
Abstract
A method combining ultrasound-assisted emulsification–microextraction (USAEME) with gas chromatography–mass spectrometry (GC–MS) was developed for simultaneous determination of four acidic pharmaceuticals, ibuprofen, naproxen, ketoprofen, and diclofenac, as well as four phenols, 4-octylphenol, 4-n-nonylphenol, bisphenol A, and triclosan in municipal wastewaters. Conditions of extraction and simultaneous derivatization were optimized with respect to such aspects as type and volume of extraction solvent, volume of derivatization reagent, kind and amount of buffering salt, location of the test tube in the ultrasonic bath, and extraction time. The average correlation coefficient of the calibration curves was 0.9946. The LOD/(LOQ) values in influent and effluent wastewater were in the range of 0.002–0.121/(0.005–0.403) μg L−1 and 0.002–0.828/(0.006–2.758) μg L−1, respectively. Quantitative recoveries (≥94 %) and satisfactory precision (average RSD 8.2 %) were obtained. The optimized USAEME/GC–MS method was applied for determination of the considered pharmaceuticals and phenols in influents and treated effluents from nine Polish municipal wastewater treatment plants. The average concentration of acidic pharmaceuticals in influent and effluent wastewater were in the range of 0.06–551.96 μg L−1 and 0.01–22.61 μg L−1, respectively, while for phenols were in the range of 0.03–102.54 μg L−1 and 0.02–10.84 μg L−1, respectively. The removal efficiencies of the target compounds during purification process were between 84 and 99 %.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Widespread accessibility and ever-increasing use of pharmaceuticals as well as cosmetics and cleaning agents have resulted in the appearance and increase in the concentration of a great number of new chemical compounds in sewage produced by humans. Under the current legal regulations, it is not obligatory to determine the content of pharmaceuticals or other organic trace contaminants in wastewater. Nor is the presence of these compounds in the wastewater taken into account in the planning and management of the sewage treatment process. As a result, different types of compounds, including pharmaceuticals and endocrine-disrupting compounds, penetrate into the environment posing a serious risk not only to aquatic organisms but also to humans. For that reason, it is essential to analyze the content of these compounds in wastewater.
Ibuprofen, naproxen, ketoprofen, and diclofenac are non-steroidal anti-inflammatory drugs (NSAIDs). They have been widely used not only to treat various inflammatory disorders and for pain relief but also for their antipyretic effect. Nearly all of these substances are available without a doctor’s prescription at lower doses. Publications regarding NSAIDs in the environment confirm their toxicity to many animal species (Oaks et al. 2004). Substances such as 4-octylphenol, 4-n-nonylphenol, bisphenol A, and triclosan are included into the group of endocrine-disrupting compounds (Esplugas et al. 2007). 4-Octylphenol and 4-n-nonylphenol are used for the production of elasticizers, technical-grade abstergents, pesticide emulsifiers, as well as trimming in spinning and weaving. Bisphenol A is a raw material for producing epoxide resins, polycarbonates, and polystyrene resins, which has been used to stabilize phenol formaldehyde. Triclosan is used as an ingredient in personal hygiene and household products such as soaps, toothpaste, mouthwash, deodorants, detergents, and disinfecting lotions.
Determining pharmaceuticals and endocrine-disrupting compounds in wastewater and other environmental samples is a very difficult task due to low concentrations and complex matrix of the substances. The highest concentrations of the analyzed compounds reaching several dozens of micrograms per liter have been recorded for wastewater entering treatment plants (Gómez et al. 2007b; Ramos Payán et al. 2010; Regueiro et al. 2009). In effluents, the concentration of particular compounds has been found to be from a few to hundreds of times lower than in the influent wastewater (Gómez et al. 2007b; Yiantzi et al. 2010). Analysis of the compounds in rivers showed the concentrations of the contaminants reaching several hundred nanograms per liter, in lake waters up to tens of nanograms per liter, and in the sea up to several nanograms per liter (Beck et al. 2005; Ozcan et al. 2010; Regueiro et al. 2009).
