
Abstract
Background
Fibroblast growth factor receptor (FGFR) 2 is overexpressed in several tumor types, including triple-negative breast cancer and gastric cancer, both of which have a high unmet medical need. Aprutumab ixadotin (BAY 1187982) is the first antibody–drug conjugate (ADC) to target FGFR2 and the first to use a novel auristatin-based payload.
Objective
This first-in-human trial was conducted to determine the safety, tolerability, and maximum tolerated dose (MTD) of aprutumab ixadotin in patients with advanced solid tumors from cancer indications known to be FGFR2-positive.
Patients and Methods
In this open-label, multicenter, phase I dose-escalation trial (NCT02368951), patients with advanced solid tumors received escalating doses of aprutumab ixadotin (starting at 0.1 mg/kg body weight), administered intravenously on day 1 of every 21-day cycle. Primary endpoints included safety, tolerability, and the MTD of aprutumab ixadotin; secondary endpoints were pharmacokinetic evaluation and tumor response to aprutumab ixadotin.
Results
Twenty patients received aprutumab ixadotin across five cohorts, at doses of 0.1–1.3 mg/kg. The most common grade ≥ 3 drug-related adverse events were anemia, aspartate aminotransferase increase, proteinuria, and thrombocytopenia. Dose-limiting toxicities were thrombocytopenia, proteinuria, and corneal epithelial microcysts, and were only seen in the two highest dosing cohorts. The MTD was determined to be 0.2 mg/kg due to lack of quantitative data following discontinuations at 0.4 and 0.8 mg/kg doses. One patient had stable disease; no responses were reported.
Conclusions
Aprutumab ixadotin was poorly tolerated, with an MTD found to be below the therapeutic threshold estimated preclinically; therefore, the trial was terminated early.
ClinicalTrials.gov Identifier
NCT02368951.
Similar content being viewed by others


Aprutumab ixadotin (BAY 1187982) is a novel conjugate of an anti-fibroblast growth factor receptor (FGFR) 2 antibody linked to an innovative auristatin W derivative toxophore. This is the first time this novel payload has been used in a clinical setting and the first time an antibody–drug conjugate has been used to target FGFR2. |
Toxicities were observed at doses lower than the predicted therapeutic dose and were unexpected based on the preclinical findings. The cause of these toxicities is not yet known but may be attributed to the unique combination of an auristatin W derivative payload with an FGFR2-targeting antibody. |
These findings highlight the need for improved preclinical models that more accurately predict the effects of novel compounds in humans, which may increase the efficiency of clinical development. |
1 Introduction
Fibroblast growth factor receptor (FGFR) 2 is a transmembrane receptor tyrosine kinase with a key role in tissue repair and embryonic development [1, 2]. Aberrations leading to constitutive activation or overexpression of FGFR2, including gene amplification, gene fusions, and single nucleotide polymorphisms, have been identified in many cancer types, including triple-negative breast cancer, pancreatic, esophageal, hepatocellular, colorectal, ovarian, gastric, non-small-cell lung cancer (NSCLC), and glioma [3,4,5,6,7,8,9,10,11,12,13,14,15,16]. Furthermore, FGFR2 overexpression has been associated with poor survival in patients with gastric cancer [7]. Although treatments are available for gastric and other cancers overexpressing FGFR2, the generally poor prognosis for patients with these tumors suggests that a high unmet medical need remains [3,4,5,6,7,8,9,10,11,12,13,14,15,16]. In contrast to typically high expression levels in tumors, FGFR2 is generally expressed at low levels in normal tissue, making it an attractive antigen for development of a targeted anticancer therapy [8, 17].
Antibody–drug conjugates (ADCs) comprise a cytotoxic payload conjugated by a linker to a monoclonal antibody directed against an antigen that is selectively expressed on the surface of tumor cells [18, 19]. This selectivity allows ADCs to be directed at tumor cells, limiting systemic exposure and off-target toxicity [20, 21]. Binding of the antibody to its target antigen triggers internalization of the ADC, after which the linker molecule is cleaved, or the antibody moiety is degraded in the lysosome (non-cleavable linker). Cleavage or degradation of the linker molecule releases the payload metabolite within the cell, resulting in cytotoxic effects [20, 21]. The ADCs brentuximab vedotin, gemtuzumab ozogamicin, inotuzumab ozogamicin, and trastuzumab emtansine have been approved based on their efficacy in late-stage clinical trials [22,23,24,25]. In addition, several ADCs are currently being investigated in clinical trials in a wide range of tumor types [26, 27]. However, ADCs targeting FGFR2 have not yet been described in any tumor type.
Aprutumab ixadotin (BAY 1187982) is a novel ADC comprising a fully human anti-FGFR2 monoclonal antibody (BAY 1179470) conjugated by lysine side chains to a non-cleavable linker and via this an innovative auristatin W derivative [28]. This novel auristatin W derivative is a highly potent microtubule-disrupting agent used for the first time in aprutumab ixadotin [28]. Lysosomal degradation of the antibody moiety releases the non-cell-permeable payload metabolite that cannot cross cell membranes due to the presence of a charged group, as previously described [28]. Results from in vitro studies indicated that aprutumab ixadotin has low nanomolar potency and suggested that high FGFR2 expression correlates with internalization and cytotoxic effects [28]. In mice, pharmacokinetic studies of aprutumab ixadotin showed that concentrations of the cytotoxic payload metabolite are enriched by more than 30-fold in FGFR2-positive tumors compared with healthy tissue [28]. Furthermore, aprutumab ixadotin treatment resulted in dose-dependent tumor regression in patient-derived xenograft (PDX) models of FGFR2-positive gastric cancer and triple-negative breast cancer, including reductions in tumor volume equivalent to partial and complete responses [28]. Results from PDX studies also indicated that FGFR2 amplification or overexpression was predictive of high antitumor activity in vivo [28]. In these preclinical studies, no evidence was found to suggest that aprutumab ixadotin would exert a bystander effect [28]. Furthermore, findings from preclinical studies have also indicated that aprutumab ixadotin was stable in the circulatory system [28].
