
ABSTRACT
Dermatomyositis is a chronic systemic autoimmune disease characterized by inflammatory infiltrates in the skin and muscle. The wide variability in clinical and serologic presentation poses a diagnostic challenge for the internist. Appreciation of the clinical variants of dermatomyositis allows for expedient diagnosis and avoidance of diagnostic error. We illustrate these challenges with the case of a 51-year-old Vietnamese-American man who initially presented with fever of unknown origin in the absence of overt skin and muscle manifestations. The diagnosis of dermatomyositis was not evident on several clinical encounters due to the absence of these hallmark symptoms. We review the variable clinical manifestations of a subtype of dermatomyositis associated with an autoantibody against melanoma differentiation-associated protein 5 (anti-MDA5) and suggest consideration of dermatomyositis as a diagnosis in patients presenting with systemic illness and markedly elevated ferritin, even in the absence of elevated muscle enzymes and classic autoantibodies.
Similar content being viewed by others


CASE PRESENTATION
A 51-year-old Vietnamese-American male industrial engineer was admitted to the medical ward for evaluation of fever of unknown origin (FUO). He reported 3 weeks of fever, fatigue, generalized weakness, and dyspnea. His past medical history included a remote 10-pack-year history of cigarette smoking and latent tuberculosis treated with 9 months of isoniazid treatment, completed 5 years prior. He had moved from Vietnam to the United States 30 years earlier and denied any recent travel, sick contacts, or insect bites. Physical examination revealed a fatigued man, with a temperature of 38.1 °C, pulse of 108 beats/minute, respiratory rate of 28 breaths/minute, and room air oxygen saturation of 96 %. Oropharyngeal, neurologic, pulmonary, cardiac, musculoskeletal, and skin evaluation were otherwise normal.
A complete blood count (CBC) revealed anemia (hemoglobin 12.3 g/dL, hematocrit 38.5 %). White blood cell counts, platelet counts, and the basic metabolic panel were normal. Erythrocyte sedimentation rate (ESR) was 97 mm/hr and C-reactive protein (CRP) was 5.7 mg/dL. Muscle enzymes, sent to evaluate his weakness in the setting of elevated inflammatory markers, showed normal creatine kinase (CK) at 44 units/liter and mildly elevated aldolase to 9.4 U/L (normal: < 7.7 U/L). Blood, urine, and sputum cultures had no growth. Chest x-ray (CXR) demonstrated low lung volumes with increased reticular markings, small bilateral pleural effusions, and bibasilar opacities. During the hospitalization, he continued to have intermittent fevers as high as 39.1 °C, generalized weakness, and dyspnea. Additionally, he developed bibasilar crackles on pulmonary exam and flat, diffuse, slightly hyperpigmented patches on his arms and chest that faded over several days. Extensive workup for infectious, inflammatory, and malignant causes of fever was unrevealing, including negative viral hepatitis serologies, human immunodeficiency virus antibody, anti-nuclear antibody (ANA), and anti-Jo1 antibodies. Serial acid-fast bacilli (AFB) smears were negative, and there was no growth of serial AFB sputum cultures. Positron emission tomography-computed tomography of the chest, abdomen, and pelvis revealed only patchy and linear opacities at the lung bases (comprehensive list of studies available in Online Appendix, Table A).
Ferritin was markedly elevated (9,354 ng/mL). A bone marrow biopsy showed mild hemophagocytosis; however, he lacked other clinical and laboratory criteria (splenomegaly, elevated soluble interleukin-2 receptor level, severe anemia) to meet the diagnosis of hemophagocytic lymphohistiocytosis (HLH) (Online Appendix, Table B).1,2 During his hospitalization, he was initially treated empirically for pneumonia with ceftriaxone and azithromycin. When he failed to clinically improve, his antimicrobial coverage was broadened to vancomycin and piperacillin-tazobactam. He had a modest symptomatic improvement and was transitioned to oral doxycycline. Prior to discharge, his fever resolved and fatigue, weakness, and dyspnea improved, though not back to baseline. A presumed diagnosis of community-acquired pneumonia was made. At the time of discharge, his ESR and CRP were normal.