The technique most extensively used for the isolation of target compounds from environmental samples is solid-phase extraction (SPE) (Ballesteros et al. 2006; Gatidou et al. 2007; Gómez et al. 2007a, b; Gracia-Lor et al. 2010; Liu et al. 2004; Yu and Wu 2013). In the case of acidic pharmaceuticals, other techniques have also been applied, such as liquid–liquid microextraction (LLME), hollow fiber liquid-phase microextraction (HF-LPME), and microwave-assisted extraction (MAE) (Antonić and Heath 2006; Quintana et al. 2004; Ramos Payán et al. 2010). To isolate target phenols, apart from SPE and solid-phase microextraction (SPME), stir bar sorptive extraction (SBSE) and LLME and its variants, dispersive liquid–liquid microextraction (DLLME) and vortex-assisted liquid–liquid microextraction (VALLME), are also applied (Kawaguchi et al. 2008; Regueiro et al. 2009; Yiantzi et al. 2010; Zgoła-Grześkowiak 2010). The final stage of the determination of pharmaceuticals and phenols in water samples requires extremely sensitive and selective techniques. Currently, for this purpose primarily chromatographic methods are used, i.e., gas chromatography (GC), high-performance liquid chromatography (HPLC), and ultrahigh-pressure liquid chromatography (UHPLC). The detector typically employed in conjunction with chromatographic techniques is mass spectrometer (MS). Another detector applied in gas chromatography assays is flame ionization detector (FID), while in HPLC the diode array detector (DAD), fluorescence detector (FLD), and ultraviolet detector (UV) are used.
Ultrasound-assisted emulsification–microextraction reported by Regueiro et al. (2008) is one of the LLME modes used in addition to the techniques such as DLLME, VALLME, HF-LPME, and single-drop microextraction (SDME). Liquid-phase microextraction techniques are now widely employed due to a great number of advantages that they offer, such as low consumption of organic solvents, simplicity of experiment, high extraction efficiency, and low costs. In USAEME, a small amount of organic solvent of the order of microliters is introduced into water sample and the system is treated with ultrasound. The organic solvent dissipates into microdroplets, thus creating a considerably large contact area of both the aqueous and organic phases. The use of ultrasound facilitates the emulsification process and speeds up the determination of equilibrium between the two phases. Immediately after the extraction, centrifugation is applied for phase separation and organic extract is analyzed. The use of ultrasound makes it possible to bypass the disadvantages connected with other microextraction techniques, such as a relatively long extraction time in VALLME, HF-LPME, and SDME as well as the necessity of using relatively large amounts of disperser solvent in DLLME. Owing to its many advantages, USAEME has gained a considerable significance since its first use 5 years ago. The technique has been used, among others, to determine polycyclic aromatic hydrocarbons (PAH), pesticides, polychlorinated biphenyls (PCBs), polybrominated diphenylethers (PBDE), haloanisoles, bisphenols, chlorinated phenoxyacetic acids as well as inorganic selenium (Fontana et al. 2009; Fontana and Altamirano 2010; Najafi et al. 2012; Ozcan et al. 2009a, b, 2010; Pizarro et al. 2012a; Viñas et al. 2012; Zhang and Lee 2012). The determination process has been mainly conducted in environmental waters (Fontana et al. 2009; Fontana and Altamirano 2010; Najafi et al. 2012; Ozcan et al. 2009a, b, 2010; Zhang and Lee 2012) and also in wine (Pizarro et al. 2012b), paper, toys, and baby products (Viñas et al. 2012). USAEME procedures generally make use of extraction solvents denser than water, e.g., chloroform (Fontana et al. 2009; Fontana and Altamirano 2010; Ozcan et al. 2009a). Utilization of solvents of lower densities than water requires special dishes or additional procedural steps to enable the separation of the solvent from water samples and consequently may lead to the deterioration of reproducibility and accuracy of final results (Pizarro et al. 2012b; Zhang and Lee 2012). The solidification of a floating drop of a low-density solvent requires employing a larger volume of extracting medium in comparison with a traditional USAEME procedure (Pizarro et al. 2012a).
In this study, a simple and sensitive analytical procedure for simultaneous determination of acidic pharmaceuticals and phenols at low concentrations has been optimized. USAEME has been used for the isolation whereas GC–MS in the selected ion monitoring (SIM) mode has been applied for the separation and determination of analytes. The effects of various extraction and derivatization parameters, i.e., the type of organic solvent and solvent volume, extraction time, location of test tube in ultrasonic bath, derivatization reagent volume, and kind and amount of buffering salt, were investigated. The developed USAEME/GC–MS method was employed to determine target compounds in influents and treated effluents from wastewater treatment plants of nine cities in Poland.
