RARS-T and are frequent in CLL [32, 33].
Finally, there is a practical consideration that distinguishes the mutations that support the diagnosis of MPN and AML from the mutations most often seen in MDS. Many AML- or MPN-related mutations are highly recurrent activating lesions such as JAK2 V617F, the MPL W515L/K mutations, internal tandem duplications of FLT3, or the frameshifts in the terminal exons of CALR and NPM1. Most of the somatic mutations in MDS are not so stereotypic. Genes can often be mutated anywhere along their length and can harbor missense mutations that can be difficult to distinguish from benign germline polymorphisms or incidental passenger mutations unrelated to the disease. This makes the interpretation of genetic tests more difficult for many MDS-related genes given our current state of knowledge.
Were these the only issues constraining the use of mutations as diagnostic biomarkers for MDS, certain mutations in specific genes might still have obvious diagnostic utility in the appropriate clinical context. For example, if a patient with ICUS (clinically significant cytopenias, morphologic criteria for MDS not met, and no alternative diagnosis evident) were found to have a typical mutation in DNMT3A, the presence of clonal hematopoiesis would be established potentially making a diagnosis of MDS more likely than a non-clonal cause of their cytopenias. However, recent findings dictate caution in making such an interpretation.
It is known that clonal hematopoiesis can occur in the absence of a clinically evident hematologic disorder. Studies of skewed X chromosome inactivation demonstrate that some women have clonal hematopoiesis and that this becomes increasingly likely with age [34, 35]. More recent work has confirmed that this is not a rare phenomenon [36]. In two studies, subjects participating in genome-wide association studies (GWAS) had their blood genotyped for single nucleotide polymorphisms (SNPs) [37, 38]. This technique found that 2–3 % of older participants carried chromosomal abnormalities likely to be acquired and therefore indicative of clonal hematopoiesis. Two subsequent GWAS studies and a study of patients with solid tumors examined the exomes of blood samples from a total of over 30,000 individuals [39••, 40, 41••]. Likely somatic mutations in several MDS-associated genes were found with a frequency that increased markedly with age. Approximately 10 % of persons aged 70–80 carried one or more of these mutations indicative of clonal hematopoiesis. The three most commonly mutated genes across these studies were DNMT3A (accounting for more than half of all cases), TET2, and ASXL1 which were closely followed by mutations in JAK2, TP53, SF3B1, SRSF2, and CBL. Only one frequently mutated gene, PPM1D, was not known to be recurrently mutated in MDS [41••]. The median age of MDS patients is in the 70–75 years range. The incidence of MDS in this age group is several orders of magnitude lower that the 10 % rate of clonal hematopoiesis detected in this population. More importantly, most of the individuals with clonal hematopoiesis had no known hematologic abnormality and there was no association between the presence of somatic mutations in these GWAS studies and clinically significant cytopenias.
The presence of a somatic mutation in these studies was not clinically benign, however. Individuals with evidence of clonal hematopoiesis had a 10–15-fold increased risk of developing a hematologic malignancy, although not necessarily MDS [39••, 41••]. This risk increased to 50-fold if the somatic mutation was present in 20 % or more nucleated blood cells (corresponding to a variant allele fraction of ≥0.10). Yet, the absolute increase in risk remained very small, and the vast majority of patients with clonal hematopoiesis never went on to develop a hematologic disorder. For this reason, incidental findings of somatic mutations have been referred to as clonal hematopoiesis of indeterminate potential, abbreviated as CHIP [42••]. The high frequency of CHIP in the elderly population most likely to have MDS places strong constraints on the diagnostic value of somatic mutations. The presence of a DNMT3A, ASXL1, or TET2 mutation typical of MDS does not clarify the diagnosis in an elderly cytopenic patient. Benign causes of cytopenias and alternative clonal disorders would still have to be excluded just as they would if these mutations were not present.