Based on these promising preclinical data, an open-label, non-randomized, first-in-human phase I dose-escalation study of aprutumab ixadotin was conducted in adult patients with advanced, refractory solid tumor indications reported to express FGFR2. Aprutumab ixadotin is the first ADC to target FGFR2 in the treatment of advanced tumors and is also the first ADC to include this novel auristatin W derivative payload. The primary objectives of this study were to determine the safety, tolerability, and maximum tolerated dose (MTD) of aprutumab ixadotin.
2 Materials and Methods
2.1 Patients
Patients aged 18 years or older with advanced solid tumors from cancer indications known to be FGFR2-positive, which were refractory to any standard therapy or had no standard therapy available, were eligible for enrollment. Patients were required to have measurable disease, Eastern Cooperative Oncology Group performance status (ECOG PS) of 0–1, an expected lifespan of at least 12 weeks, and a formalin-fixed paraffin-embedded (FFPE) tumor tissue sample from either archival tissues or fresh biopsy for intended retrospective FGFR2 expression analyses. Patients were excluded if they had a history of allergic reactions to monoclonal antibody therapy, anticancer chemotherapy, immunotherapy, or experimental cancer therapy (including clinical trials) within 3 weeks prior to the first dose of study drug. Full eligibility criteria are reported in the Electronic Supplementary Material Appendix.
2.2 Objectives
The primary objectives were to determine the safety, tolerability, and MTD of aprutumab ixadotin. Secondary objectives included evaluation of pharmacokinetics, pharmacodynamics, immunogenicity, and tumor response.
2.3 Study Design
This was a phase I, open-label, first-in-human, non-randomized, multicenter trial. The trial was planned to include two phases, the first being a dose-escalation phase to determine the MTD of aprutumab ixadotin, and the second an expansion phase to further evaluate safety, pharmacokinetics, and clinical activity at the MTD.
Aprutumab ixadotin was administered as an intravenous infusion over 1 h on day 1 of every 21-day cycle, with the first infusion given on cycle 1, day 1. Treatment continued until evidence of disease progression, unacceptable toxicity, withdrawal of consent, or patient withdrawal from the trial. The aprutumab ixadotin dose could be reduced or delayed to manage dose-limiting toxicities (DLTs) or adverse events (AEs). Treatment was stopped if more than two dose reductions were required.
Dose escalation and MTD determination were conducted using a previously described adaptive dose-escalation trial design [29]. The MTD was defined as the maximum dose at which the incidence of DLTs during cycle 1 was below 20%, or the maximum dose administered, whichever was achieved first during dose escalation. DLTs were defined as any of the following AEs occurring during cycle 1 and considered related to study drug: grade 4 absolute neutrophil count decrease for ≥ 7 days; grade ≥ 3 febrile neutropenia; grade 4 thrombocytopenia for > 1 day; grade 4 anemia or anemia requiring blood transfusion; grade ≥ 3 hemorrhage; grade ≥ 3 thrombotic event; any other non-hematological toxicity considered to be drug-related, excluding grade ≥ 3 nausea and vomiting controllable by anti-emetics within 3 days; grade 3 fatigue lasting ≤ 72 h; isolated change in laboratory biochemical values; and grade 3 infusion-related reactions resolving within 6 h and controlled by medical management.
Figure 1 shows the patient disposition of the dose-escalation cohorts. Five dose-escalation cohorts of at least three patients were enrolled (where multiple sites conducted the dose escalation, there was an option to enroll four patients). The first four patients of each cohort were enrolled with a safety interval of at least 48 h between drug administrations on cycle 1, day 1. The starting dose was 0.1 mg/kg body weight, with doses increased in two-fold increments up to 0.8 mg/kg, after which the dose was escalated in 0.5 mg/kg increments. All participants in a dose-level cohort must have received one complete infusion of study drug and have safety data available for cycle 1, or had discontinued early, before the next cohort was initiated. Modeling of DLTs was carried out after at least one DLT was reported or grade ≥ 2 drug-related AE (except asymptomatic changes in biochemistry laboratory values) was reported in two different patients in the first 21 days of treatment in cycle 1. The five dose-escalation cohorts enrolled were 0.1, 0.2, 0.4, 0.8, and 1.3 mg/kg.

Patient disposition and analysis population
2.4 Safety Assessments
Patients were assessed for safety on days 1, 2, 3, 5, 8, 15, and 21 of cycles 1 and 3, and days 1, 8, 15, and 21 of cycle 2, cycle 4, and subsequent cycles. End-of-treatment visits were also conducted at the time of study drug discontinuation or 14 days after last treatment, with an end-of-study visit or phone call taking place 30 days after the last treatment with study drug. Patients had ophthalmic evaluation prior to initiation of study drug and on day 15 of each cycle. AEs were reported using the National Cancer Institute Common Terminology Criteria for AEs (NCI-CTCAE) version 4.03. The study protocol was amended to include dose modification as well as remedial treatment guidelines for corneal toxicity.