Experimental
Reagents and solvents
Ibuprofen (IBP), naproxen (NAP), ketoprofen (KET), diclofenac (DIC), 4-octylphenol (OP), 4-n-nonylphenol (NP), bisphenol A (BPA), pyrene (PYR), 1,2-dichlorobenzene (1,2-DCB), and decane (DEC) were obtained from Sigma-Aldrich (Germany). Triclosan (TRC) was obtained from Fluka (Italy). Carbon tetrachloride (CTC) was purchased from Merck (Germany). Chloroform (CHL), toluene (TOL), acetic anhydride, sodium hydrogen phosphate (V), and methanol were obtained from POCH (Poland). Series of C10–C22 n-alkanes used to determine retention indices of target compounds were purchased from Fluka (Switzerland).
Stock solutions containing all analytes (at 1 mg mL−1 of each) were prepared in methanol and stored at −18 °C for not longer than 1 month. Working solutions were prepared by diluting the stock standard solution in methanol and stored at −18 °C for not longer than 2 weeks. Deionized water was from purification system (Milli-Q RG, Millipore, USA) and was stored in a glass bottle.
Wastewater samples
The wastewater samples were obtained from wastewater treatment plants (WWTPs) in nine cities in Poland. In these plants, the treatment process includes both mechanical purification and biological purification with the use of activated sludge. Figure 1 shows the locations and average daily capacity \( \left({Q}_{\overline{\mathrm{d}}}\right) \) of the mentioned wastewater treatment plants. Average daily samples of influents and treated effluents were collected in glass bottles and transported to the laboratory. Upon arrival, the samples were filtered through a membrane filter with 0.45 μm pore size and acidified with concentrated hydrochloric acid to pH = 2. Later, the wastewater samples were stored at −18 °C.

Location and average daily capacity \( \left({\mathrm{Q}}_{\overline{\mathrm{d}}}\right) \) of the studied wastewater treatment plants
The procedure of ultrasound-assisted emulsification–microextraction (USAEME) with in situ derivatization
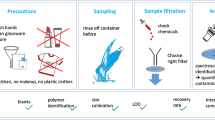
For the simultaneous USAEME and derivatization, aliquots of 5-mL water samples were placed in 10-mL glass centrifuge tubes containing previously weighed 0.1 g of sodium hydrogen phosphate. The extraction solvent (carbon tetrachloride, 40 μL) containing pyrene (5 μg L−1) as an internal standard and the derivatization reagent (acetic anhydride, 300 μL) were added to the water sample and mixed. Immediately after, the tube was immersed in an ultrasonic Unitra Unima (Poland) water bath in such a way that the levels of both liquids (i.e., the bath and the sample) were equal. Extractions were performed at 42 kHz of ultrasound frequency and 230 W of power for the duration of 5 min at room temperature. Emulsions were disrupted by centrifugation at 4,000 rpm/min for 5 min in an MPW-250 Med Instruments (Poland) laboratory centrifuge. In effect, the organic phase settled at the bottom of the conical tube. After centrifugation, carbon tetrachloride was removed using a 100-μL Hamilton syringe (USA) and transferred into a 150-μL microvial with integrated insert. GC–MS analysis was then performed as described in “GC–MS conditions”. Consecutive steps taken to isolate the target compounds are shown in Fig. 2.

Schematic diagram of isolation of acidic pharmaceuticals and phenols by USAEME with in situ derivatization
GC–MS conditions
Analysis was performed with a HP 6890 gas chromatograph with a mass spectrometric detector MSD5973 and HP 7673 autosampler (Agilent Technologies, USA). This device was equipped with HP-5MS (5 % phenylmethylsiloxane) size 30 m length × 0.25 mm, i.e., coated with 0.25 μm film thickness and split/splitless injector. The injector worked in splitless mode. Helium of purity 99.999 % was used as carrier gas at a flow rate of 1 mL min−1. The injector temperature was 250 °C. The oven temperature was programmed from 150 °C, increased at 5 °C/min, to 250 °C. The total run time was 20 min.
The MS detector worked in selected ion monitoring mode. The electron impact source temperature was 230 °C with electron energy of 70 eV. The quadrupole temperature was 150 °C, and the GC interface temperature was 280 °C. The retention times of target compounds are shown in Table 1 together with the quantification and identification of ions. The mass spectra of acetylated derivatives of the target compounds are presented in Fig. 3. The registered mass spectra of acetylated IBP, NAP, KET, DIC, TRC, and BPA are compatible with those in literature (NIST Chemistry WebBook 2011). As for the mass spectra of OP and NP acetylated derivatives, the perusal of relevant literature proved that they have not been published until now. Because of the absence of these data in the NIST MS database, computer software wrongly indicated “octylphenol” as a result of determination of octylphenol acetylated derivative, and likewise, “nonylphenol” as a result of determination of nonylphenol acetylated derivative. The presence of peaks corresponding to the masses 42 units greater (i.e., 248, 262) than molar masses of octylphenol and nonylphenol in the mass spectra was proof that these compounds existed in the forms of the acetylated derivatives. Another evidence confirming the effectiveness of the conducted derivatization is the increase of retention times at which the peaks corresponding to octylphenol and nonylphenol were registered after the acetylation process.