Two months later, the patient presented again with similar but worsened constitutional and respiratory symptoms, with cough, sore throat, dysphagia, and arthralgias in the bilateral wrists and right knee and ankle. Physical examination was remarkable for an ill and uncomfortable appearance, temperature of 38.4 °C, superficial erosions in his posterior oropharynx, and synovitis in his bilateral wrists, elbows, and knees. Workup for fever was again initiated (Online Appendix A, Table A). Blood, urine, and sputum cultures had no growth, and CXR was unchanged with persistent bibasilar opacities. Ferritin was elevated at 2,351 ng/mL. Arthrocentesis was deferred due to the small volume of joint effusion. He was started on colchicine for inflammatory arthritis of unclear etiology and was discharged after symptomatic improvement.
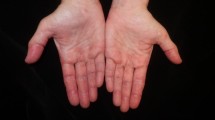
Ten days later, the patient returned with worsening pharyngitis, dysphagia, fatigue, and arthritis refractory to colchicine and analgesic therapy. He had also developed new symptoms including rash, alopecia, lip edema, and xerostomia. Skin examination was notable for poikiloderma in a V-sign (Fig. 1), edema of the nasal root, and erythema of the inner canthi. Neurologic examination was notable for reduced strength (4/5) of hip flexion bilaterally. CK and aldolase were normal. Skin biopsy demonstrated vacuolar changes with interface dermatitis consistent with, but not diagnostic for, dermatomyositis. Magnetic resonance imaging was performed, due to the marked weakness and to identify a site for muscle biopsy; this showed muscle inflammation concerning for myositis (Fig. 2). A muscle biopsy was performed which revealed membrane-attack complex staining and endothelial tubuloreticular inclusions, consistent with dermatomyositis (Fig. 3). The patient was treated with high-dose prednisone with significant improvement in pain, swelling, rash, and functional status. Given his unusual presentation, ethnicity, and negative ANA and anti-Jo1 autoantibodies, further screening for other dermatomyositis autoantibodies was performed. Immunoprecipitation blot screen testing, performed by an outside clinical immunology laboratory, indicated the presence of anti-MDA5 autoantibodies. He was discharged home.

Patient skin examination. On third hospital admission, the patient presented with a rash on sun-exposed areas, most notably over the face, neck, and upper chest and back in a shawl distribution.

Femur MRI. MRI of the right femur demonstrated muscle inflammation in the anterior compartment muscles, most pronounced in the distal aspect of the vastus intermedialis muscle, as seen by the increased signal (arrow).

Muscle biopsy. The patient’s muscle biopsy stained positive for membrane attack complex (left panel, arrow), and electron microscopy demonstrated endothelial tubuloreticular inclusions (right panel, arrow), consistent with dermatomyositis. Membrane attack complex is involved in the pathogenesis of dermatomyositis and its deposition serves as a marker to distinguish dermatomyositis from other idiopathic inflammatory myopathies.37
Despite his initial improvement, the patient’s arthralgias, dysphagia, dyspnea, and weakness worsened over the weeks following discharge. Pulmonary function testing showed a severe restrictive ventilatory defect with greatly reduced diffusing capacity. High-resolution chest computed tomography (HRCT) showed significant volume loss, peripheral consolidation in the right upper lobe, and bilateral lower lobe ground glass opacities and bronchial dilatation with peribronchial consolidation (Fig. 4). He was diagnosed with interstitial lung disease (ILD) with radiographic findings consistent with non-specific interstitial pneumonia. No evidence of malignancy was found with laryngoscopy, colonoscopy, and computed tomography of the chest, abdomen, and pelvis.