2.5 Pharmacokinetics
Serial blood samples for pharmacokinetic characterization of aprutumab ixadotin (total drug), total antibody, and toxophore metabolite BAY 1159184 were collected during cycles 1 and 3 at the following timepoints: pre-dose, 0.5, 1 (end-of-infusion sample), 1.5, 2, 3, 5, 8, 24, 48, 96, 168, 336, and 504 h after the start of infusion, with the 504-h sample being collected before the start of cycles 2 and 4, respectively. Plasma concentrations were determined by enzyme immunoassay (EIA) for aprutumab ixadotin and total antibody and by liquid chromatography–tandem mass spectrometry (LC–MS/MS) for the toxophore metabolite BAY 1159184. If a patient discontinued study after cycle 3, day 21, the 504-h blood sample was collected on cycle 3, day 21. Starting with cycle 5, day 1, samples were collected before infusion with aprutumab ixadotin and at the end of infusion, in every second cycle. Tumor tissue samples were planned to be analyzed for FGFR2 overexpression; however, this was not conducted.
2.6 Tumor Response Assessment
Tumors were evaluated using computed tomography or magnetic resonance imaging; response was assessed using Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1.
3 Results
3.1 Patient Characteristics
Twenty patients were enrolled at three sites in the USA and one site in Korea, and treated with aprutumab ixadotin. The median age was 52 years (range 24–76 years) and 55% were female (n = 11). The four most common tumor types were colorectal cancer (n = 5; 25%), cholangiocarcinoma (n = 4; 20%), gastric cancer (n = 2; 10%), and hepatocellular carcinoma (n = 2; 10%). Most patients had an ECOG PS of 1 (n = 14; 70%). The median number of prior systemic therapies was eight (range 1–23). Four patients had FGFR2 amplification according to institution-led DNA copy number analysis. Patient demographics and characteristics are described in Table 1.
3.2 Dose Escalation
All patients enrolled were assigned to one of five dose-escalation cohorts (0.1 mg/kg [n = 4], 0.2 mg/kg [n = 3], 0.4 mg/kg [n = 4], 0.8 mg/kg [n = 4], or 1.3 mg/kg [n = 5]) and completed the study. Fifteen patients discontinued treatment due to progressive disease (75%) and five discontinued due to AEs (25%). The longest treatment duration observed was 13 cycles, which included one dose reduction from 0.8 to 0.4 mg/kg after the first cycle due to grade 4 thrombocytopenia and one dose interruption between cycles 10 and 11 due to reactivation of hepatitis B that was controlled.
Patients in the lower dose cohorts of 0.1, 0.2, and 0.4 mg/kg did not experience any DLTs. DLTs were observed in one patient in the 0.8 mg/kg group and three of five patients in the 1.3 mg/kg group. The patient in the 0.8 mg/kg group had grade 4 thrombocytopenia; of the three patients in the 1.3 mg/kg group, one patient had grade 3 proteinuria, one patient had grade 3 proteinuria and grade 4 thrombocytopenia, and a third patient had grade 3 corneal epithelial microcysts and grade 2 blurred vision.
After three of five patients in the 1.3 mg/kg group and one patient in the 0.8 mg/kg group experienced DLTs, it was decided that new patients should only be enrolled at a starting dose of 0.2 mg/kg or lower due to the high degree of uncertainty regarding the time course and degree of reversibility of these toxicities. Since two of the patients in the 1.3 mg/kg group experienced grade 3 proteinuria, dose levels of 0.4 and 0.8 mg/kg were not considered safe due to lack of quantitative data for protein in urine and limited exposure as the majority of patients discontinued; therefore, a dose of 0.2 mg/kg was determined to be the MTD. At 0.2 mg/kg, the efficacy profile of the drug did not favor further development; therefore, the study was discontinued by the sponsor and the dose-expansion phase was not carried out.
3.3 Safety
All patients had at least one treatment-emergent AE (TEAE) of any grade (100%), with at least one grade ≥ 3 TEAE reported in 15 patients (75%; Table 2). Grade 3 TEAEs occurred in 13 patients (65%), and grade 4 TEAEs in two patients (10%). The most common grade 3 TEAEs were thrombocytopenia, increased aspartate transaminase (AST), increased blood alkaline phosphatase (ALP) levels, proteinuria, and anemia. The only grade 4 TEAE was thrombocytopenia in two patients (10%); no grade 5 TEAEs were reported.
TEAEs determined to be drug-related were reported by 17 patients (85%), and nine patients (45%) reported grade ≥ 3 drug-related TEAEs. The most common of these were thrombocytopenia (15%), anemia (10%), increased AST (10%), and proteinuria (10%). Other grade 3 drug-related TEAEs were hypertension, increased alanine aminotransferase (ALT), decreased blood albumin, increased blood ALP levels, dehydration, decreased neutrophil count, and corneal epithelial microcysts, which occurred in one patient each (5%). Grade 4 drug-related thrombocytopenia was reported in two patients (10%).
Treatment-emergent serious AEs (SAEs) were reported in eight patients (40%), the most common being anorexia, thrombocytopenia, and proteinuria, each reported in two patients (10%). Drug-related SAEs were proteinuria and thrombocytopenia, both reported in two patients (10%), and anorexia and nausea in one patient each (5%).
One death due to colorectal cancer disease progression was reported during follow-up, after discontinuation of aprutumab ixadotin. The patient experienced two occurrences of grade 3 ascites that were considered to be unrelated to the study drug.
Five patients discontinued treatment due to one or more TEAEs (proteinuria, thrombocytopenia, dehydration, anemia, fatigue, worsening chronic renal disease, corneal epithelial microcysts, and blurred vision). Three patients had treatment interruptions due to TEAEs (proteinuria, gastroparesis, corneal epithelial microcysts, and blurred vision). Two patients had dose reductions due to thrombocytopenia.