Section III—Utility of DNA Sequencing at the Time of Diagnosis
Given the many caveats discussed above, it may appear that molecular genetic tests have no role in the diagnostic setting for MDS. However, there are several scenarios in which somatic mutations can clarify the diagnosis and, in the future, might inform a molecular definition of disease. CHIP was described in various populations that were not suspected of having a hematologic disorder. In practice, these are not the people subjected to molecular genetic testing. Instead, it is the cytopenic patients for which MDS is in the differential diagnosis that are more likely to undergo sequencing of their blood or bone marrow. Finding a somatic mutation typical for MDS in this clinical context may have different implications. Currently, patients with unexplained cytopenias that do not meet the diagnostic criteria for MDS are labeled as having ICUS [7, 43•, 44]. The eventual outcome for patients with ICUS is not well understood nor have clear risk features for progressive disease been established. Small studies have suggested that clonality may be one adverse risk factor [45]. Recent studies have demonstrated that a subset of ICUS patients carry somatic mutations in MDS-related genes indicative of clonal hematopoiesis [46, 47]. This may not seem surprising given the high rate of CHIP in the age range typical for MDS. However, ICUS patients with clonal hematopoiesis, henceforth referred to as having clonal cytopenias of undetermined significance (CCUS), have other features that distinguish them from people with non-cytopenic CHIP [42••].
Two studies presented at the American Society of Hematology Annual Meeting in 2014 examined diagnostic material from cytopenic patients suspected of having MDS. One was a prospective examination of 146 patients [46]. Based on their bone marrow findings, 21 patients were diagnosed with MDS, 22 as ICUS with some dysplastic features not meeting criteria for MDS, and 103 as having no morphologic features of MDS at all. A panel of 21 frequently mutated MDS genes was examined by next-generation sequencing in each patient. In patients with a clear diagnosis of MDS, 76 % had at least one typical somatic mutation with more than a third having three or more such mutations. The mutation rate was 55 % in the ICUS group with some evidence of MDS and 22 % in cytopenic patients with no evidence of MDS at all. The fraction of CCUS patients across the two ICUS arms was 28 %, which is much higher than the 10–15 % of CHIP expected in an aged matched “normal” population. Strikingly, the number of CCUS patient was nearly 1.5-fold greater than the number of patients that met the diagnostic criteria for MDS, suggesting that the incidence of CCUS may be large and comparable to that of MDS under its current diagnostic definition.
The second study examined 250 sequential cases of ICUS collected over 6 months at a commercial laboratory and compared them to 90 cases of lower risk MDS diagnosed in the same time frame [47]. The results were similar to the prior study in that 33 % of ICUS patients carried at least one somatic mutation. In both studies, the genes mutated in ICUS and MDS were similar with the exception of SF3B1 which was rarely mutated in CCUS and common in MDS, particularly lower risk MDS. Interestingly, the median variant allele frequencies for MDS and CCUS were similar at about 30 %. More than 80 % of mutations were present at an allele frequency of at least 10 %, the median described for CHIP in population studies. The likelihood of having clonal hematopoiesis in a cytopenic patient was greater than the background rate of CHIP, and mutations were more abundant on average suggesting a continuum between CHIP, CCUS, and MDS. Unfortunately, we still lack data on outcomes in patients with CCUS and how clinical and molecular features could help us further refine disease risk in this group.
In absence of validated outcomes data, a Bayesian approach could help support the diagnosis of clinically meaningful cytopenias even if they are not labeled as MDS. For example, persons with CHIP were mostly over age 50 and typically had one MDS-related mutation at a low (<10 %) variant allele frequency. If a 45 year-old patient with ICUS were found to carry two or more mutated genes with a variant allele frequency of 30–40 %, it would seem much less likely that this person had CHIP and coincidentally, an overlooked benign cause for their cytopenias. Similarly, if a 73 year-old cytopenic patient has no somatic mutations detected in a large panel of genes, alternatives to MDS should be strongly considered.
We may learn that certain mutations have different degrees of diagnostic utility. For example, DNMT3A, TET2, and ASXL1 are so frequent in CHIP and so prevalent in disorders other than MDS that an isolated mutation in one of these genes likely cannot be considered diagnostically helpful (Table 2). Some of the less frequently mutated CHIP genes, like U2AF1, RUNX1, and TP53, may retain some specificity for MDS in the appropriate clinical context.