High-Resolution Computed Tomography (HRCT) of chest. The patient’s HRCT showed bilateral lower lobe bronchial dilatation with peribronchial consolidation and significant volume loss, most prominent in the left lower lobe. Ground glass opacities were seen in the lower lobes. Imaging of the right upper lobe revealed peripheral consolidation (arrow).
The patient was started on monthly intravenous immunoglobulin therapy, mycophenolate mofetil, and high-dose prednisone with subsequent clinical improvement, though he remains limited by his poor pulmonary function.
DISCUSSION
Dermatomyositis is a systemic autoimmune disease that primarily affects the muscle and skin; other prominent associations are dysphagia, ILD, and malignancy. In recent years, there has been a growing understanding of several myositis-specific antibodies that are associated with specific clinical features of dermatomyositis and are usually mutually exclusive. This evolving body of evidence suggests that the dermatomyositis patient population is heterogeneous with multiple subsets. In association with different autoantibodies, these subsets vary in rates of skin involvement, muscle involvement, ILD, and underlying malignancy (Table 1).3–5 Although many of these antibodies are not yet available for routine clinical testing, knowledge of the clinical variants can assist clinicians with the diagnosis of dermatomyositis in patients who present without classic features.
Anti-MDA5-positive-dermatomyositis (anti-MDA5-positive-DM) was first described in a group of patients who lacked clinical signs of muscle involvement (clinically amyopathic dermatomyositis); however, subsequent studies have also found patients with this antibody who have classic muscle involvement with weakness and elevated muscle enzymes.6–11 Anti-MDA5-positive-DM patients typically present with prominent skin manifestations, including heliotrope rash, Gottron’s papules as well as cutaneous ulceration and tender palmar papules, the latter of which are unique to anti-MDA5-positive-DM (Table 1).6,12–14 Oral manifestations—including oral ulceration, mucosal pain, and hoarseness—are common, as are arthritis, arthralgia, and fever.6,7,9,15 This patient initially had fever and later developed hoarseness, pharyngeal ulceration, arthritis, and arthralgia.
Laboratory testing in patients with anti-MDA5-positive-DM may reveal normal or mildly elevated muscle enzymes. Patients commonly have markedly elevated ferritin levels, often over 1,000 ng/mL.14,16 ANA and anti-Jo1 antibodies are frequently negative.6,7,12,17 This patient presented with markedly elevated ferritin, normal CK, mildly elevated aldolase, and negative ANA and anti-Jo1 antibodies. There is currently no commercially available test to detect anti-MDA5 antibodies, though testing can be done in specialized research laboratories.
Most patients with anti-MDA5-positive-DM develop ILD, with studies reporting a 67–100 % incidence.6,7,12,14,16 In comparison to other patients with dermatomyositis and ILD, anti-MDA5-positive-DM patients are more likely to have rapidly progressive disease.7,8 The development and rapid progression of ILD may account for the significantly higher mortality observed in anti-MDA5-positive-DM patients as compared to serologically negative cohorts.6,14,18,19 Generally, skin manifestations precede ILD in these patients.14 Previous studies have shown a correlation between ferritin levels and ILD activity, as well as an association between elevated ferritin levels and mortality in patients with anti-MDA5- positive-DM.14,16 This patient did develop severe ILD.