3.4 Key Safety Observations
Proteinuria or nephrotic syndrome was reported in two of five patients after the cycle 1 at the 1.3 mg/kg dose level, with a time to onset of approximately 20 days. Proteinuria worsened in consecutive cycles, and both patients developed nephrotic syndrome. Both patients also showed abnormally high fibrinogen and D-dimer concentrations, while serum blood urea nitrogen-to-creatinine ratios were normal. Following these findings at the 1.3 mg/kg dose level, dipstick values from previous cohorts at the 0.1, 0.2, 0.4, and 0.8 mg/kg dose levels (where available) were retrospectively reviewed and plotted to analyze intensity and kinetics. Based on these dipstick test results, proteinuria was also observed in three of four patients at the 0.8 mg/kg dose level and in all four patients at the 0.4 mg/kg dose level. However, discontinuation of patients in cycle 2 due to early tumor progression precluded further characterization of the observed proteinuria. Due to the lack of quantitative values, the course and clinical relevance of proteinuria in these patients could not be interpreted. Additionally, the limited drug exposure and insufficient observation period in these patients precluded assessment of the nephrotoxic potential of aprutumab ixadotin at dose ranges of 0.4–0.8 mg/kg. Therefore, the 0.2 mg/kg dose level was regarded as non-nephrotoxic based on available data from three patients at the 0.2 mg/kg dose level. No signs of hematuria were observed using a dipstick test. Signs of recovery were observed 20 days after discontinuation of the study drug.
Ocular events occurred in eight patients (40%) across all dose groups, and included corneal epithelial microcysts, blurred vision, corneal deposits, retinal hemorrhage, and ocular discomfort. The ocular events manifested within the first two cycles and did not typically result in dosage changes, except in one patient for whom treatment was discontinued due to dose-limiting grade 3 corneal epithelial microcysts and grade 1 blurred vision. Ocular events considered to be drug-related included corneal deposits in four patients (deposits of lipids or calcium that build up on the cornea in layers resulting in blurred vision; 20%), corneal epithelial microcysts in three patients (incompletely formed cells in the epithelia that can cause vision hazing; 15%), and blurred vision in one patient (5%). These drug-related events were reported in two patients (50%) in the 0.8 mg/kg group (time to onset: 36 days) and in all five patients in the 1.3 mg/kg group (time to onset: 6–22 days). Remedial treatment for ocular events consisted of carmellose ophthalmic drops in concert with polyacrylic acid, difluprednate, or fluorometholone drops. Ocular symptoms had fully resolved for one patient, appeared to be improving in two patients, and showed no further worsening in three patients at the time of last study visit.
A transient decrease in platelet count was observed in one patient at the 0.1 mg/kg dose level during cycle 1. Thrombocytopenia was subsequently observed in all patients at the 0.4, 0.8, and 1.3 mg/kg dose levels. Thrombocytopenia was reported as a DLT in one patient each at both the 0.8 and 1.3 mg/kg dose levels. The time to onset and time to recovery of thrombocytopenia was consistent in consecutive cycles at all dose levels.
Increased serum transaminase levels were detected in cycle 1 at the 0.2 mg/kg dose level. The intensity and frequency of abnormally high transaminase levels tended to increase with the dose of aprutumab ixadotin. The highest levels were observed in one patient in each of the 0.8 and 1.3 mg/kg cohorts, with levels that were five times higher than the upper limit of normal. Elevated transaminase levels typically resolved within the same treatment cycle.
3.5 Pharmacokinetics
Initial pharmacokinetic analyses of 18 patients showed that the pharmacokinetic profile of the toxophore metabolite BAY 1159184 appeared approximately dose-proportional at doses from 0.4 to 1.3 mg/kg (Fig. 2). Dose-proportional increases in exposure to aprutumab ixadotin (total drug) at doses of 0.1–1.3 mg/kg were also observed (Fig. 2). The pharmacokinetic profiles of total antibody and FGFR2-ADC (aprutumab ixadotin, i.e., total drug) were similar, which may indicate that aprutumab ixadotin would be stable in plasma.

Geometric mean plasma concentrations of a aprutumab ixadotin, b total antibody, and c toxophore metabolite (BAY 1159184) during cycle 1 for the 0.1 (n = 4), 0.2, 0.4 (n = 3), 0.8 (n = 4), and 1.3 (n = 5) mg/kg dose cohorts. Data for the toxophore metabolite (BAY 1159184) are only available for the 0.4, 0.8, and 1.3 mg/kg dose cohorts. *Data are presented individually in the 0.2 mg/kg dose cohort due to the low number of patients with available pharmacokinetic data (n = 2)
3.6 Tumor Response
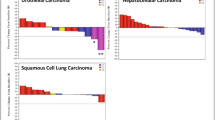
Stable disease was observed in one patient with tongue base adenoid cystic carcinoma assigned to the 0.8 mg/kg group whose dose was reduced after cycle 1–0.4 mg/kg (Fig. 3). Disease progression occurred in this patient during cycle 13. No partial or complete responses were observed in any patients.