Mass spectra of acetylated derivatives of the target compounds
Determination of retention indices
To determine the retention indices of analytes, the mixture of C10–C22 n-alkanes was analyzed in the same GC–MS conditions as target compounds. The values of retention times were used to calculate linear retention indices from Eq. (1):
where t x , t n , and t n + 1 are the retention times of compound x, and n-alkanes with the number of carbon atoms in the molecule n and n + 1, respectively (t n < t x < t n + 1) (van Den Dool and Kratz 1963). The retention indices of target compounds are shown in Table 1.
Results and discussion
Optimization of extraction and derivatization procedure
All tests during the optimization process had been carried out using distilled water containing 100 μg L−1 of each target compound.
Organic solvent selection
Properties of extraction solvent have crucial impact on the efficiency of the liquid–liquid extraction process. The organic solvent must have low water solubility and high affinity with the isolated compounds. In the USAEME technique, the choice of the solvent is also dictated by the possibility of forming emulsion during the extraction procedure. Accordingly, five different solvents, 1,2-dichlorobenzene, carbon tetrachloride, chloroform, n-decane, and toluene, were examined in the preliminary experiments. In their course, 100 μL of solvent was added to 5-mL aliquots of the target compounds solution, and the samples were placed in an ultrasonic bath for 5 min. Emulsification was observed in all the cases. After centrifugation, the extraction solvents with density higher than water (1,2-dichlorobenzene, carbon tetrachloride, and chloroform) were removed with a syringe from the conical bottom of the test tube. n-Decane and toluene, which have lower density than water, were collected from the bottom of the tube after the removal of water. In the cases of DIC, KET, OP, NP, TRC, and BPA, the largest peak areas were obtained using chloroform, whereas in the case of IBP and NAP, the largest peak areas were obtained using carbon tetrachloride as the extraction solvent. During these experiments, it was noted that introducing chloroform into the chromatographic system results in considerable elevation of the baseline on the chromatograms, and at the same time reduces repeatability of IBP, NAP, and KET peak areas. That is why eventually carbon tetrachloride was selected as the optimum extraction solvent for further experiments.
Effect of solvent volume
The amount of solvent used in extraction has a direct impact on the condensation of the analyte: the lower the volume of organic phase, the greater the concentration of the analyte, which effects in lowering the limit of quantitation (LOQ). What is more, using the smallest possible amounts of toxic solvents conforms with the guidelines of “green chemistry”.
In order to obtain the highest extraction efficiency of the USAEME procedure, the volume of extraction solvent had to be optimized. For this purpose, different volumes of carbon tetrachloride in the range of 40–100 μL were examined (Fig. 4). The analysis showed that reduction of solvent volume is accompanied by growth of peak areas of the analyzed compounds. The amount of solvent recovered after the extraction process conducted with the use of 40 μL of carbon tetrachloride ranged between 25 and 30 μL, and the used volume was the smallest which allowed the introduction of the sample into chromatograph with autosampler. All in all, the volume of carbon tetrachloride equal to 40 μL was chosen as the optimal volume for further investigations.

The influence of solvent volume on extraction efficiency of target pharmaceuticals and phenols (n = 3)
Effect of manual shaking of sample
Manual shaking was used to ensure that the extraction solvent and aqueous sample were adequately mixed before the ultrasound treatment. Manual shaking was conducted for 10 s after injection of extraction solvent and derivatization agent but before the sonication stage. It was observed that the extraction efficiency and reproducibility improved when this step of the procedure was added. Thus, manual shaking was used in subsequent optimization experiments.
Effect of the test-tube material
Furthermore, the impact of extraction vessel material on the efficiency of the reaction was also investigated. For this purpose, the extraction procedure was conducted in both glass and polypropylene test tubes. In the course of the investigation, it was observed that the greater efficiency of the process with all the compounds was achieved when using glass tubes. It can be speculated that the analyzed compounds are subject to adsorption on polypropylene surface, which leads to a fall in efficiency of the extractions proceeding in polypropylene test tubes. Therefore, glass test tubes were selected for further experiments.