All patients with anti-MDA5-positive-DM should undergo screening for ILD and malignancy. Evidence suggests that anti-MDA5-positive-DM is more common in Asian populations, particularly in Japanese and Chinese individuals.7,9,20 Patients with anti-MDA5-positive-DM have a malignancy risk similar to the general dermatomyositis population (3–6 fold increased risk compared to the general population), and thus should undergo age-appropriate workup for malignancy, including screening for nasopharyngeal carcinoma in patients of Asian descent.7,14,21–24 Treatment of anti-MDA5-positive-DM consists of potent immunosuppressive therapy, especially in the presence of underlying rapidly progressive ILD.14
This patient had an unusual presentation of dermatomyositis, and several diagnostic errors contributed to a delay in his diagnosis (Table 2).25 He initially presented with FUO, dyspnea, and an abnormal CXR, and over the course of several months developed more classic symptoms such as arthritis, rash, oral ulcers, and objective weakness. Availability heuristics likely led to the clinical diagnosis of pneumonia at initial presentation when he presented with fever, tachycardia, dyspnea, tachypnea, and an abnormal CXR. Anchoring heuristics may have contributed to the failure to consider other diagnoses, such as ILD or pneumonia superimposed on ILD, and therefore, to the neglect to perform further workup such as HRCT. Furthermore, his initial presenting symptom of subjective weakness was attributed to systemic illness from pneumonia, and anchoring may have also led to failure to attribute clinical significance to his initial elevated aldolase.
Multiple hospitalizations and numerous diagnostic tests may have also led to information overload, resulting in decreased cognitive assimilation of data, exacerbation of working memory capacity, increased distractibility, and decreased situational awareness—all of which can impair decision-making and cognitive work.26–33 This may subsequently have distracted clinicians from re-examining the patient’s entire clinical presentation and caused inattentional blindness, information avoidance, and increased dependence on other cognitive heuristics.34– For example, information overload may have contributed to overreliance on normal results of classic serum muscle enzymes (such as CK) and autoantibodies (such as ANA and anti-Jo1), while information avoidance led to false reassurance despite the elevated aldolase. Moreover, blind obedience towards the negative ANA and anti-Jo1 autoantibodies likely resulted in premature closure, leading clinicians to exclude dermatomyositis from the differential diagnosis.
Systems issues also affected clinical decision-making. Multiple physicians cared for the patient over several hospitalizations with variable continuity of care. While this could lead to a “fresh” perspective, it also hindered the clinicians’ ability to assess changes over time and likely decreased the quality of information transfer.36 Moreover, numerous hospitalizations and diagnostic tests made the patient’s medical record long and cumbersome to review, potentially contributing to communication breakdown. It also likely led subsequent providers to miss key details earlier in the patient’s presentation, potentially excluding unrecognized pertinent information. For instance, the evanescent rash that the patient experienced on his first hospitalization was excluded from the patient’s discharge summary.
Finally, lack of knowledge about this variant of dermatomyositis contributed to the delay in diagnosis. In retrospect, the patient’s markedly elevated ferritin was consistent with anti-MDA5-positive-DM; however, availability heuristic and lack of familiarity with anti-MDA5-positive-DM resulted in greater consideration towards other causes of hyper-ferritinemia such as HLH, infection, and liver disease. The patient’s overall clinical presentation of fever, weakness, dysphagia, and dyspnea in the presence of hyperferritinemia, normal or mildly elevated muscle enzymes, and negative ANA and anti-Jo1 autoantibodies suggested anti-MDA5-positive-DM as a possibility that was unrecognized due to lack of awareness of this clinical variant.
CONCLUSION
Appreciation of the clinical variants of dermatomyositis and their presenting symptoms can reduce diagnostic errors. Patients with anti-MDA5-positive-DM often present differently than patients with classic dermatomyositis, as anti-MDA5-positive-DM patients may lack muscle involvement or elevated muscle enzymes and may have unusual skin manifestations such as ulceration and palmar papules. Laboratory clues to the diagnosis may include a markedly elevated ferritin or elevated aldolase despite other normal muscle enzymes. Importantly, these patients may present, as this patient did, with signs of systemic illness to the internist. We suggest that the internist keep anti-MDA5-positive-DM on the short differential diagnosis of a markedly elevated ferritin and not be unsettled by a normal CK, negative ANA, and negative anti-Jo1 antibody. Despite the diagnostic challenge, early diagnosis is important given the need for expedient initiation of potent immunosuppressive therapy and the association with progressive interstitial lung disease and higher mortality. Unfortunately, clinical availability of testing for anti-MDA5 and other dermatomyositis-associated antibodies is limited; therefore, the clinical syndrome must be recognized.