Several genes are mutated much more frequently in disorders that can mimic MDS. For example, a BRAF mutation in a cytopenic patient without marked dysplasia should prompt careful examination for features of hairy cell leukemia, a rare cause of cytopenias that nearly always carries an activating BRAF mutation [48]. Similarly, recurrent mutations of STAT3 and STAT5B have been associated with clonal large granular lymphocyte leukemia, a disorder of T or NK cells that can present like MDS, but is treated very differently [49, 50]. Just as t(8;21), inv(16), t(16;16), and t(15;17) define specific subtypes of AML regardless of bone marrow blast proportion, there may be mutated genes that are de facto evidence of leukemia. These might include small FLT3-ITD mutant subclones or more abundant mutations of IDH1, IDH2, or NPM1, all of which are very rare in both CHIP and MDS compared with AML [51, 52].
Rare cytopenic patients may have germline mutations in genes associated with an increased risk of developing MDS or AML. The genes involved can include RUNX1, CEBPA, GATA2, ETV6, DDX41, TERT, DKC1, and TP53, among others [53–56]. Patients may have a suggestive family history, earlier age at diagnosis, and clinical manifestations specific to the congenital mutation involved. However, patients can present later in life with bone marrow failure as their only prominent clinical finding, and germline mutations can occur de novo in individuals with no relevant family history [57, 58]. Often, patients may have a prodromal stage of abnormal blood counts without clear evidence of MDS or AML. This is common in patients with germline RUNX1, ETV6, or GATA2 mutations, for example [54, 55]. Genetic testing at diagnosis may identify these individuals earlier in the course of disease and help screen siblings if related donor allogeneic stem cell transplant is being considered.
Finally, DNA sequencing at the time of diagnosis may identify somatic mutations that help classify patients with traditionally defined MDS. Currently, only patients with isolated del(5q) are classified based on a genetic abnormality. These patients share a clinical phenotype including a more favorable prognosis and a striking response to treatment with lenalidomide [59]. However, there are likely similar phenotypic subtypes of MDS that might be defined by somatic mutations. The most evident are those patients with SF3B1 mutations who commonly have ring sideroblasts [31•, 60, 61]. In the current schema, patients with >15 % ring sideroblasts, <5 % myeloblasts, and isolated erythroid dysplasia are assigned the RARS subtype. This threshold for ring sideroblasts is arbitrarily defined and has no biologic significance. It may be better to define a subtype based on SF3B1 mutation status independent of ring sideroblast percentage [62]. Several studies suggest that this would create a more uniform subset of patients [63, 64•]. On the other end of the risk spectrum, patients with TP53 mutations may comprise another MDS subtype. Patients with TP53 mutations have fewer mutations in other genes and often carry multiple chromosomal lesions that can include del(5q), monosomy 7, or an abnormal chromosome 17 [14••, 19•, 20•, 65, 66]. Clinically, these patients are more likely to have thrombocytopenia, excess bone marrow blasts, and shorter overall survival. Mutations in other genes, or recurrent combinations of genes, may define additional clinically significant MDS subtypes in the future [15••, 16••].
Conclusion
Current evidence does not support the use of somatic mutations as presumptive evidence of MDS absent traditional diagnostic criteria [42••]. However, genetic testing at the time of initial evaluation can aid in establishing a diagnosis and can provide additional clinically relevant information. In cases that meet morphologic criteria for MDS, typical somatic mutations strongly support the diagnosis. Several of these lesions have demonstrated prognostic significance that can refine risk stratification and impact treatment decisions. As with the del(5q) chromosomal abnormality, some gene mutations may be strongly associated with clinical features and thus be used to help classify MDS subtypes in patients that meet the classical diagnostic criteria.
In cytopenic cases that are diagnostically unclear, somatic mutations must be interpreted more cautiously [8]. Genetic mutations recurrently identified in MDS patients lack the frequency and specificity required to serve as presumptive evidence of the disease, particularly due to high rate of CHIP defined by the same abnormalities in patients without a hematologic disorder. Even in this setting, somatic mutations can be useful. Finding more than one typical mutation at higher allele frequencies may raise the likelihood that the mutations and cytopenias are linked, particularly in younger patients where the rate of incidental CHIP is low. Other mutations, if present, may raise the likelihood of diagnoses other than MDS or identify patients with a congenital mutation predisposing to MDS/AML. And, the absence of mutations in a sufficiently broad gene panel may identify patients with lower risk of progression or in which an eventual diagnosis of MDS is less likely.