Effect of location of the test tube in the ultrasonic bath
In conducting the optimization process, it was noticed that the ultrasonic bath was not homogeneous and two sources of ultrasound waves could be observed. According this, we placed the test tubes in the left source and in the right source of the ultrasound. Holding the test tube in the right source gave a higher extraction efficiency. Therefore, placing of test tubes in the right source was selected for further optimization experiments.
Effect of kind and amount of buffering salt
In the in situ derivatization technique with the use of acetic anhydride, it is necessary to use an addition of buffer salt. This is because stable alkaline reaction environment has to be ensured in order to prevent the reaction equilibrium shift towards the substrates and to ensure greater efficiency of the process. In acylation processes conducted with the use of acetic anhydride, sodium hydrogen carbonate is the most frequently used buffer salt. But in this particular case when sodium hydrogen carbonate was used, carbon dioxide bubbles interfering with the collection of the organic phase appeared. They were produced as a consequence of decomposition of carbonic acid generated by carbonates in the presence of the acetic acid formed from the anhydride hydrolysis.
For this reason, in the described experiments sodium hydrogen phosphate was used. To study the effects of the various amounts of added sodium hydrogen phosphate on the derivatization process, experiments were performed with different quantities of this salt (0.1–0.4 g) per 5 mL of wastewater sample. Figure 5 illustrates the influence of added quantity of sodium hydrogen phosphate on extraction efficiency of the target compounds. It can be noticed that the results obtained with different concentrations of the salt were very similar; hence, the amount of 0.1 g was used in subsequent experiments.

The influence of salting out effect on extraction efficiency of target compounds (n = 3)
Effect of derivatization reagent volume
The influence of the volume of acetic anhydride on the relative peak area was studied in the range of 200–350 μL (Fig. 6). The results indicated that the volume of acetic anhydride equal to 300 μL should be chosen as optimal.

The influence of derivatization reagent volume on extraction efficiency of target compounds (n = 3)
Effect of the extraction time
The time of isolation is of great significance in all extraction procedures. In USAEME, extraction time is defined as the time between injection of extraction solvent and the end of the sonication stage (Ma et al. 2009). To determine the influence of the extraction time on isolation efficiency, the parameter was examined in the range of 5–15 min. The highest values of IBP, NAP, and KET peak areas were achieved during the extraction which lasted 15 min, but the differences between individual results obtained with different times were not statistically significant. In the cases of OP, NP, DIC, TRC, and BPA, equally high levels of extraction efficiency were achieved when the process continued for 5 and 10 min, while the results obtained after 15 min were considerably lower. Presumably, under the prolonged influence of ultrasound, the equilibrium of derivatization shifts towards the substrates, resulting in the decrease of efficiency of the concurrently proceeding extraction and derivatization processes. Keeping that in mind, 5-min extraction time was chosen as the most suitable for further studies.
Method validation
Tables 2 and 3 present an overview of the method’s performance, including linearity, limits of quantification (LOQ) and detection (LOD), repeatability, and recovery for these analytes when using deionized water as the sample matrix (Table 2) and influent and effluent wastewater as the sample matrix (Table 3). Calibration curves were obtained by spiking the wastewater samples with eight concentration and performing the extraction. Calibration curves were linear within the studied concentration range, with coefficients of correlation greater than 0.987 for all the target compounds. RSD of the determination ranged from 2.0 to 16.8 %, depending on the analyzed compound and the kind of matrix used. The LOQ, determined as the analyte concentration corresponding to a signal/noise ratio of 10, ranged from 0.005 to 2.758 μg L−1. The LOD, defined as the concentration that corresponds to three times the standard deviation of blanks, were between 0.002 and 0.828 μg L−1. Recoveries were evaluated by spiking matrix before extraction and compare the signal obtained with the extract spiked after. Where the target compounds were initially present in the wastewater samples, the areas of peaks registered during blank analysis were subtracted from the corresponding peak areas of spiked samples. Recoveries were obtained from influent and effluent wastewater samples at concentrations of 1 μg L−1 in the cases of OP, NP, TRC, BPA, and DIC, and at 10 μg L−1 in the cases of IBP, NAP, and KET; these values are shown in Table 3. In the case when influent wastewater was used as the sample matrix, recoveries were between 94 and 106 %. When using treated effluents as the sample matrix, the recoveries were between 96 and 108 %. The comparison of the performance of proposed method with methods usually used for the analysis of target compounds in wastewater samples shows many advantages of USAEME/GC–MS procedure. The recoveries obtained in the presented work are better than those determined by SPE/GC–MS and SPE/LC–MS methods, which are usually involved in such determinations, while repeatability is on similar level. The sensitivity of USAEME/GC–MS method, expressed as LOD values, is better than those obtained using SPE technique for phenols, while for pharmaceuticals it is comparable or slightly worse (Ballesteros et al. 2006; Gatidou et al. 2007; Gómez et al. 2007a, b; Gracia-Lor et al. 2010; Yu and Wu 2013).