REFERENCES
Jordan MB, Allen CE, Weitzman S, Filipovich AH, Mcclain KL. How I treat How I treat hemophagocytic lymphohistiocytosis. 2011;118:4041–4052.
Henter J-I, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–131.
Gunawardena H, Betteridge ZE, McHugh NJ. Myositis-specific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatology. 2009;48:607–612.
Fujikawa K, et al. Association of distinct clinical subsets with myositis-specific autoantibodies towards anti-155/140-kDa polypeptides, anti-140-kDa polypeptides, and anti-aminoacyl tRNA synthetases in Japanese patients with dermatomyositis: a single-centre, cross-sectional study. Scand J Rheumatol. 2009;38:263–267.
Targoff IN. Idiopathic inflammatory myopathy: autoantibody update. Curr Rheumatol Rep. 2002;4:434–441.
Fiorentino D, Chung L, Zwerner J, Rosen A, Casciola-Rosen L. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25–34.
Chen Z, et al. Utility of anti-melanoma differentiation-associated gene 5 antibody measurement in identifying patients with dermatomyositis and a high risk for developing rapidly progressive interstitial lung disease: a review of the literature and a meta-analysis. Arthritis Care Res. 2013;65:1316–1324.
Gono T, et al. Anti-MDA5 antibody, ferritin and IL-18 are useful for the evaluation of response to treatment in interstitial lung disease with anti-MDA5 antibody-positive dermatomyositis. Rheumatol. (United Kingdom). 2012;51:1563–1570.
Sato S, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum. 2005;52:1571–1576.
Sato S, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: association with rapidly progressive interstitial lung disease. Arthritis Rheum. 2009;60:2193–2200.
Sato S, Kuwana M. Clinically amyopathic dermatomyositis. Curr Opin Rheumatol. 2010;22:639–643.
Cao H, et al. Clinical manifestations of dermatomyositis and clinically amyopathic dermatomyositis patients with positive expression of anti-melanoma differentiation-associated gene 5 antibody. Arthritis Care Res. 2012;64:1602–1610.
Narang NS, Casciola-Rosen L, Li S, Chung L, Fiorentino DF. Cutaneous ulceration in dermatomyositis: association with anti-melanoma differentiation-associated gene 5 antibodies and interstitial lung disease. Arthritis Care Res. 2015; 67:667-672.
Koga T, et al. The diagnostic utility of anti-melanoma differentiation-associated gene 5 antibody testing for predicting the prognosis of Japanese patients with DM. Rheumatol. (United Kingdom). 2012;51:1278–1284.
Hamaguchi Y, et al. Clinical correlations with dermatomyositis-specific autoantibodies in adult Japanese patients with dermatomyositis: a multicenter cross-sectional study. Arch Dermatol. 2011;147:391–398.
Gono T, et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology. 2010;49:1713–1719.
Hall JC, et al. Anti-melanoma differentiation-associated protein 5-associated dermatomyositis: expanding the clinical spectrum. Arthritis Care Res. 2013;65:1307–1315.
Nakashima R, et al. The RIG-I-like receptor IFIH1/MDA5 is a dermatomyositis-specific autoantigen identified by the anti-CADM-140 antibody. Rheumatology. 2009;49:433–440.
Tanizawa K, et al. HRCT features of interstitial lung disease in dermatomyositis with anti-CADM-140 antibody. Respir Med. 2011;105:1380–1387.
Ikeda N, et al. Analysis of dermatomyositis-specific autoantibodies and clinical characteristics in Japanese patients. J Dermatol. 2011;38:973–979.
Ponyi A, et al. Cancer-associated myositis: clinical features and prognostic signs. Ann N Y Acad Sci. 2005;1051:64–71.
