
Abstract
Purpose of Review
This review focuses on immunologic findings, relationships among immunologic findings and associated conditions of autoimmunity and atopy, and management of immunologic disease in chromosome 22q11.2 deletion syndrome (22q11.2DS, historically known as DiGeorge syndrome).
Recent Findings
The implementation of assessment of T cell receptor excision circles (TRECs) in newborn screening has led to increased detection of 22q11.2 deletion syndrome. While not yet applied in clinical practice, cell-free DNA screening for 22q11.2DS also has the potential to improve early detection, which may benefit prompt evaluation and management. Multiple studies have further elucidated phenotypic features and potential biomarkers associated with immunologic outcomes, including the development of autoimmune disease and atopy.
Summary
The clinical presentation of 22q11.2DS is highly variable particularly with respect to immunologic manifestations. Time to recovery of immune system abnormalities is not well-defined in current literature. An understanding of the underlying causes of immunologic changes found in 22q11.2DS, and the progression and evolution of immunologic changes over the lifespan have expanded over time and with improved survival. An included case highlights the variability of presentation and potential severity of T cell lymphopenia in partial DiGeorge syndrome and demonstrates successful spontaneous immune reconstitution in partial DiGeorge syndrome despite initial severe T cell lymphopenia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chromosome 22q11.2 deletion syndrome (22q11.2DS), historically known as DiGeorge syndrome, was described by Angelo DiGeorge in 1968 in a series of patients with absent thymic tissue and parathyroid glands [1]. 22q11.2DS is the most common microdeletion syndrome in humans, occurring in approximately 1:4000 live births. Abnormal development of the third and fourth pharyngeal pouches leads to the hallmark features of 22q11.2DS, including cardiac anomalies, hypoparathyroidism, and thymic hypoplasia or aplasia. Other phenotypic features can include palatal defects, feeding and swallowing abnormalities, neuropsychiatric disorders, renal anomalies, and others. There is wide variability of phenotypic features. Up to 75–80% of those with 22q11.2DS exhibit derangements of the immune system, which can lead to increased susceptibility to infections, autoimmune disease, and atopy. 22q11.2DS can be grouped into two general categories, complete DiGeorge syndrome and partial DiGeorge syndrome. Individuals with complete absence of the thymus are considered to have complete DiGeorge syndrome. This phenomenon occurs in less than 1.5% of those born with 22q11.2 deletion and may be treated with thymic transplantation [2]. In contrast, those with partial DiGeorge syndrome have thymic tissue present, sometimes in the form of microscopic rests. Partial DiGeorge syndrome has wide variability in immune deficits. Recently updated terminology refers to these categories as 22q11.2del with T cell lymphopenia (mild, significant) and 22q11.2del with congenital athymia, with the additional categories of 22q11.2del without T cell lymphopenia and 22q11.2del with congenital athymia and autologous immune dysregulation (Omenn-like syndrome, previously atypical complete DiGeorge) [3••]. This literature review highlights the immunologic features, diagnosis, and management of 22q11.2DS. A case report highlights successful spontaneous immune reconstitution in partial DiGeorge syndrome despite initial severe T cell lymphopenia.
Case Report
A 3-year-old female with a history of severe T cell lymphopenia related to chromosome 22q11.2 deletion syndrome was born at 39-week gestation to a G2P2 mother via repeat Cesarean section at a regional hospital following an uncomplicated pregnancy. She was intubated shortly after birth due to hypoxia and transferred to a tertiary medical center pediatric intensive care unit (PICU); an echocardiogram revealed tetralogy of Fallot with a partially absent and dysplastic pulmonic valve, for which she underwent surgical repair at 2 weeks of age. The absence of an identifiable thymus was noted during this procedure. Her postoperative course was complicated by pulmonary hemorrhage, chylothorax, severe bronchomalacia requiring tracheostomy and intermittent mechanical ventilation, feeding difficulty temporarily requiring G-tube placement, and hypocalcemia.
When immunologically evaluated at 1 month of age, she was noted to have abnormally low T cell receptor excision circle levels (TREC) on newborn metabolic screen, collected on day 3 of life, and later repeated for confirmation (see Table 1). Fluorescence in situ hybridization (FISH), collected on day 1 of life, identified a loss of TUPLE1 at chromosome 22q11.2. Initial immunologic studies were delayed due to frequent blood transfusions during management of cardiac complications. Flow cytometry obtained at 2 and 3 months of age demonstrated profound T cell lymphopenia (Table 1). T cell enumeration further demonstrated profoundly decreased total CD3 T cell, CD4 T cell, and CD8 T cell counts for age, but no overt anomaly in T cell subset distribution in either the CD4 or CD8 T cell compartments. Despite initial concerns for complete DiGeorge due to the severity of T cell lymphopenia, the normal ratio of naïve to memory T cells, the production of naïve T cells (CD45RA+), and the presence of a normal percentage of CD4+ recent thymic emigrants revealed this to be a severe case of partial DiGeorge syndrome (Table 1). Mitogen stimulation testing showed normal proliferative responses to pokeweed mitogen (PWM) and phytohemagglutinin (PHA). Throughout her initial hospitalization for the first 7.5 months of life, the total lymphocyte count ranged from 600 to 2600. Her lymphocyte subsets measured by flow cytometry demonstrated markedly low T, B, and NK cells (see Fig. 1). Only at 28 months of age were her immunoglobulin levels, CD4 lymphocyte, and NK cell counts all normal for age. The total CD3 count remains low for age but continues to increase [4].

Lymphocyte subset absolute counts
In addition to T cell lymphopenia, she demonstrated evidence of humoral immunodeficiency. Immunoglobulin levels obtained at 3 months were markedly low (Table 1). The initial severe hypogammaglobulinemia was also partially attributable to gastrointestinal protein loss via chronic diarrhea, as her albumin was markedly decreased, consistent with protein-losing enteropathy. Her total IgG level improved but did not normalize despite eventual resolution of the diarrhea and other signs of enteropathy, suggesting that she also had underlying primary hypogammaglobulinemia. She received immunoglobulin replacement through age 19 months. Her IgG level continued to spontaneously increase after discontinuation of replacement therapy.