Treatment duration in patients treated with aprutumab ixadotin. Dose reductions are indicated with a black arrow; dose reduction occurred a on day 22 from 0.8 to 0.4 mg/kg and b on day 23 from 1.3 to 0.8 mg/kg
4 Discussion
FGFR2 has been identified as a promising drug target because it is overexpressed in various tumor types [7,8,9, 11]. This phase I trial was initiated to investigate the safety of the anti-FGFR2 ADC aprutumab ixadotin (BAY 1187982), following positive in vitro and in vivo data [28], and to determine the MTD for subsequent dose-expansion cohorts. This study represented the first time an ADC had been used to target FGFR2 in the treatment of advanced tumors. Despite promising results in preclinical studies, safety data collected during this trial suggested that in a heavily pretreated population of patients with advanced solid tumors, aprutumab ixadotin was poorly tolerated with a MTD of 0.2 mg/kg every 3 weeks The activity of aprutumab ixadotin has been evaluated in a range of preclinical models, including assessment of antitumor activity and pharmacokinetics in mouse xenograft tumor models [28]. The pharmacokinetics of aprutumab ixadotin were also assessed in cynomolgus monkeys after single and repeat dosing, and a rat model was used to assess the toxophore-linker metabolite [30]. Results from preclinical mouse xenograft studies and pharmacokinetic modeling predicated that the minimum doses required for stable disease, partial response, and complete response were 0.4, 0.5, and 1.2 mg/kg, respectively [31]. As the MTD of aprutumab ixadotin in this trial was below the predicted minimum therapeutic dose established during PDX studies, the sponsor discontinued the trial after enrollment and treatment of 20 patients [31].
In non-clinical safety studies in cynomolgus monkey models, aprutumab ixadotin treatment led to increased ALT and AST levels; extrapolation of these data to humans led to an estimated highest non-severely toxic dose of aprutumab ixadotin of 1.48 mg/kg when administered every 3 weeks, as was applied in this trial. The predicted therapeutic index in humans was approximately 3.5, calculated using the highest non-severely toxic dose and the dose required to achieve stable disease [31]. These data supported the decision to further investigate aprutumab ixadotin in a first-in-human phase I trial.
The N-terminal epitope of FGFR2 is conserved between humans, monkeys, rats, and mice. The antibody moiety of aprutumab ixadotin binds to recombinant FGFR2 protein with a similar EC50 (half-maximal effective concentration) across these species, making any of them relevant for toxicity testing. Toxicity studies in rats and cynomolgus monkeys revealed effects typical of ADC-mediated toxicity related to toxophore activity. This was observed in the form of morphological degeneration/regeneration and inflammation in lung, liver, kidneys, cornea, and the lympho-hematopoietic system at sufficient multiples of exposure compared with the expected human efficacious exposure to suggest an acceptable therapeutic index; most effects were reversible and no functional organ toxicity was detected. Data from cynomolgus monkeys showed that aprutumab ixadotin had a small effect on blood clot formation in the form of a small and transient decrease in platelets that was restricted to the first treatment cycle; additionally, increases in thrombin time and d-dimer levels were observed. These effects were fully reversible. Similar effects on blood clot formation were induced by an isotype control ADC with an identical toxophore payload. No adverse effects were observed after administration of the payload metabolite alone, including in the coagulation system, indicating that the combination of the payload-linker metabolite and an antibody moiety may be the causative factor for the coagulation effects. To our knowledge, similar coagulation effects have not been seen in clinical trials of other ADCs or toxophores [32,33,34,35].
The most common TEAEs that occurred in patients treated with aprutumab ixadotin in this phase I trial included thrombocytopenia and increased levels of AST, which occurred in 50% and 60% of patients, respectively, with a further eight TEAEs occurring in 25% or more of patients across all dose cohorts. The safety data indicate the following undesirable effects of aprutumab ixadotin: proteinuria/nephrotic syndrome; thrombocytopenia; increased serum ALT and AST levels; and corneal epithelial microcysts. Patients treated with aprutumab ixadotin in this study were heavily pretreated, with a median number of prior therapies of eight (range 1–23), which may have contributed to the observed poor tolerance.
Patients in the 1.3 mg/kg dose cohort had d-dimer levels notably higher in the second cycle than during the first cycle of treatment with aprutumab ixadotin. d-Dimer elevation is thought to be caused by kidney damage leading to nephrotic syndrome [36]; during the second cycle it was concluded that kidney recovery did not seem to balance the kidney damage. Furthermore, patients with nephrotic syndrome have a significantly higher risk of thrombotic events and renal failure [37]. Proteinuria and nephrotic syndrome may be considered as toxicities specifically associated with aprutumab ixadotin treatment, as similar toxicities have not been previously reported in clinical trials of other ADCs that carry a range of payloads [38,39,40,41]. AEs reported for pan-FGFR inhibitors and FGFR1–3 specific inhibitors include hyperphosphatemia, stomatitis, mucosal dryness, nail changes with onycholysis, hair modifications, and ocular disorders (dry eye and keratitis); asymptomatic retinal pigment epithelial detachment has also been described for the FGFR1–3 inhibitor BGJ398. Thus, while dry eye and corneal toxicity has been reported with FGFR inhibitors, kidney toxicity has not been previously observed [42,43,44,45,46]. The similar temporal pattern of the course of proteinuria in patients in this trial and the lack of alternative explanation for this abnormality support the causal relationship between observed proteinuria/nephrotic syndrome and aprutumab ixadotin, although this association was not seen in non-clinical safety studies conducted in rats and cynomolgus monkeys.
Ocular toxicities seem to be a class effect of ADCs with microtubule-disrupting payloads, with corneal epitheliopathy reported during treatment with both maytansinoid (DM4 payload metabolite) and auristatin-containing (monomethylauristatin F payload metabolite) ADCs [39,40,41, 47, 48]. As observed in the current study, corneal epitheliopathy typically resolves with supportive care and treatment interruption or discontinuation.
Although efficacy was not formally evaluated during the trial, one patient with tongue base adenoid cystic carcinoma had a best response of stable disease and remained on treatment until disease progression during cycle 14. This patient was enrolled in the 0.8 mg/kg dose cohort and had a dose reduction to 0.4 mg/kg to manage toxicities during cycle 2. No other responses were observed in the four patients (two cases of gastric cancer and one case each of cholangiocarcinoma and colorectal cancer) with known FGFR2 amplifications. Most patients discontinued treatment because of disease progression.