The key to these approaches will be to start with a careful appreciation of the clinical context. Low frequency somatic mutations in patients with mild cytopenias and only marginal suspicion for MDS should be given little significance. In fact, these patients should likely not be tested for mutations at all. Patients with more profound cytopenias in which alternative diagnoses have been carefully explored may be more likely to harbor clinical relevant mutations. These CCUS patients may merit closer observation, or if severely affected, might be treated similarly to patients with lower risk, transfusion dependent MDS. As it stands today, there is insufficient evidence regarding the long-term outcomes of patients with CCUS to merit redefining the diagnostic criteria for MDS. With this in mind, an added benefit of sequencing cytopenic patients at the time of diagnosis is that it will allow us to recognize and study CCUS patients longitudinally to determine the impact of mutations on their eventual outcome.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Liesveld JL, Lichtman MA. Myelodysplastic syndromes. In: Kaushansky K, Lichtman MA, Seligsohn U, et al., editors. Williams hematology. Eighthth ed. New York: McGraw-Hill Medical; 2010. p. 1249–76.
Font P, Loscertales J, Soto C, et al. Interobserver variance in myelodysplastic syndromes with less than 5 % bone marrow blasts: unilineage vs. multilineage dysplasia and reproducibility of the threshold of 2 % blasts. Ann Hematol. 2015;94:565–73.
Font P, Loscertales J, Benavente C, et al. Inter-observer variance with the diagnosis of myelodysplastic syndromes (MDS) following the 2008 WHO classification. Ann Hematol. 2013;92:19–24.
Bejar R. Clinical and genetic predictors of prognosis in myelodysplastic syndromes. Haematologica. 2014;99:956–64.
Greenberg PL, Stone RM, Bejar R, et al. Myelodysplastic syndromes, version 2.2015. J Natl Compr Cancer Netw: JNCCN. 2015;13:261–72.
Malcovati L, Hellstrom-Lindberg E, Bowen D, et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood. 2013;122:2943–64.
Valent P. Low blood counts: immune mediated, idiopathic, or myelodysplasia. Hematol / Edu Program Am Soc Hematol Am Soc Hematol Edu Program. 2012;2012:485–91.
Della Porta MG, Travaglino E, Boveri E, et al. Minimal morphological criteria for defining bone marrow dysplasia: a basis for clinical implementation of WHO classification of myelodysplastic syndromes. Leukemia. 2014;20:161.
DePalma L, Delgado P, Werner M. Diagnostic discrimination and cost effective assay strategy for leukocyte alkaline phosphatase. Clin Chim Acta. 1996;244:83–90.
Neumann F, Herold C, Hildebrandt B, et al. Quantitative real-time reverse-transcription polymerase chain reaction for diagnosis of BCR-ABL positive leukemias and molecular monitoring following allogeneic stem cell transplantation. Eur J Haematol. 2003;70:1–10.
Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937–51.
Pemmaraju N, Moliterno AR. From Philadelphia-negative to JAK2-positive: effect of genetic discovery on risk stratification and management. Am Soc Clin Oncol Educ Book. 2015;35:139–45.
Lindsley RC, Ebert BL. Molecular pathophysiology of myelodysplastic syndromes. Annu Rev Pathol. 2013;8:21–47. Excellent review on somatic mutations and the molecular pathology of MDS.
Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364:2496–506. This is one of three large studies to demonstrate that somatic mutations have prognostic significance independent of the IPSS.
Papaemmanuil E, Gerstung M, Malcovati L, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122:3616–27. This is one of three large studies that examined patterns of mutations and their prognostic value in a large cohort of MDS patients.
Haferlach T, Nagata Y, Grossmann V, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leuk: Off J Leuk Soc Am, Leuk Res Fund, UK. 2014;28:241–7. This is one of three large studies that examined patterns of mutations and their prognostic value in a large cohort of MDS patients.
Itzykson R, Kosmider O, Cluzeau T, et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia. 2011;25:1147–52.
Traina F, Visconte V, Elson P, et al. Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia. 2014;28:78–87.
Bejar R, Stevenson KE, Caughey B, et al. Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem-cell transplantation. J Clin Oncol: Off J Am Soc Clin Oncol. 2014;32:2691–8. This article demonstrates the predictive value of somatic mutations in MDS patients undergoing allogeneic stem cell transplant.