During the initial hospitalization, she was administered prophylaxis for Mycobacterium avium complex, Pneumocystis pneumonia, and invasive candidiasis. Mycobacterium prophylaxis was discontinued prior to discharge due to clinical improvement. Other prophylaxis was continued until after she achieved immune reconstitution. She has now received both inactivated and live vaccines, without complication. Her specific antibody levels measured 10 months after her last dose of IVIG were protective for tetanus, diphtheria, and 18 of 22 measured Streptococcus pneumoniae serotypes. Her initial hospital course was complicated by intermittent febrile episodes; workup for underlying infection was unrevealing except for separate instances of bacterial tracheitis and bacterial urinary tract infection which were appropriately treated. Between her initial hospital discharge and first birthday, she was hospitalized multiple times for suspected upper and lower respiratory tract infections. Bacterial tracheitis was identified on one occasion, but no causative organisms were identified during the other hospital admissions. To date, she has continued to have viral respiratory infections at a normal frequency for age, with intermittent brief hospital observation. She was infected with SARS-CoV-2 at ages 25 months and 42 months. She developed a fever without other symptoms with the initial SARS-CoV-2 infection and was asymptomatic with the second infection. She has had no opportunistic infections. Written parental consent and a waiver from the institutional IRB were obtained regarding publishing this data.
Genetic Basis of 22q11.2DS
A hemizygous microdeletion on chromosome 22 leads to the classic clinical phenotype in approximately 35–90% of 22q11.2DS cases [5–7]. The deletion occurs due to erroneous chromosomal rearrangement during meiosis involving low copy repeats (LCRs) distributed along a segment of chromosome 22q11.2. The most common 3 Mb deletion spans LCR22A-D, accounting for 84% of cases [8]. Smaller, sometimes nested deletions have also been identified. Of over 30 involved genes, the T-Box Transcription Factor 1 (TBX1) gene is of particular interest in relation to immune deficiency in 22q11.2DS. TBX1 is involved in regulation of expression of transcription factors and indirectly affects neural crest migration in the process of thymus development [9].
In a minority of cases, no chromosome 22 deletion is identified. Multiple other etiologies of DGS have been described. Teratogen exposures, including ethanol, retinoic acid, and maternal diabetes, have led to phenotypic features of DGS [10–14]. Although the causative mechanisms from the noted teratogens have not been fully elucidated, alterations of the retinoic acid pathway in the case of either exogenous retinoic acid or pregestational diabetes may be involved [15, 16•]. The retinoic acid signaling pathway is important in thymus development and is also involved in regulation of TBX1 expression. Epigenetic factors may also contribute to the DGS phenotype in maternal diabetes [13, 16•].
Other chromosomal anomalies with phenotypic features overlapping with DGS have been identified [17–22]. CHARGE syndrome, caused by CHD7 gene mutations, can similarly present with facial and palatal abnormalities, cardiac malformations, developmental delay, and rarely immunologic deficits [23, 24]. Genome-wide array technologies have been particularly useful in identifying other potentially causative genetic syndromes.
Cellular Immunity in 22q11.2DS
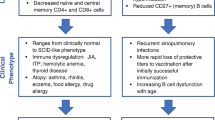
In most cases of partial DiGeorge syndrome, T cell subset counts are mild to moderately decreased compared to age-matched normal populations. T cell lymphopenia may persist beyond infancy, although the magnitude of the difference between 22q11.2DS patients and controls decreases over time [25]. Typically, lymphocyte subset counts are closer to that of controls by 1 year of age [26]. In normal children, T cell counts decline within the first 1 to 2 years of life due to thymic involution. 22q11.2DS patients demonstrate a slower rate of decline in T cell counts compared to healthy controls [25, 27, 28•]. Thymic function in 22q11.2DS patients is comparable to healthy controls, with a similar ability to generate new lymphocytes per gram of tissue, which suggests lymphopenia in 22q11.2DS is related to the lack of thymic tissue rather than abnormal function, and favors the concept that the less pronounced decline in T cell numbers is a compensatory mechanism serving to sustain this cell population [29]. Additionally, increased secretion of IL-7, which is involved in growth and differentiation of T cells, may lead to increased thymic output and increased peripheral proliferation of T cells in 22q11.2DS patients [30]. Another proposed mechanism to explain the apparent improvement or normalization of T cell counts is homeostatic proliferation to compensate for poor thymic output. Evidence in support of homeostatic proliferation includes accelerated conversion of naïve to memory T cells, shortened telomeres, and increased oligoclonality of the T cell repertoire [31]. Normal or near normal peripheral blood T cell counts in 22q11.2DS patients may belie an impaired ability to respond to antigens due to this restricted T cell repertoire. T cell subset maturation may also be affected in 22q11.2DS. There are smaller proportions of naïve regulatory T cells and larger proportions of activated or memory regulatory T cells, particularly in 22q11.2DS patients with abnormal TREC counts [30, 32••]. T cell proliferative responses to mitogens are typically normal [25, 26].
A number of studies have attempted to find genetic and clinical associations with T cell lymphopenia in 22q11.2DS. It is not yet known if very low T cell numbers in infancy predict worse immunologic defects in adulthood [33]. Chromosomal deletions involving the TBX1 gene (LCR22A-B, LCR22A-C, and LCR22A-D) are associated with decreased CD3 and CD4 counts, although the size of the chromosomal deletion has not otherwise been found to associate with clinical phenotype [3••, 34]. Phenotypic features including hypocalcemia and palatal abnormalities are not associated with the frequency or severity of immunocompromise in 22q11.2DS patients [35]. The presence of cardiac anomalies has not been associated with severity of T cell lymphopenia [35, 36]. Additionally, low lymphocyte counts in 22q11.2DS are not attributable to the stress of cardiac anomalies [26]. In contrast, Gul et al. report occurrence of cardiac defects correlates with low TREC quartiles, which may reflect the relationship of TBX1 expression to development of both the thymus and the heart [37].