Occurrence of target phenols and pharmaceuticals in municipal wastewaters from Polish WWTPs
The developed USAEME/GC–MS method was applied for determination of ibuprofen, naproxen, ketoprofen, diclofenac, octylphenol, nonylphenol, triclosan, and bisphenol A in influents and effluents from municipal wastewater treatment plants located in nine cities of Poland (see Fig. 1). The chromatograms of influent (a) and effluent wastewater samples (b) from WWTP B are shown in Fig. 7. The occurrence of target compounds and levels of contamination in the analyzed wastewater samples are summarized in Table 4.

The chromatograms registered during analysis of influent (a) and effluent (b) wastewater samples from WWTP B
All studied compounds were found in the influents; in the effluents, all compounds besides KET were registered. The concentrations of determined pollutants in influent wastewaters varied from values below LOD to several hundreds of micrograms per liter, while in treated effluents were between LOD and several dozens of micrograms per liter. The target pharmaceuticals were found with frequency of 56 % for IBP, 44 % for NAP, 30 % for DIC, and 21 % for KET in influent wastewater and 14 % for DIC, 12 % for IBP, and 2 % for NAP in effluent wastewater. The highest average values were found for NAP (552 μg L−1) and KET (234 μg L−1) in WWTP I. In 32 cases out of 172 (4 pharmaceuticals × 43 samples), measured concentration was above 10 μg L−1 in influent wastewater. It happened 21 times for IBP, seven times for NAP, and four times for KET. In seven cases out of 172, measured concentration was above 1 μg L−1 in effluent wastewater. It happened five times for IBP and NAP and one time for DIC. The target phenols were found with frequency of 65 % for NP, 47 % for OP and BPA, and 44 % for TRC in influent wastewater and 26 % for TRC, 23 % for OP, 19 % for BPA, and 14 % for NP in effluent wastewater. The highest values were found for NP (24 μg L−1) and OP (5.7 μg L−1) in influent wastewater. In four cases out of 172 (4 phenols × 43 samples), measured concentration was above 10 μg L−1 in influent wastewater. It was registered only for NP. In 10 cases out of 172, measured concentration was above 1 μg L−1 in effluent wastewater. It happened six times for BPA, three times for OP, and one time for NP.
The concentrations of target phenols and pharmaceuticals were similar in samples collected from all the wastewater treatment plants except of WWTPI, where the registered concentrations were much greater than in other WWTPs. The WWTPI is a small municipal wastewater treatment plant; the influents are transported into this plant from households mostly by special waste-removal trucks.
In most cases, concentrations of the target compounds registered in effluents were much lower than those found in influent wastewaters. Nevertheless, the concentrations of OP and BPA registered in WWTPE were higher in effluents than influents. This phenomenon might be explained by the fact that the samples of both influent and effluent wastewaters were taken at the same time, meaning that they did not originate from the same portion of wastewater. Analogous situation was previously reported during determination of naproxen, diclofenac, and ibuprofen in municipal wastewaters (Hernando et al. 2006). In order to determine the effectiveness of the wastewater purification, collection of the influent and effluent samples from the same wastewater portion was done four times in WWTPB. It was achieved by collecting the effluents 24 h after influent taking. Table 5 shows the average concentrations of target pharmaceuticals and phenols determined in the influents and effluents from the same wastewater portion and the removal efficiencies were calculated. The obtained values of removal efficiency rates were very high and exceed 90 % for all compounds except DIC.
Conclusions
In the present study, a new analytical methodology based on ultrasound-assisted emulsification–microextraction followed by GC–MS determination has been proposed for the determination of acidic pharmaceuticals and phenols. In situ derivatization with acetic anhydride, conducted simultaneously with extraction, was successful under the optimized conditions. Scrutiny of the available literature sources shows that the present work is first to describe acylation with the use of acetic anhydride applied for determination of acidic pharmaceuticals and OP, NP, and BPA in water samples.