5 Conclusion
The ADC aprutumab ixadotin was found to be poorly tolerated in this phase I first-in-human trial, with an MTD of 0.2 mg/kg, which is below the estimated minimum therapeutic dose predicted in preclinical studies. The safety profile of this ADC in humans also differed markedly from that observed in animal models, with a high rate of proteinuria and nephropathy. These results underline the need to develop new methods to predict the effects of investigational ADCs and their metabolites in humans during preclinical development.
References
Wesche J, Haglund K, Haugsten EM. Fibroblast growth factors and their receptors in cancer. Biochem J. 2011;437(2):199–213. https://doi.org/10.1042/bj20101603.
De Moerlooze L, Spencer-Dene B, Revest JM, Hajihosseini M, Rosewell I, Dickson C. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal–epithelial signalling during mouse organogenesis. Development. 2000;127(3):483–92.
Matsuda Y, Yoshimura H, Suzuki T, Uchida E, Naito Z, Ishiwata T. Inhibition of fibroblast growth factor receptor 2 attenuates proliferation and invasion of pancreatic cancer. Cancer Sci. 2014;105(9):1212–9. https://doi.org/10.1111/cas.12470.
Hattori Y, Itoh H, Uchino S, Hosokawa K, Ochiai A, Ino Y, et al. Immunohistochemical detection of K-sam protein in stomach cancer. Clin Cancer Res. 1996;2(8):1373–81.
Carter EP, Fearon AE, Grose RP. Careless talk costs lives: fibroblast growth factor receptor signalling and the consequences of pathway malfunction. Trends Cell Biol. 2015;25(4):221–33. https://doi.org/10.1016/j.tcb.2014.11.003.
Andre F, Cortes J. Rationale for targeting fibroblast growth factor receptor signaling in breast cancer. Breast Cancer Res Treat. 2015;150(1):1–8. https://doi.org/10.1007/s10549-015-3301-y.
Deng N, Goh LK, Wang H, Das K, Tao J, Tan IB, et al. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut. 2012;61(5):673–84. https://doi.org/10.1136/gutjnl-2011-301839.
Dienstmann R, Rodon J, Prat A, Perez-Garcia J, Adamo B, Felip E, et al. Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors. Ann Oncol. 2014;25(3):552–63. https://doi.org/10.1093/annonc/mdt419.
Kim S, Dubrovska A, Salamone RJ, Walker JR, Grandinetti KB, Bonamy GM, et al. FGFR2 promotes breast tumorigenicity through maintenance of breast tumor-initiating cells. PLoS One. 2013;8(1):e51671. https://doi.org/10.1371/journal.pone.0051671.
Martignetti JA, Camacho-Vanegas O, Priedigkeit N, Camacho C, Pereira E, Lin L, et al. Personalized ovarian cancer disease surveillance and detection of candidate therapeutic drug target in circulating tumor DNA. Neoplasia. 2014;16(1):97–103.
Turner N, Lambros MB, Horlings HM, Pearson A, Sharpe R, Natrajan R, et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene. 2010;29(14):2013–23. https://doi.org/10.1038/onc.2009.489.
Lee HJ, Kang HJ, Kim KM, Yu ES, Kim KH, Kim SM, et al. Fibroblast growth factor receptor isotype expression and its association with overall survival in patients with hepatocellular carcinoma. Clin Mol Hepatol. 2015;21(1):60–70. https://doi.org/10.3350/cmh.2015.21.1.60.
Matsuda Y, Ishiwata T, Yamahatsu K, Kawahara K, Hagio M, Peng WX, et al. Overexpressed fibroblast growth factor receptor 2 in the invasive front of colorectal cancer: a potential therapeutic target in colorectal cancer. Cancer Lett. 2011;309(2):209–19. https://doi.org/10.1016/j.canlet.2011.06.009.
Nomura S, Yoshitomi H, Takano S, Shida T, Kobayashi S, Ohtsuka M, et al. FGF10/FGFR2 signal induces cell migration and invasion in pancreatic cancer. Br J Cancer. 2008;99(2):305–13. https://doi.org/10.1038/sj.bjc.6604473.
Ohashi R, Matsuda Y, Ishiwata T, Naito Z. Downregulation of fibroblast growth factor receptor 2 and its isoforms correlates with a high proliferation rate and poor prognosis in high-grade glioma. Oncol Rep. 2014;32(3):1163–9. https://doi.org/10.3892/or.2014.3283.
Tokunaga R, Imamura Y, Nakamura K, Ishimoto T, Nakagawa S, Miyake K, et al. Fibroblast growth factor receptor 2 expression, but not its genetic amplification, is associated with tumor growth and worse survival in esophagogastric junction adenocarcinoma. Oncotarget. 2016;7(15):19748–61. https://doi.org/10.18632/oncotarget.7782.
Parker BC, Engels M, Annala M, Zhang W. Emergence of FGFR family gene fusions as therapeutic targets in a wide spectrum of solid tumours. J Pathol. 2014;232(1):4–15.
Sievers EL, Senter PD. Antibody–drug conjugates in cancer therapy. Annu Rev Med. 2013;64:15–29. https://doi.org/10.1146/annurev-med-050311-201823.
Mack F, Ritchie M, Sapra P. The next generation of antibody drug conjugates. Semin Oncol. 2014;41(5):637–52. https://doi.org/10.1053/j.seminoncol.2014.08.001.
Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat Rev Drug Discov. 2017;16(5):315–37. https://doi.org/10.1038/nrd.2016.268.
Lambert JM, Morris CQ. Antibody-drug conjugates (ADCs) for personalized treatment of solid tumors: a review. Adv Ther. 2017;34(5):1015–35. https://doi.org/10.1007/s12325-017-0519-6.