Mammen AL. Autoimmune myopathies: autoantibodies, phenotypes and pathogenesis. Nat Rev Neurol. 2011;7:343–354.
Hu WJ, Chen DL, Min HQ. Study of 45 cases of nasopharyngeal carcinoma with dermatomyositis. Am J Clin Oncol. 1996;19:35–38.
Huang YL, et al. Malignancies associated with dermatomyositis and polymyositis in Taiwan: a nationwide population-based study. Br J Dermatol. 2009;161:854–860.
Redelmeier DA. Improving patient care. The cognitive psychology of missed diagnoses. Ann Intern Med. 2005;142:115–120.
Endsley MR. Toward a theory of situation awareness in dynamic systems. Hum Factors J Hum Factors Ergon Soc. 1995;37:32–64.
Smith K, Hancock PA. Situation awareness is adaptive, externally directed consciousness. Hum Factors J Hum Factors Ergon Soc. 1995;37:137–148.
Hazlehurst B, McMullen CK, Gorman PN. Distributed cognition in the heart room: how situation awareness arises from coordinated communications during cardiac surgery. J Biomed Inform. 2007;40:539–551.
Stanton NA, Chambers PRG, Piggott J. Situational awareness and safety. Saf Sci. 2001;39:189–204.
Stanton NA, et al. Distributed situation awareness in dynamic systems: theoretical development and application of an ergonomics methodology. Ergonomics. 2007;49:1288–1311.
Hancock PA, Szalma JL. Operator stress and display design. Ergon Des. 2003;11:13–18.
Schiff GD, et al. Diagnosing diagnosis errors: lessons from a multi-institutional collaborative project. Advances in Patient Safety: From Research to Implementation, Agency for Healthcare Research and Quality, Rockville: MD (2007) vol 2. February 2005. AHRQ Publication No. 050021. Available at: http://www.ahrq.gov/downloads/pub/advances/vol2/schiff.pdf./. Accessed 16 June
Fukukura J, Ferguson MJ, Fujita K. Psychological distance can improve decision making under information overload via gist memory. J Exp Psychol Gen. 2012;142:658–665.
Bawden D, Robinson L. The dark side of information: overload, anxiety and other paradoxes and pathologies. J Inf Sci. 2008;35:180–191.
Case DO, Andrews JE, Johnson JD, Allard SL. Avoiding versus seeking: the relationship of information seeking to avoidance, blunting, coping, dissonance, and related concepts. J Med Libr Assoc. 2005;93:353–62.
Singh H, Naik AD, Rao R, Petersen LA. Reducing diagnostic errors through effective communication: harnessing the power of information technology. J Gen Intern Med. 2008;23:489–494.
Jain A, et al. Detection of the membrane attack complex as a diagnostic tool in dermatomyositis. Acta Neurol Scand. 2011;123:122–129.
Nishikai M, Reichlin M. Heterogeneity of precipitating antibodies characterization of the Jo- 1 antibody system. Arthritis Rheum. 1980;23:881–888.
Yoshida S, et al. The precipitating antibody to an acidic nuclear protein antigen, the Jo-1, in connective tissue diseases. A marker for a subset of polymyositis with interstitial pulmonary fibrosis. Arthritis Rheum. 1983;26:604–611.
Targoff IN. Update on myositis-specific and myositis-associated autoantibodies. Curr Opin Rheumatol. 2000;12:475–481.
Gunawardena H. The clinical features of myositis-associated autoantibodies: a review. Clin Rev Allergy Immunol. 2015. doi:10.1007/s12016-015-8513-8.
Nakashima R, Mimori T. Clinical and pathophysiological significance of myositis-specific and myositis-associated autoantibodies. Int J Clin Rheumatol. 2010;5:523–536.
Targoff IN, Reichlin M. The association between Mi-2 antibodies and dermatomyositis. Arthritis Rheum. 1985;28:796–803.