Humoral Immunity in 22q11.2DS
While 22q11.2DS has primarily been associated with cellular immune defects, there is substantial evidence of humoral involvement. Humoral deficits found in 22q11.2DS include hypogammaglobulinemia, poor specific antibody responses, and altered B cell maturation [28•, 38–43]. Up to 6% of 22q11.2DS have low IgG levels (defined as < 500 mg/dL) after 3 years of age [38]. Selective IgA deficiency is present in up to 13% of 22q11.2DS [44]. Selective IgM deficiency is less common but has also been reported in association with 22q11.2DS [45, 46]. In one cohort, low IgM was the most common humoral defect [28•]. Switched and unswitched memory B cells are diminished in adult 22q11.2DS patients [28•, 32••, 39]. The underlying cause of abnormal B cell maturation is suspected to be impaired T cell help and not due to B cell proliferation pressure or exhaustion [39]. Kappa-deleting recombination excision circles (KRECs) are circular DNA segments generated in B cells during their maturation in the bone marrow [47]. KREC levels in 22q11.2DS patients are not significantly different from healthy controls, suggesting normal bone marrow output of B cells [48, 49]. However, TREC and KREC levels are correlated in 22q11.2DS patients but not healthy controls, which underscores the importance of T cells in the activation and regulation of B cells.
Vaccine responses to polysaccharide antigens may be inadequate [40, 50, 51]. In a cohort of 53 subjects with available results, 39.6% of 22q11.2DS subjects had low specific antibody levels for Streptococcus pneumoniae. Vaccine responses to protein antigens, on the other hand, are adequate in most cases [50, 51].
Outcomes
A majority of 22q11.2DS patients may experience recurrent infections, most commonly viral respiratory infections, otitis, and sinopulmonary infections [41]. Most do not experience opportunistic infections even in the setting of persistent T lymphopenia [26, 32••]. The underlying immune deficits in 22q11.2DS are a risk factor for recurrent infection. However, 22q11.2DS patients with recurrent infections do not always present with identifiable immune defects, highlighting the importance of non-immunologic features of 22q11.2DS that increase risk of infections [25]. Anatomical palatal abnormalities are associated with respiratory tract infections and recurrent otitis media. Other comorbidities, including bronchomalacia, gastroesophageal reflux, asthma, and rhinitis, may also predispose 22q11.2DS patients to respiratory infections [28•].
As management and surgical techniques for congenital heart disease have improved, the survival rate in infancy has improved. Campbell et al. reported a 4% overall mortality rate, with most deaths related to complex congenital heart disease [8]. The median age at death was 5 months. Studies of those with 22q11.2DS surviving to adulthood are limited but will be of increasing importance as the 22q11.2DS population ages. One cohort study of adults with 22q11.2DS found an increased risk of sudden death, with an average age of death of 41.5 years [52].
Autoimmune Disease
Multiple autoimmune conditions have been associated with 22q11.2DS, including autoimmune cytopenias, juvenile idiopathic arthritis, and autoimmune thyroid disease [41, 53]. Macrothrombocytopenia is a common finding in 22q11.2DS due to the hemizygous deletion of the GPIbβ gene on chromosome 22, which encodes a subunit of the platelet GPIb-IX-V receptor involved in platelet adhesion [54]. Baseline thrombocytopenia may potentially confound evaluation of immune thrombocytopenia.
In general, low absolute CD3+ T cell counts have been associated with higher odds of autoimmunity and/or asthma in 22q11.2DS patients [53]. Low naïve T cell counts have been proposed as a potential biomarker for autoimmunity in 22q11.2DS patients [28•, 53]. Odds ratios of immune cytopenias are estimated to be 14.0 for CD4+ naïve T cells of ≤ 30% and 44.0 for switched memory B cells of ≤ 2% [42]. Persistent hypocalcemia was a clinical feature that was more commonly associated with autoimmune cytopenia [42]. A recent study has suggested that mechanisms of autoimmune cytopenias and solid-organ autoimmunity may be distinct, as low naïve T cell counts were associated with idiopathic thrombocytopenia purpura (ITP), while autoimmune thyroid disease was associated with low CD4 T cells but not specifically CD4/CD45RA+ T cells [55]. A decrease in regulatory T cells has also been proposed as a contributor to the development of autoimmune disease [56].
Atopy
Atopic disease is commonly seen in 22q11.2DS patients. Cytokine studies have shown initial Th1 skewing of cytokine production in young children with 22q11.2DS, which then evolves towards a Th2 phenotype in adults with 22q11.2DS [43]. There have been conflicting results regarding potential biomarkers for increased risk of atopy. Low CD3 counts in infancy were associated with 2.56-fold increased risk of atopy compared to a normal CD3 count [33]. However, there is no association with low CD4 or CD4/CD45RA+ T cell counts [55]. Giardino et al. reported an association between allergy and persistently low IgM levels [28•].
Diagnosis of 22q11.2DS
Due to the large phenotypic variability in 22q11.2DS, prompt diagnosis requires a high degree of clinical suspicion and careful examination of potentially subtle features. The median age at diagnosis was 360 days in one cohort of 22q11.2DS patients, although there have been instances of diagnosis in adulthood [8]. The presence of congenital heart disease is the most common instigating factor for diagnostic evaluation for 22q11.2DS and thus is associated with earlier age at diagnosis. Early diagnosis of 22q11.2DS is favorable for multiple reasons, including early surveillance and treatment of associated conditions and appropriate genetic counseling for parents. While the majority of cases occur due to de novo mutations, approximately 7% of 22q11.2DS have an inherited deletion from a parent [8]. With regard to immunologic status, early recognition may help with early implementation of antibiotic prophylaxis and immunoglobulin replacement, if appropriate, and precautions for infection avoidance, transfusion of blood products, and vaccination with live agents.