LoRusso PM, Weiss D, Guardino E, Girish S, Sliwkowski MX. Trastuzumab emtansine: a unique antibody–drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clin Cancer Res. 2011;17(20):6437–47. https://doi.org/10.1158/1078-0432.ccr-11-0762.
Moskowitz CH, Nademanee A, Masszi T, Agura E, Holowiecki J, Abidi MH, et al. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2015;385(9980):1853–62. https://doi.org/10.1016/s0140-6736(15)60165-9.
Castaigne S, Pautas C, Terre C, Raffoux E, Bordessoule D, Bastie JN, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet. 2012;379(9825):1508–16. https://doi.org/10.1016/s0140-6736(12)60485-1.
Kantarjian HM, DeAngelo DJ, Stelljes M, Martinelli G, Liedtke M, Stock W, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740–53. https://doi.org/10.1056/NEJMoa1509277.
Hedrich WD, Fandy TE, Ashour HM, Wang H, Hassan HE. Antibody-drug conjugates: pharmacokinetic/pharmacodynamic modeling, preclinical characterization, clinical studies, and lessons learned. Clin Pharmacokinet. 2018;57(6):687–703. https://doi.org/10.1007/s40262-017-0619-0.
Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat Rev Drug Discov. 2018;17(3):197–223. https://doi.org/10.1038/nrd.2017.227.
Sommer A, Kopitz C, Schatz CA, Nising CF, Mahlert C, Lerchen HG, et al. Preclinical efficacy of the auristatin-based antibody–drug conjugate BAY 1187982 for the treatment of FGFR2-positive solid tumors. Cancer Res. 2016;76(21):6331–9. https://doi.org/10.1158/0008-5472.CAN-16-0180.
Tibaldi C, Vasile E, Antonuzzo A, Di Marsico R, Fabbri A, Innocenti F, et al. First line chemotherapy with planned sequential administration of gemcitabine followed by docetaxel in elderly advanced non-small-cell lung cancer patients: a multicenter phase II study. Br J Cancer. 2008;98(3):558–63. https://doi.org/10.1038/sj.bjc.6604187.
Data on file, Bayer AG (2019)
Wittemer-Rump S, Sommer A, Kopitz C, Huynh H, Schatz C, Zierz R, et al. Pharmacokinetic/pharmacodynamic (PK/PD) and toxicokinetic/toxicodynamic (TK/TD) modeling of preclinical data of FGFR2-ADC (BAY 1187982) to guide dosing in phase 1 [abstract]. In: Proceedings of the 106th annual meeting of the American Association for Cancer Research, 18–22 Apr 2015, Philadelphia, PA. Philadelphia, PA: AACR; Cancer Res 2015;75(15 Suppl):Abstract nr 1683. https://doi.org/10.1158/1538-7445.AM2015-1683
Younes A, Gopal AK, Smith SE, Ansell SM, Rosenblatt JD, Savage KJ, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J Clin Oncol. 2012;30(18):2183–9. https://doi.org/10.1200/jco.2011.38.0410.
Amadori S, Suciu S, Selleslag D, Aversa F, Gaidano G, Musso M, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol. 2016;34(9):972–9. https://doi.org/10.1200/jco.2015.64.0060.
Bross PF, Beitz J, Chen G, Chen XH, Duffy E, Kieffer L, et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res. 2001;7(6):1490–6.
Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367(19):1783–91. https://doi.org/10.1056/NEJMoa1209124.
Sexton DJ, Clarkson MR, Mazur MJ, Plant WD, Eustace JA. Serum D-dimer concentrations in nephrotic syndrome track with albuminuria, not estimated glomerular filtration rate. Am J Nephrol. 2012;36(6):554–60. https://doi.org/10.1159/000345475.
Mahmoodi BK, ten Kate MK, Waanders F, Veeger NJ, Brouwer JL, Vogt L, et al. High absolute risks and predictors of venous and arterial thromboembolic events in patients with nephrotic syndrome: results from a large retrospective cohort study. Circulation. 2008;117(2):224–30. https://doi.org/10.1161/circulationaha.107.716951.
Stagg NJ, Shen BQ, Brunstein F, Li C, Kamath AV, Zhong F, et al. Peripheral neuropathy with microtubule inhibitor containing antibody drug conjugates: challenges and perspectives in translatability from nonclinical toxicology studies to the clinic. Regul Toxicol Pharmacol. 2016;82:1–13. https://doi.org/10.1016/j.yrtph.2016.10.012.
Eaton JS, Miller PE, Mannis MJ, Murphy CJ. Ocular adverse events associated with antibody–drug conjugates in human clinical trials. J Ocul Pharmacol Ther. 2015;31(10):589–604. https://doi.org/10.1089/jop.2015.0064.
Younes A, Kim S, Romaguera J, Copeland A, Farial Sde C, Kwak LW, et al. Phase I multidose-escalation study of the anti-CD19 maytansinoid immunoconjugate SAR3419 administered by intravenous infusion every 3 weeks to patients with relapsed/refractory B-cell lymphoma. J Clin Oncol. 2012;30(22):2776–82. https://doi.org/10.1200/jco.2011.39.4403.
Tannir NM, Forero-Torres A, Ramchandren R, Pal SK, Ansell SM, Infante JR, et al. Phase I dose-escalation study of SGN-75 in patients with CD70-positive relapsed/refractory non-Hodgkin lymphoma or metastatic renal cell carcinoma. Investig New Drugs. 2014;32(6):1246–57. https://doi.org/10.1007/s10637-014-0151-0.