Iaccarino L, et al. The clinical features, diagnosis and classification of dermatomyositis. J Autoimmun. 2014;48–49:122–127.
Love LA, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore). 1991;70:360–374.
Betteridge ZE, et al. Autoantibodies to the p140 autoantigen NXP-2 in adult dermatomyositis. Arthritis Rheum. 2009;60:815.
Fiorentino DF, et al. Most patients with cancer-associated dermatomyositis have antibodies to nuclear matrix protein NXP-2 or transcription intermediary factor 1γ. Arthritis Rheum. 2013;65:2954–62.
Ichimura Y, et al. Anti-NXP2 autoantibodies in adult patients with idiopathic inflammatory myopathies: possible association with malignancy. Ann Rheum Dis. 2012;71:710–713.
Betteridge Z, Gunawardena H, North J, Slinn J, McHugh N. Identification of a novel autoantibody directed against small ubiquitin-like modifier activating enzyme in dermatomyositis. Arthritis Rheum. 2007;56:3132–7.
Tarricone E, et al. Anti-SAE antibodies in autoimmune myositis: identification by unlabelled protein immunoprecipitation in an Italian patient cohort. J Immunol Methods. 2012;384:128–134.
Ghirardello A, et al. Myositis autoantibodies and clinical phenotypes. Auto Immun Highlight. 2014;5:69–75.
Betteridge ZE, et al. Clinical and human leucocyte antigen class II haplotype associations of autoantibodies to small ubiquitin-like modifier enzyme, a dermatomyositis-specific autoantigen target, in UK Caucasian adult-onset myositis. Ann Rheum Dis. 2009;68:1621–5.
Fujimoto M, et al. Autoantibodies to small ubiquitin-like modifier activating enzymes in Japanese patients with dermatomyositis. Arthritis Rheum. 2012;64:S97.
Targoff IN, et al. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum. 2006;54:3682–3689.
Fujimoto M, et al. Myositis-specific anti-155/140 autoantibodies target transcription intermediary factor 1 family proteins. Arthritis Rheum. 2012;64:513–22.
Trallero-Araguás E, et al. Usefulness of anti-p155 autoantibody for diagnosing cancer-associated dermatomyositis: a systematic review and meta-analysis. Arthritis Rheum. 2012;64:523–532.
Trallero-Araguás E, et al. Cancer-associated myositis and anti-p155 autoantibody in a series of 85 patients with idiopathic inflammatory myopathy. Medicine (Baltimore). 2010;89:47–52.
Gunawardena H, et al. Clinical associations of autoantibodies to a p155/140 kDa doublet protein in juvenile dermatomyositis. Rheumatology (Oxford). 2008;47:324–8.
Mack A, Rock I. Inattentional Blindness. In: Malik J, Perona P, eds. Cambridge MA, MIT Press; 1998:7, 287
Acknowledgements
The authors would like to thank Dr. Frank Chen for his dedicated clinical care for the patient, Dr. Lorinda Chung and Dr. David Florentino for their guidance in researching the manuscript, Dr. Hannes Vogel for his assistance in obtaining and interpreting the pathology images, and Dr. Khalil Jivraj for his assistance in obtaining and interpreting the radiology images. A.P. receives research support from NIH grant 5T32AR050942-10. An earlier version of this work was presented as a poster presentation at the American College of Physicians Northern California Chapter meeting on 22 November 2014 and as a poster presentation at the Society of General Internal Medicine, California–Hawaii 2015 Regional meeting, on 10 January 2015.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of Interest
The authors have no conflicts of interest to disclose.
Disclaimers
None.
Sources of Support
None.
Rights and permissions
About this article

Cite this article
Lee, L.W., Narang, N.S., Postolova, A. et al. Anti-MDA5-Positive Dermatomyositis Presenting as Fever of Unknown Origin. J GEN INTERN MED 31, 1530–1536 (2016). https://doi.org/10.1007/s11606-016-3769-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11606-016-3769-0