Fluorescence in situ hybridization (FISH) remains a common method of diagnosis for 22q11.2DS with a detection rate of approximately 95%. FISH can identify deletions in the proximal part of the critical region, including the typical LCR22A-D deletion. However, FISH is unable to recognize some atypical or nested deletions, such as LCR22B-D or LCR22C-D. Other approaches for diagnosis of 22q11.2DS include multiplex ligation-dependent probe amplification (MLPA) and array-based comparative genomic hybridization analysis [57]. MLPA has the advantages of a potentially higher detection rate and results suggesting the extent of the deletion [58].
T cell receptor excision circle (TREC)-based newborn screening has now been implemented into the standard newborn metabolic screen in all 50 states. While the primary purpose of this screening is early identification of severe combined immunodeficiency (SCID), the test does not differentiate the underlying cause of low TREC counts and thus has also identified cases of 22q11.2DS with T lymphopenia. TREC screening does lead to diagnosis of additional cases of 22q11.2DS that would not have otherwise been identified, absent other noted anomalies [59]. Additionally, the time to diagnosis was substantially shortened compared to patients who did not have abnormal TREC screening (25.1 days vs 3.8 years). However, only an estimated 3–15% of infants with 22q11.2DS would have abnormal newborn screening based on current reference range cutoffs. Notably, 22q11.2DS patients with decreased TREC counts demonstrate long-term impairment of thymic output and were significantly more prone to viral infections, as may be expected in the setting of more severe T cell lymphopenia [32••, 49]. The association of lower TREC levels with recurrent infections continues regardless of age [48]. TREC counts are not affected by cardiac surgery, which may potentially involve thymectomy, so the results remain valid in patients undergoing treatment of associated cardiac disease [48].
Diagnostic studies are performed at the discretion of the patient’s medical team, as there is currently no widespread screening available specifically directed at identifying 22q11.2DS. At present, direct newborn screening for 22q11.2DS by FISH, MLPA, or chromosomal microarray is not cost efficient. Liao et al. have proposed that reflex testing could be performed in the case of abnormal TREC screening using the same blood sample [57]. This method may hasten the specific diagnosis of 22q11.2DS in the subset of cases with abnormal TREC screening but would miss the majority of 22q11.2DS patients with normal TREC counts. Efforts to develop widespread newborn screening specifically targeting 22q11.2DS have not yet come to fruition.
Multiple recent studies have investigated the possibility of prenatal screening for 22q11.2DS via cell-free DNA testing, as is performed for trisomy 13, 18, and 21. Trials of cell-free DNA testing targeted for fetal 22q11.2 deletion have demonstrated specificity > 99% [60, 61]. A large multicenter prospective observational study of single-nucleotide polymorphism (SNP)-based prenatal cell-free DNA testing found a sensitivity of 75%, specificity of 99.84%, positive predictive value of 23.7%, and negative predictive value of 99.98% [62]. The PPV and false positive rate are comparable to cell-free DNA screening currently used in clinical practice for some of the aneuploidies. Cell-free DNA testing for 22q11.2DS is currently being performed in a research capacity and has not yet been implemented into general clinical practice but may be a viable method for screening in the future.
Management
Approaches to management of 22q11.2DS have varied, as formal guidelines regarding management have only recently been published [3••]. Previous t recommendations include the following immunologic evaluation at diagnosis: CBC with differential, lymphocyte phenotyping, quantitative immunoglobulin levels, lymphocyte proliferation testing (if available, and T cell count is low), and post-immunization or Hib antibodies [63••, 64]. Current guidelines emphasize obtaining lymphocyte phenotyping, including naïve and memory T cell subsets at diagnosis [3••]. T cell phenotyping, in particular assessment of naïve CD45RA+ T cells, is useful in distinguishing complete DiGeorge syndrome, which should then prompt an urgent referral for thymus transplant evaluation [65]. Recommendations for subsequent evaluation include lymphocyte phenotyping, quantitative immunoglobulin levels and tetanus IgG (at least 3 weeks after the third DTaP), and prior to 12 months of age (assuming initial diagnosis is made early in life) [3••]. At age 4–5 years (following the 4–5-year booster series), it is recommended to obtain quantitative immunoglobulin levels and tetanus IgG and consider lymphocyte phenotyping and pneumococcal serotypes. Lymphocyte phenotyping may not be necessary if laboratory assessment in the first year of life meets criteria for safe live vaccine administration and the patient’s infectious history remains unremarkable. At age 10, it is recommended to obtain immunoglobulin levels and tetanus IgG and consider pneumococcal serotypes if humoral assessments are abnormal and/or infectious history is significant. Every 5–10 years thereafter, repeat assessment of immunoglobulin levels and consideration of tetanus IgG and pneumococcal serotypes are advised. In the event of recurrent or severe infections or evidence of significant immune deficiency, additional or more frequent follow-up may be needed [3••].
Special attention should be paid to immunization of 22q11.2DS patients. 22q11.2DS patients should receive routine vaccination with non-live vaccines. Two doses of the 23-valent pneumococcal polysaccharide vaccine (PPSV23) are recommended for children with T lymphocyte deficiencies, with the first administered at least 8 weeks following any PCV-13 vaccination and the second administered at least 5 years after the first PPSV23. For age 18 and over, PPSV23 is not indicated if the PCV-20 has been administered [3••]. 22q11.2DS patients are known to have potentially poor responses to vaccines, even in cases with normal lymphocyte counts and immunoglobulin levels. One proposed recommendation is to obtain post-vaccination serologies to document protective responses and identify patients for whom revaccination should be considered [51]. Live virus vaccination in 22q11.2DS patients with mild-to-moderate immunosuppression has been well-tolerated [66]. Habel et al. recommended administering MMR even with CD4 T cell lymphopenia, but not administering varicella vaccine below a CD4 count of 200 cells/mL [63••]. Current guidelines recommend administration of live vaccines if the following criteria are met: CD4 ≥ cells/mm3 (absolute), CD8 ≥ 200 cells/mm3 (absolute), and tetanus IgG protective (3 + weeks after dose 3) or hepatitis B IgG surface antibody protective, if tetanus IgG assay is not available [3••]. A greater percentage of CD45RA+CD3+/4+ to CD45RO+CD3+/4+ cells at earliest assessment, normal TREC assay results on newborn screening, or flow cytometry confirming recent thymic emigrants (CD31) may also be used to support the decision to administer live virus vaccines [3••].