The proposed USAEME technique with in situ derivatization step offers several advantages in terms of simplicity, low cost, minimal solvent consumption, and very short time of sample preparation. The method has been successfully used to determine ibuprofen, naproxen, ketoprofen, diclofenac, octylphenol, nonylphenol, triclosan, and bisphenol A in influent and effluent municipal wastewater samples obtained from nine wastewater treatment plants located in Poland. All studied pharmaceuticals and phenols were found in the influents from Polish WWTPs; in the effluents, all compounds besides KET were registered. The concentrations of pollutants in influent wastewaters varied from values below LOD to several hundreds of micrograms per liter, while in treated effluents concentrations were between LOD and several dozens of micrograms per liter. Average elimination rates of pharmaceuticals and phenols during purification process carried in municipal WWTP are 93 % and 97 %, respectively.
References
Ahel M, Giger W (1993) Partitioning of alkylphenols and alkylphenol polyethoxylates between water and organic solvents. Chemosphere 26:1471–1478
Antonić J, Heath E (2006) Determination of NSAIDs in river sediment samples. Anal Bioanal Chem 387:1337–1342
Ballesteros O, Zafra A, Navalón A, Vílchez JL (2006) Sensitive gas chromatographic–mass spectrometric method for the determination of phthalate esters, alkylphenols, bisphenol A and their chlorinated derivatives in wastewater samples. J Chromatogr A 1121:154–162
Beck I-C, Bruhn R, Gandrass J, Ruck W (2005) Liquid chromatography–tandem mass spectrometry analysis of estrogenic compounds in coastal surface water of the Baltic Sea. J Chromatogr A 1090:98–106
Błędzka D, Gryglik D, Olak M et al (2010) Degradation of n-butylparaben and 4-tert-octylphenol in H2O2/UV system. Radiat Phys Chem 79:409–416
Esplugas S, Bila DM, Krause LGT, Dezotti M (2007) Ozonation and advanced oxidation technologies to remove endocrine disrupting chemicals (EDCs) and pharmaceuticals and personal care products (PPCPs) in water effluents. J Hazard Mater 149:631–642
Fontana AR, Altamirano JC (2010) Sensitive determination of 2,4,6-trichloroanisole in water samples by ultrasound assisted emulsification microextraction prior to gas chromatography–tandem mass spectrometry analysis. Talanta 81:1536–1541
Fontana AR, Wuilloud RG, Martínez LD, Altamirano JC (2009) Simple approach based on ultrasound-assisted emulsification–microextraction for determination of polybrominated flame retardants in water samples by gas chromatography–mass spectrometry. J Chromatogr A 1216:147–153
Gatidou G, Thomaidis NS, Stasinakis AS, Lekkas TD (2007) Simultaneous determination of the endocrine disrupting compounds nonylphenol, nonylphenol ethoxylates, triclosan and bisphenol A in wastewater and sewage sludge by gas chromatography–mass spectrometry. J Chromatogr A 1138:32–41
Gómez MJ, Agüera A, Mezcua M et al (2007a) Simultaneous analysis of neutral and acidic pharmaceuticals as well as related compounds by gas chromatography–tandem mass spectrometry in wastewater. Talanta 73:314–320
Gómez MJ, Martínez Bueno MJ, Lacorte S et al (2007b) Pilot survey monitoring pharmaceuticals and related compounds in a sewage treatment plant located on the Mediterranean coast. Chemosphere 66:993–1002
Gracia-Lor E, Sancho JV, Hernández F (2010) Simultaneous determination of acidic, neutral and basic pharmaceuticals in urban wastewater by ultra high-pressure liquid chromatography–tandem mass spectrometry. J Chromatogr A 1217:622–632
Hernando MD, Heath E, Petrovic M, Barceló D (2006) Trace-level determination of pharmaceutical residues by LC–MS/MS in natural and treated waters. A pilot-survey study. Anal Bioanal Chem 385:985–991
Kawaguchi M, Ito R, Honda H et al (2008) Stir bar sorptive extraction and thermal desorption–gas chromatography–mass spectrometry for trace analysis of triclosan in water sample. J Chromatogr A 1206:196–199
Liu R, Zhou J, Wilding A (2004) Simultaneous determination of endocrine disrupting phenolic compounds and steroids in water by solid-phase extraction–gas chromatography–mass spectrometry. J Chromatogr A 1022:179–189
Ma JJ, Du X, Zhang JW et al (2009) Ultrasound-assisted emulsification–microextraction combined with flame atomic absorption spectrometry for determination of trace cadmium in water samples. Talanta 80:980–984