Bejar R, Lord A, Stevenson K, et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood. 2014;124:2705–12. This article demonstrates the predictive value of somatic mutations in MDS patients treated with hypomethylating agents.
Gondek LP, DeZern AE. I walk the line: how to tell MDS from other bone marrow failure conditions. Curr Hematol Malig Rep. 2014;9:389–99.
Kohlmann A, Bacher U, Schnittger S, et al. Perspective on how to approach molecular diagnostics in acute myeloid leukemia and myelodysplastic syndromes in the era of next-generation sequencing. Leuk Lymphoma. 2014;55:1725–34.
Valent P, Horny HP, Bennett JM, et al. Definitions and standards in the diagnosis and treatment of the myelodysplastic syndromes: Consensus statements and report from a working conference. Leuk Res. 2007;31:727–36.
Komrokji RS, Corrales-Yepez M, Al Ali N, et al. Validation of the MD Anderson prognostic risk model for patients with myelodysplastic syndrome. Cancer. 2012;118:2659–64.
Garcia-Manero G, Shan J, Faderl S, et al. A prognostic score for patients with lower risk myelodysplastic syndrome. Leukemia. 2008;22:538–43.
Bejar R, Stevenson KE, Caughey BA, et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J Clin Oncol: Off J Am Soc Clin Oncol. 2012;30:3376–82.
Carbuccia N, Murati A, Trouplin V, et al. Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia. 2009;23:2183–6.
Gelsi-Boyer V, Trouplin V, Adelaide J, et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol. 2009;145:788–800.
Kulasekararaj AG, Jiang J, Smith AE, et al. Somatic mutations identify a subgroup of aplastic anemia patients who progress to myelodysplastic syndrome. Blood. 2014;124:2698–704.
Couronne L, Bastard C, Bernard OA. TET2 and DNMT3A mutations in human T-cell lymphoma. N Engl J Med. 2012;366:95–6.
Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–9. This article describes somatic mutations in 8 splicing factor genes and their frequency in myeloid malignancies.
Damm F, Mylonas E, Cosson A, et al. Acquired initiating mutations in early hematopoietic cells of CLL patients. Cancer Discov. 2014;4:1088–101.
Wang L, Lawrence MS, Wan Y, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365:2497–506.
Busque L, Mio R, Mattioli J, et al. Nonrandom X-inactivation patterns in normal females: lyonization ratios vary with age. Blood. 1996;88:59–65.
Busque L, Paquette Y, Provost S, et al. Skewing of X-inactivation ratios in blood cells of aging women is confirmed by independent methodologies. Blood. 2009;113:3472–4.
Busque L, Patel JP, Figueroa ME, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012;44:1179–81.
Jacobs KB, Yeager M, Zhou W, et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet. 2012;44:651–8.
Laurie CC, Laurie CA, Rice K, et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet. 2012;44:642–50.
Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–98. This is one of two whole exome sequencing studies that describe age-related somatic mutations in the blood of GWAS participants.
Xie M, Lu C, Wang J, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20:1472–8.
Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–87. This is one of two whole exome sequencing studies that describe age-related somatic mutations in the blood of GWAS participants.
Steensma DP, Bejar R, Jaiswal S, et al.: Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood, 2015. Timely perspective article describing the implications of CHIP and CCUS on the use of somatic mutations for the diagnosis of MDS.
Steensma DP. Dysplasia has a differential diagnosis: distinguishing genuine myelodysplastic syndromes (MDS) from mimics, imitators, copycats and impostors. Curr Hematol Malig Rep. 2012;7:310–20. An excellent guide to the challenges associated with the diagnosis of MDS.
Valent P, Bain BJ, Bennett JM, et al. Idiopathic cytopenia of undetermined significance (ICUS) and idiopathic dysplasia of uncertain significance (IDUS), and their distinction from low risk MDS. Leuk Res. 2012;36:1–5.
Schroeder T, Ruf L, Bernhardt A, et al. Distinguishing myelodysplastic syndromes (MDS) from idiopathic cytopenia of undetermined significance (ICUS): HUMARA unravels clonality in a subgroup of patients. Ann Oncol. 2010;21:2267–71.
Hall J, Al Hafidh J, Balmert E, et al. Somatic mutations indicative of clonal hematopoiesis are present in a large fraction of cytopenic patients who lack diagnostic evidence of MDS. Blood. 2014;124:3272.