Various in-depth immunologic evaluations that may help to determine risk of morbidity and mortality have been suggested. Longitudinal monitoring of naïve T cells or TRECs could be helpful to determine patients at high risk of non-cardiac death [67]. The prognostic value of low TRECs in 22q11.2DS infants without athymia remains unclear, but T lymphopenia is associated with increased risk of autoimmune disease. Thus, immunophenotyping may potentially be helpful in risk stratification regarding the development of autoimmune cytopenias [32••, 42].
Few specific treatments are available for 22q11.2DS. In the case of complete DiGeorge, thymus transplant results in development of host-derived naïve T cells with a normal T cell receptor repertoire [3••]. Hematopoietic stem cell transplant has also been performed but is not preferred due to poorer outcomes. In patients with low IgG levels, immunoglobulin replacement can be considered. Immunoglobulin replacement therapy may currently be underutilized, as one study found a significant gap between 22q11.2DS patients demonstrating a humoral deficiency and those being treated for it [68]. Depending upon the severity of immune defects, antibiotic prophylaxis may be utilized. Mycobacterium avium prophylaxis should be considered in cases of congenital athymia [3••]. Pneumocystis jirovecii pneumonia prophylaxis can be considered in the setting of low CD4 counts, typically < 200 cells/mm3. For individuals presenting with recurrent upper respiratory tract infection, antibiotic prophylaxis during peak viral seasons may be beneficial in reducing subsequent bacterial infections and supporting school attendance [63••].
Conclusion
While there is wide phenotypic variability in the presentation of DiGeorge syndrome, immune deficiency and recurrent infections are a common feature. In the case presented, a patient identified as having 22q11.2DS shortly after birth was found to have profoundly low T, B, and NK cell counts. T cell lymphopenia in partial DiGeorge syndrome typically improves over time due to a blunted rate of T cell decline compared to heathy controls and through homeostatic expansion of T cells, although the age at which immune reconstitution is typically achieved is not well-defined. This case is noteworthy as the patient did achieve eventual immune reconstitution, but not until approximately 28 months of age. Immunologists play an important role in the evaluation and management of these patients due to the frequency of immune defects and potential need for treatment, such as immunoglobulin replacement or antibiotic prophylaxis. Formal guidelines are still needed for management of immunologic disease in DiGeorge syndrome. Efforts are currently ongoing to improve early diagnosis. While there are still advances to be made, significant progress has shaped our understanding of the genetics and underlying mechanisms leading to immunodeficiency, autoimmunity, and atopy in 22q11.2 DS/DiGeorge syndrome.
Data Availability
The data are PHI, only accessible via EMR in our institution.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
DiGeorge AM, editor. Congenital absence of the thymus and its immunologic consequences: concurrence with congenital hypothyroidism. Birth defects. Vol IV(1). White Plains, New York: March of Dimes-Birth Defects Foundation, 1968.
Davies EG. Immunodeficiency in DiGeorge syndrome and options for treating cases with complete athymia. Front Immunol. 2013;4:322. https://doi.org/10.3389/fimmu.2013.00322.
•• Mustillo PJ, Sullivan KE, Chinn IK, et al. Clinical practice guidelines for the immunological management of chromosome 22q11.2 deletion syndrome and other defects in thymic development. J Clin Immunol. 2023;43(2):247–70. https://doi.org/10.1007/s10875-022-01418-y. This recent article provides guidelines for initial and longitudinal assessment of immunologic status in 22q11.2DS. The article also reviews the common immunologic findings in 22q11.2DS and the genetic and non-genetic causes of DGS.
Shearer WT, Rosenblatt HM, Gelman RS, et al. Lymphocyte subsets in healthy children from birth through 18 years of age: the Pediatric AIDS Clinical Trials Group P1009 study. J Allergy Clin Immunol. 2003;112(5):973–80. https://doi.org/10.1016/j.jaci.2003.07.003.
Driscoll DA, Salvin J, Sellinger B, et al. Prevalence of 22q11 microdeletions in DiGeorge and velocardiofacial syndromes: implications for genetic counselling and prenatal diagnosis. J Med Genet. 1993;30(10):813–7. https://doi.org/10.1136/jmg.30.10.813.
Katzman PJ, Smoot LB, Cox GF. Cardiac registry screening for DiGeorge critical region deletion using loss of heterozygosity analysis. Pediatr Dev Pathol. 2006;9:266–79.
Rope AF, Cragun DL, Saal HM, Hopkin RJ. DiGeorge anomaly in the absence of chromosome 22q11.2 deletion. J Pediatr. 2009;155(4):560–5. https://doi.org/10.1016/j.jpeds.2009.04.010.
Campbell IM, Sheppard SE, Crowley TB, et al. What is new with 22q? An update from the 22q and You Center at the Children’s Hospital of Philadelphia. Am J Med Genet A. 2018;176(10):2058–69. https://doi.org/10.1002/ajmg.a.40637.
Calmont A, Ivins S, Van Bueren KL, et al. Tbx1 controls cardiac neural crest cell migration during arch artery development by regulating Gbx2 expression in the pharyngeal ectoderm. Development. 2009;136(18):3173–83. https://doi.org/10.1242/dev.028902.