Najafi NM, Tavakoli H, Abdollahzadeh Y, Alizadeh R (2012) Comparison of ultrasound-assisted emulsification and dispersive liquid–liquid microextraction methods for the speciation of inorganic selenium in environmental water samples using low density extraction solvents. Anal Chim Acta 714:82–88, 3
NIST Chemistry WebBook (2011) National Institute of Standards and Technology, Gaithersburg, MD. http://webbook.nist.gov/chemistry/. Accessed 31 Oct 2012
Oaks JL, Gilbert M, Virani MZ et al (2004) Diclofenac residues as the cause of vulture population decline in Pakistan. Nature 427:630–633
Ozcan S, Tor A, Aydin ME (2009a) Application of ultrasound-assisted emulsification–micro-extraction for the analysis of organochlorine pesticides in waters. Water Res 43:4269–4277
Ozcan S, Tor A, Aydin ME (2009b) Determination of selected polychlorinated biphenyls in water samples by ultrasound-assisted emulsification–microextraction and gas chromatography–mass-selective detection. Anal Chim Acta 647:182–188
Ozcan S, Tor A, Aydin ME (2010) Determination of polycyclic aromatic hydrocarbons in waters by ultrasound-assisted emulsification–microextraction and gas chromatography–mass spectrometry. Anal Chim Acta 665:193–199
Pizarro C, Sáenz-González C, Pérez-del-Notario N, González-Sáiz JM (2012a) Simultaneous determination of cork taint and Brett character responsible compounds in wine using ultrasound-assisted emulsification microextraction with solidification of floating organic drop. J Chromatogr A 1249:54–61
Pizarro C, Sáenz-González C, Pérez-del-Notario N, González-Sáiz JM (2012b) Optimisation of a sensitive method based on ultrasound-assisted emulsification–microextraction for the simultaneous determination of haloanisoles and volatile phenols in wine. J Chromatogr A 1244:37–45
Quintana JB, Rodil R, Reemtsma T (2004) Suitability of hollow fibre liquid-phase microextraction for the determination of acidic pharmaceuticals in wastewater by liquid chromatography–electrospray tandem mass spectrometry without matrix effects. J Chromatogr A 1061:19–26
Ramos Payán M, Bello López MÁ, Fernández-Torres R et al (2010) Application of hollow fiber-based liquid-phase microextraction (HF-LPME) for the determination of acidic pharmaceuticals in wastewaters. Talanta 82:854–858
Regueiro J, Llompart M, Garcia-Jares C et al (2008) Ultrasound-assisted emulsification–microextraction of emergent contaminants and pesticides in environmental waters. J Chromatogr A 1190:27–38
Regueiro J, Becerril E, Garcia-Jares C, Llompart M (2009) Trace analysis of parabens, triclosan and related chlorophenols in water by headspace solid-phase microextraction with in situ derivatization and gas chromatography–tandem mass spectrometry. J Chromatogr A 1216:4693–4702
LOGKOW-Sangster Research Laboratories (2013) A databank of evaluated octanol-water partition coefficients (Log P). http://logkow.cisti.nrc.ca/logkow/. Accessed 30 Oct 2012
Van Den Dool H, Kratz P (1963) A generalization of the retention index system including linear temperature programmed gas–liquid partition chromatography. J Chromatogr A 11:463–471
Viñas P, López-García I, Campillo N et al (2012) Ultrasound-assisted emulsification microextraction coupled with gas chromatography–mass spectrometry using the Taguchi design method for bisphenol migration studies from thermal printer paper, toys and baby utensils. Anal Bioanal Chem 404:671–678
Yiantzi E, Psillakis E, Tyrovola K, Kalogerakis N (2010) Vortex-assisted liquid–liquid microextraction of octylphenol, nonylphenol and bisphenol-A. Talanta 80:2057–2062
Yu Y, Wu L (2013) Application of graphene for the analysis of pharmaceuticals and personal care products in wastewater. Anal Bioanal Chem 405:4913–4919
Zgoła-Grześkowiak A (2010) Dispersive liquid–liquid microextraction applied to isolation and concentration of alkylphenols and their short-chained ethoxylates in water samples. J Chromatogr A 1217:1761–1766
Zhang Y, Lee HK (2012) Application of ultrasound-assisted emulsification microextraction based on applying low-density organic solvent for the determination of organochlorine pesticides in water samples. J Chromatogr A 1252:67–73
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Philippe Garrigues
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Kotowska, U., Kapelewska, J. & Sturgulewska, J. Determination of phenols and pharmaceuticals in municipal wastewaters from Polish treatment plants by ultrasound-assisted emulsification–microextraction followed by GC–MS. Environ Sci Pollut Res 21, 660–673 (2014). https://doi.org/10.1007/s11356-013-1904-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-013-1904-6