Chae YK, Ranganath K, Hammerman PS, Vaklavas C, Mohindra N, Kalyan A, et al. Inhibition of the fibroblast growth factor receptor (FGFR) pathway: the current landscape and barriers to clinical application. Oncotarget. 2017;8(9):16052–74. https://doi.org/10.18632/oncotarget.14109.
Nogova L, Sequist LV, Perez Garcia JM, Andre F, Delord JP, Hidalgo M, et al. Evaluation of BGJ398, a fibroblast growth factor receptor 1–3 kinase inhibitor, in patients with advanced solid tumors harboring genetic alterations in fibroblast growth factor receptors: results of a global phase I, dose-escalation and dose-expansion study. J Clin Oncol. 2017;35(2):157–65. https://doi.org/10.1200/JCO.2016.67.2048.
Paik PK, Shen R, Berger MF, Ferry D, Soria JC, Mathewson A, et al. A phase Ib open-label multicenter study of AZD4547 in patients with advanced squamous cell lung cancers. Clin Cancer Res. 2017;23(18):5366–73. https://doi.org/10.1158/1078-0432.CCR-17-0645.
Touat M, Ileana E, Postel-Vinay S, Andre F, Soria JC. Targeting FGFR signaling in cancer. Clin Cancer Res. 2015;21(12):2684–94. https://doi.org/10.1158/1078-0432.CCR-14-2329.
Markham A. Erdafitinib: first global approval. Drugs. 2019;79(9):1017–21. https://doi.org/10.1007/s40265-019-01142-9.
Donaghy H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody–drug conjugates. MAbs. 2016;8(4):659–71. https://doi.org/10.1080/19420862.2016.1156829.
Masters JC, Nickens DJ, Xuan D, Shazer RL, Amantea M. Clinical toxicity of antibody drug conjugates: a meta-analysis of payloads. Investig New Drugs. 2018;36(1):121–35. https://doi.org/10.1007/s10637-017-0520-6.
Acknowledgements
The authors thank the patients, their families, and all investigators involved in this study. We would also like to thank Ruprecht Zierz of Bayer AG for providing the preclinical toxicology data and for his review and feedback on this manuscript.
Author information
Authors and Affiliations
Contributions
Conception and design: S-BK and FR. Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): FM-B, S-BK, AK, and JB. Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): FM-B, S-BK, and TT. Writing, review, and/or revision of the manuscript: S-BK, FM-B, AK, AB, RL, TT, AS, MO, FR, DL, SW-R, and JB. Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): S-BK and TT.
Corresponding author
Ethics declarations
Funding
This study was funded by Bayer Healthcare. Medical writing support, including assisting authors with the development of the initial draft and incorporation of comments, was provided by Karl Kemp-O’Brien, Ph.D, and editorial support, including referencing, formatting, and proofreading, was provided by Ian Norton, Ph.D and Annabel Ola, MSc, all of Scion, London, UK, supported by Bayer HealthCare according to Good Publication Practice guidelines. The Sponsor was involved in the study design, collection, analysis, and interpretation of data, as well as data checking of information provided in the manuscript. However, ultimate responsibility for opinions, conclusions, and data interpretation lies with the authors.
Conflict of interest
A. Sommer, F. Reetz, A. Babich, S. Wittemer-Rump, and R. Liu are shareholders and employees of Bayer AG. T. Tanigawa is an employee of Bayer Yakuhin. M. Osada and D. Laurent were employees of Bayer AG, Berlin, Germany, during the conduct of the study. M. Osada now works at Merck Serono, Tokyo, Japan. D. Laurent now works at Berlin-Chemie, Berlin, Germany. S.-B. Kim reports receiving institutional research funding from Novartis, Sanofi-Genzyme, Kyowa Kirin Inc., and Dongkook Pharma Co Ltd. F. Meric-Bernstam reports receiving commercial research grants from Novartis, AstraZeneca, Taiho, Genentech, Calithera, Debio International Group, Bayer, PUMA, Aileron, Jounce, CytoMx, Effector, Zymeworks, Curis, and Pfizer, and is a consultant/advisory board member for Dialecta, Sumitomo Dainippon, Pieris Pharmaceuticals, Darwin Health, Samsung Bioepis, Aduro, Spectrum, OrigiMed, Debiopharm International, Inflection Biosciences, Xencor, and Genentech. J. Berlin reports consultancy fees and institutional research funding from Genentech, EMD Serono, 5Prime, BeiGene, Karyopharm, and Symphogen, honoraria from Nestlé Health Science, consultancy fees from Celgene, Cornerstone, Exelis, Gritstone Oncology, ERYTECH Pharma, AstraZeneca, Arno Therapeutics, Symphogen, Abbvie, and Eisai, and institutional research funding from Immunomedics, Gilead, Taiho, Loxo, Bayer, Incyte, and Pharmacyclics. A. Kalyan reports advisory boards for BMS, Exelis, Ipsen, and Eisai.
Ethical approval and consent to participate
All patients provided written informed consent. The study was conducted in accordance with the Declaration of Helsinki. The protocol was approved by the institutional review boards (IRBs) of the participating centers: Asan Medical Center IRB (Republic of Korea), Chesapeake IRB, Vanderbilt IRB, Vanderbilt University Human Research Protection Program, and University of Texas, MD Anderson Cancer Center IRB (USA).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kim, SB., Meric-Bernstam, F., Kalyan, A. et al. First-in-Human Phase I Study of Aprutumab Ixadotin, a Fibroblast Growth Factor Receptor 2 Antibody–Drug Conjugate (BAY 1187982) in Patients with Advanced Cancer. Targ Oncol 14, 591–601 (2019). https://doi.org/10.1007/s11523-019-00670-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-019-00670-4