Sulik KK, Johnston MC, Daft PA, Russell WE, Dehart DB. Fetal alcohol syndrome and DiGeorge anomaly: critical ethanol exposure periods for craniofacial malformations as illustrated in an animal model. Am J Med Genet Suppl. 1986;2:97–112. https://doi.org/10.1002/ajmg.1320250614.
Ammann AJ, Wara DW, Cowan MJ, Barrett DJ, Stiehm ER. The DiGeorge syndrome and the fetal alcohol syndrome. Am J Dis Child. 1982;136(10):906–8.
Coberly S, Lammer E, Alashari M. Retinoic acid embryopathy: case report and review of literature. Pediatr Pathol Lab Med. 1996;16(5):823–36.
Cirillo E, Giardino G, Gallo V, et al. DiGeorge-like syndrome in a child with a 3p12.3 deletion involving MIR4273 gene born to a mother with gestational diabetes mellitus. Am J Med Genet A. 2017;173(7):1913–8. https://doi.org/10.1002/ajmg.a.38242.
Wilson TA, Blethen SL, Vallone A, et al. DiGeorge anomaly with renal agenesis in infants of mothers with diabetes. Am J Med Genet. 1993;47(7):1078–82. https://doi.org/10.1002/ajmg.1320470729.
Roberts C, Ivins SM, James CT, Scambler PJ. Retinoic acid down-regulates Tbx1 expression in vivo and in vitro. Dev Dyn. 2005;232(4):928–38. https://doi.org/10.1002/dvdy.20268.
• Du Q, de la Morena MT, van Oers NSC. The genetics and epigenetics of 22q11.2 deletion syndrome. Front Genet. 2020;10:1365. Published 2020 Feb 6. https://doi.org/10.3389/fgene.2019.01365. This article reviews the genetic origins of 22q11.2DS and the pathogenesis of disease, which helps to explain the variability in phenotype.
Greenberg F, Courtney KB, Wessels RA, et al. Prenatal diagnosis of deletion 17p13 associated with DiGeorge anomaly. Am J Med Genet. 1988;31(1):1–4. https://doi.org/10.1002/ajmg.1320310102.
Cirillo E, Prencipe MR, Giardino G, et al. Clinical phenotype, immunological abnormalities, and genomic findings in patients with DiGeorge spectrum phenotype without 22q11.2 deletion. J Allergy Clin Immunol Pract. 2020;8(9):3112–20. https://doi.org/10.1016/j.jaip.2020.06.051.
Fernández L, Lapunzina P, Pajares IL, et al. Unrelated chromosomal anomalies found in patients with suspected 22q11.2 deletion. Am J Med Genet A. 2008;146A(9):1134–41. https://doi.org/10.1002/ajmg.a.32256.
Gottlieb S, Driscoll DA, Punnett HH, Sellinger B, Emanuel BS, Budarf ML. Characterization of 10p deletions suggests two nonoverlapping regions contribute to the DiGeorge syndrome phenotype. Am J Hum Genet. 1998;62(2):495–8. https://doi.org/10.1086/301718.
Koczkowska M, Wierzba J, Śmigiel R, et al. Genomic findings in patients with clinical suspicion of 22q11.2 deletion syndrome. J Appl Genet. 2017;58(1):93–8. https://doi.org/10.1007/s13353-016-0366-1.
Snijders Blok C, Corsten-Janssen N, FitzPatrick DR, et al. Definition of 5q11.2 microdeletion syndrome reveals overlap with CHARGE syndrome and 22q11 deletion syndrome phenotypes. Am J Med Genet A. 2014;164A(11):2843–8. https://doi.org/10.1002/ajmg.a.36680.
Jyonouchi S, McDonald-McGinn DM, Bale S, Zackai EH, Sullivan KE. CHARGE (coloboma, heart defect, atresia choanae, retarded growth and development, genital hypoplasia, ear anomalies/deafness) syndrome and chromosome 22q11.2 deletion syndrome: a comparison of immunologic and nonimmunologic phenotypic features. Pediatrics. 2009;123(5):e871–7. https://doi.org/10.1542/peds.2008-3400.
Corsten-Janssen N, Saitta SC, Hoefsloot LH, et al. More clinical overlap between 22q11.2 deletion syndrome and CHARGE syndrome than often anticipated. Mol Syndromol. 2013;4(5):235–45. https://doi.org/10.1159/000351127.
Jawad AF, McDonald-Mcginn DM, Zackai E, Sullivan KE. Immunologic features of chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). J Pediatr. 2001;139(5):715–23. https://doi.org/10.1067/mpd.2001.118534.
Sullivan KE, McDonald-McGinn D, Driscoll DA, Emanuel BS, Zackai EH, Jawad AF. Longitudinal analysis of lymphocyte function and numbers in the first year of life in chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Clin Diagn Lab Immunol. 1999;6(6):906–11. https://doi.org/10.1128/CDLI.6.6.906-911.1999.
Chinen J, Rosenblatt HM, Smith EO, Shearer WT, Noroski LM. Long-term assessment of T-cell populations in DiGeorge syndrome. J Allergy Clin Immunol. 2003;111(3):573–9. https://doi.org/10.1067/mai.2003.165.
• Giardino G, Radwan N, Koletsi P, et al. Clinical and immunological features in a cohort of patients with partial DiGeorge syndrome followed at a single center. Blood. 2019;133(24):2586–96. https://doi.org/10.1182/blood.2018885244. This recent article provides clinical and immunological data from a large clinical cohort of patients that helps to identify risk factors for infections, atopy, and autoimmunity.
Ferrando-Martínez S, Lorente R, Gurbindo D, et al. Low thymic output, peripheral homeostasis deregulation, and hastened regulatory T cells differentiation in children with 22q11.2 deletion syndrome. J Pediatr. 2014;164(4):882–9. https://doi.org/10.1016/j.jpeds.2013.12.013.