Kwok B, Reddy P, Lin K, et al. Next-generation sequencing (NGS)-based profiling of idiopathic cytopenia of undetermined significance (ICUS) identifies a subset of patients with genomic similarities to lower-risk myelodysplastic syndrome (MDS). Blood. 2014;124:166.
Tiacci E, Trifonov V, Schiavoni G, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med. 2011;364:2305–15.
Andersson EI, Rajala HL, Eldfors S, et al. Novel somatic mutations in large granular lymphocytic leukemia affecting the STAT-pathway and T-cell activation. Blood Cancer J. 2013;3:e168.
Rajala HL, Eldfors S, Kuusanmaki H, et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood. 2013;121:4541–50.
Genovese G, Jaiswal S, Ebert BL, et al. Clonal hematopoiesis and blood-cancer risk. N Engl J Med. 2015;372:1071–2.
Cancer Genome Atlas Research N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74.
Polprasert C, Schulze I, Sekeres MA, et al. Inherited and somatic defects in DDX41 in myeloid neoplasms. Cancer Cell. 2015;27:658–70.
Zhang MY, Churpek JE, Keel SB, et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet. 2015;47:180–5.
West AH, Godley LA, Churpek JE. Familial myelodysplastic syndrome/acute leukemia syndromes: a review and utility for translational investigations. Ann N Y Acad Sci. 2014;1310:111–8.
Talwalkar SS, Yin CC, Naeem RC, et al. Myelodysplastic syndromes arising in patients with germline TP53 mutation and Li-Fraumeni syndrome. Arch Pathol Lab Med. 2010;134:1010–5.
Link DC, Schuettpelz LG, Shen D, et al. Identification of a novel TP53 cancer susceptibility mutation through whole-genome sequencing of a patient with therapy-related AML. JAMA. 2011;305:1568–76.
Zatterale A, Calzone R, Renda S, et al. Identification and treatment of late onset Fanconi’s anemia. Haematologica. 1995;80:535–8.
List A, Dewald G, Bennett J, et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med. 2006;355:1456–65.
Papaemmanuil E, Cazzola M, Boultwood J, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365:1384–95.
Damm F, Thol F, Kosmider O, et al. SF3B1 mutations in myelodysplastic syndromes: clinical associations and prognostic implications. Leukemia. 2012;26:1137–40.
Juneja SK, Imbert M, Sigaux F, et al. Prevalence and distribution of ringed sideroblasts in primary myelodysplastic syndromes. J Clin Pathol. 1983;36:566–9.
Malcovati L, Papaemmanuil E, Ambaglio I, et al. Driver somatic mutations identify distinct disease entities within myeloid neoplasms with myelodysplasia. Blood. 2014;26:2014–03.
Malcovati L, Karimi M, Papaemmanuil E, et al.: SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood, 2015. This study demonstrates how somatic mutations in SF3B1 identify a molecular subtype of MDS with share clinical features.
Volkert S, Kohlmann A, Schnittger S, et al. Association of the type of 5q loss with complex karyotype, clonal evolution, TP53 mutation status, and prognosis in acute myeloid leukemia and myelodysplastic syndrome. Genes, Chromosomes Cancer. 2014;3:22151.
Sebaa A, Ades L, Baran-Marzack F, et al. Incidence of 17p deletions and TP53 mutation in myelodysplastic syndrome and acute myeloid leukemia with 5q deletion. Genes, Chromosomes Cancer. 2012;51:1086–92.
Compliance with Ethics Guidelines
Conflict of Interest
Rafael Bejar reports personal fees from Genoptix Inc, personal fees from Celgene Inc, and personal fees from Alexion Inc. In addition, Dr. Bejar has a patent MDS Mutation Signature Associated with increased disease risk licensed to Genoptix Inc and a patent MDS Mutations associated with response to hypomethylating agents pending.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is a part of the Topical Collection on Myelodysplastic Syndromes.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bejar, R. Myelodysplastic Syndromes Diagnosis: What Is the Role of Molecular Testing?. Curr Hematol Malig Rep 10, 282–291 (2015). https://doi.org/10.1007/s11899-015-0270-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11899-015-0270-5