Tantibhaedhyangkul U, Davis CM, Noroski LM, Hanson IC, Shearer WT, Chinen J. Role of IL-7 in the regulation of T-cell homeostasis in partial DiGeorge syndrome. J Allergy Clin Immunol. 2009;123(4):960-2.e2. https://doi.org/10.1016/j.jaci.2009.01.002.
Piliero LM, Sanford AN, McDonald-McGinn DM, Zackai EH, Sullivan KE. T-cell homeostasis in humans with thymic hypoplasia due to chromosome 22q11.2 deletion syndrome. Blood. 2004;103(3):1020–5. https://doi.org/10.1182/blood-2003-08-2824.
•• Framme JL, Lundqvist C, Lundell AC, et al. Long-term follow-up of newborns with 22q11 deletion syndrome and low TRECs. J Clin Immunol. 2022;42(3):618–33. https://doi.org/10.1007/s10875-021-01201-5. This article details persistent immunologic aberrations in a subset of patients with 22q11.2DS, which emphasizes the role of expert immunologic evaluation and the importance of continued monitoring.
Morsheimer M, Brown Whitehorn TF, Heimall J, Sullivan KE. The immune deficiency of chromosome 22q11.2 deletion syndrome. Am J Med Genet A. 2017;173(9):2366–72. https://doi.org/10.1002/ajmg.a.38319.
Crowley B, Ruffner M, McDonald McGinn DM, Sullivan KE. Variable immune deficiency related to deletion size in chromosome 22q11.2 deletion syndrome. Am J Med Genet A. 2018;176(10):2082–6. https://doi.org/10.1002/ajmg.a.38597.
Sullivan KE, Jawad AF, Randall P, et al. Lack of correlation between impaired T cell production, immunodeficiency, and other phenotypic features in chromosome 22q11.2 deletion syndromes. Clin Immunol Immunopathol. 1998;86(2):141–6. https://doi.org/10.1006/clin.1997.4463.
Sullivan KE, Crowley TB, Maurer K, et al. T-cell lymphopenia in 22q11.2 deletion syndrome: Relationship to cardiac disease. J Allergy Clin Immunol Pract. 2018;6(2):690–1. https://doi.org/10.1016/j.jaip.2017.08.028.
Gul KA, Øverland T, Osnes L, et al. Neonatal levels of T-cell receptor excision circles (TREC) in patients with 22q11.2 deletion syndrome and later disease features. J Clin Immunol. 2015;35(4):408–15. https://doi.org/10.1007/s10875-015-0153-5.
Patel K, Akhter J, Kobrynski L, et al. Immunoglobulin deficiencies: the B-lymphocyte side of DiGeorge syndrome. J Pediatr. 2012;161(5):950–3. https://doi.org/10.1016/j.jpeds.2012.06.018.
Derfalvi B, Maurer K, McDonald McGinn DM, et al. B cell development in chromosome 22q11.2 deletion syndrome. Clin Immunol. 2016;163:1–9. https://doi.org/10.1016/j.clim.2015.12.004.
Finocchi A, Di Cesare S, Romiti ML, et al. Humoral immune responses and CD27+ B cells in children with DiGeorge syndrome (22q11.2 deletion syndrome). Pediatr Allergy Immunol. 2006;17(5):382–8. https://doi.org/10.1111/j.1399-3038.2006.00409.x.
Gennery AR, Barge D, O'Sullivan JJ, Flood TJ, Abinun M, Cant AJ. Antibody deficiency and autoimmunity in 22q11.2 deletion syndrome. Arch Dis Child. 2002;86(6):422–425. https://doi.org/10.1136/adc.86.6.422.
Montin D, Marolda A, Licciardi F, et al. Immunophenotype anomalies predict the development of autoimmune cytopenia in 22q11.2 deletion syndrome. J Allergy Clin Immunol Pract. 2019;7(7):2369–76. https://doi.org/10.1016/j.jaip.2019.03.014.
Zemble R, Luning Prak E, McDonald K, McDonald-McGinn D, Zackai E, Sullivan K. Secondary immunologic consequences in chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Clin Immunol. 2010;136(3):409–18. https://doi.org/10.1016/j.clim.2010.04.011.
Smith CA, Driscoll DA, Emanuel BS, McDonald-McGinn DM, Zackai EH, Sullivan KE. Increased prevalence of immunoglobulin A deficiency in patients with the chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Clin Diagn Lab Immunol. 1998;5(3):415–7. https://doi.org/10.1128/CDLI.5.3.415-417.1998.
Al-Herz W, McGeady SJ, Gripp KW. 22q11.2 deletion syndrome and selective IgM deficiency: an association of a common chromosomal abnormality with a rare immunodeficiency. Am J Med Genet A. 2004;127A(1):99–100. https://doi.org/10.1002/ajmg.a.20639.
Kung SJ, Gripp KW, Stephan MJ, Fairchok MP, McGeady SJ. Selective IgM deficiency and 22q11.2 deletion syndrome. Ann Allergy Asthma Immunol. 2007;99(1):87–92. https://doi.org/10.1016/S1081-1206(10)60627-8.
Serana F, Chiarini M, Zanotti C, et al. Use of V(D)J recombination excision circles to identify T- and B-cell defects and to monitor the treatment in primary and acquired immunodeficiencies. J Transl Med. 2013;11:119. https://doi.org/10.1186/1479-5876-11-119.
Dar N, Gothelf D, Korn D, et al. Thymic and bone marrow output in individuals with 22q11.2 deletion syndrome. Pediatr Res. 2015;77(4):579–85. https://doi.org/10.1038/pr.2015.14.
Lingman Framme J, Borte S, von Döbeln U, Hammarström L, Oskarsdóttir S. Retrospective analysis of TREC based newborn screening results and clinical phenotypes in infants with the 22q11 deletion syndrome. J Clin Immunol. 2014;34(4):514–9. https://doi.org/10.1007/s10875-014-0002-y.
Davis CM, Kancherla VS, Reddy A, et al. Development of specific T-cell responses to Candida and tetanus antigens in partial DiGeorge syndrome. J Allergy Clin Immunol. 2008;122(6):1194–9. https://doi.org/10.1016/j.jaci.2008.06.039.
Iroh Tam PY, McAllister SC. Vaccine responses and immunologic characteristics of pediatric patients with DiGeorge Syndrome. Clin Pediatr (Phila). 2015;54(13):1290–2. https://doi.org/10.1177/0009922814565885.
Bassett AS, Chow EW, Husted J, et al. Premature death in adults with 22q11.2 deletion syndrome. J Med Genet. 2009;46(5):324–30. https://doi.org/10.1136/jmg.2008.063800.
Deshpande DR, Demirdag YY, Marsh RA, Sullivan KE, Orange JS, USIDNET Consortium. Relationship between severity of T cell lymphopenia and immune dysregulation in patients with DiGeorge syndrome (22q11.2 deletions and/or related TBX1 Mutations): a USIDNET Study. J Clin Immunol. 2021;41(1):29–37. https://doi.org/10.1007/s10875-020-00854-y.
Rosa RF, Rosa RC, Dos Santos PP, Zen PR, Paskulin GA. Hematological abnormalities and 22q11.2 deletion syndrome. Rev Bras Hematol Hemoter. 2011;33(2):151–4. https://doi.org/10.5581/1516-8484.20110037.
Crowley TB, Campbell IM, Liebling EJ, et al. Distinct immune trajectories in patients with chromosome 22q11.2 deletion syndrome and immune-mediated diseases. J Allergy Clin Immunol. 2022;149(1):445–50. https://doi.org/10.1016/j.jaci.2021.06.007.
Sullivan KE, McDonald-McGinn D, Zackai EH. CD4(+) CD25(+) T-cell production in healthy humans and in patients with thymic hypoplasia. Clin Diagn Lab Immunol. 2002;9(5):1129–31. https://doi.org/10.1128/cdli.9.5.1129-1131.2002.
Liao HC, Liao CH, Kao SM, Chiang CC, Chen YJ. Detecting 22q11.2 deletion syndrome in newborns with low T cell teceptor excision circles from severe combined immunodeficiency screening. J Pediatr. 2019;204:219-224.e1. https://doi.org/10.1016/j.jpeds.2018.08.072.
Sørensen KM, Agergaard P, Olesen C, et al. Detecting 22q11.2 deletions by use of multiplex ligation-dependent probe amplification on DNA from neonatal dried blood spot samples. J Mol Diagn. 2010;12(02):147–51.
Barry JC, Crowley TB, Jyonouchi S, et al. Identification of 22q11.2 deletion syndrome via newborn screening for severe combined immunodeficiency. J Clin Immunol. 2017;37(5):476–85. https://doi.org/10.1007/s10875-017-0403-9.
Bevilacqua E, Jani JC, Chaoui R, et al. Performance of a targeted cell-free DNA prenatal test for 22q11.2 deletion in a large clinical cohort. Ultrasound Obstet Gynecol. 2021;58(4):597–602. https://doi.org/10.1002/uog.23699.
Ravi H, McNeill G, Goel S, et al. Validation of a SNP-based non-invasive prenatal test to detect the fetal 22q11.2 deletion in maternal plasma samples. PLoS ONE. 2018;13(2):e0193476. https://doi.org/10.1371/journal.pone.0193476.
Dar P, Jacobsson B, Clifton R, et al. Cell-free DNA screening for prenatal detection of 22q11.2 deletion syndrome [published online ahead of print, 2022 Jan 13]. Am J Obstet Gynecol. 2022;S0002-9378(22)00006-0. https://doi.org/10.1016/j.ajog.2022.01.002.
•• Habel A, Herriot R, Kumararatne D, et al. Towards a safety net for management of 22q11.2 deletion syndrome: guidelines for our times. Eur J Pediatr. 2014;173(6):757–65. https://doi.org/10.1007/s00431-013-2240-z. This article provides systems-based guidance on clinical management of patients with 22q11.2DS.
Bassett AS, McDonald-McGinn DM, Devriendt K, et al. Practical guidelines for managing patients with 22q11.2 deletion syndrome. J Pediatr. 2011;159(2):332-9.e1. https://doi.org/10.1016/j.jpeds.2011.02.039.
Knutsen AP, Baker MW, Markert ML. Interpreting low T-cell receptor excision circles in newborns with DiGeorge anomaly: importance of assessing naive T-cell markers. J Allergy Clin Immunol. 2011;128(6):1375–6. https://doi.org/10.1016/j.jaci.2011.08.019.
Hofstetter AM, Jakob K, Klein NP, et al. Live vaccine use and safety in DiGeorge syndrome. Pediatrics. 2014;133(4):e946–54. https://doi.org/10.1542/peds.2013-0831.
Eberle P, Berger C, Junge S, et al. Persistent low thymic activity and non-cardiac mortality in children with chromosome 22q11.2 microdeletion and partial DiGeorge syndrome. Clin Exp Immunol. 2009;155(2):189–98. https://doi.org/10.1111/j.1365-2249.2008.03809.x.
Soshnick SH, Joseph T, Bennett NJ. Humoral immunodeficiency and immune globulin replacement therapy (IGRT) usage in DiGeorge syndrome. J Clin Immunol. 2021;41(6):1208–12. https://doi.org/10.1007/s10875-021-01012-8.
Acknowledgements
We thank Dr. Anahita Mahmoudabadi for her contribution to manuscript preparation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Biggs, S.E., Gilchrist, B. & May, K.R. Chromosome 22q11.2 Deletion (DiGeorge Syndrome): Immunologic Features, Diagnosis, and Management. Curr Allergy Asthma Rep 23, 213–222 (2023). https://doi.org/10.1007/s11882-023-01071-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11882-023-01071-4