
Abstract
Atherosclerosis is the main pathological basis of cardiovascular disease and involves damage to vascular endothelial cells (ECs) that results in endothelial dysfunction (ED). The vascular endothelium is the key to maintaining blood vessel health and homeostasis. ED is a complex pathological process involving inflammation, shear stress, vascular tone, adhesion of leukocytes to ECs, and platelet aggregation. The activation of P2X4, P2X7, and P2Y2 receptors regulates vascular tone in response to shear stress, while activation of the A2A, P2X4, P2X7, P2Y1, P2Y2, P2Y6, and P2Y12 receptors promotes the secretion of inflammatory cytokines. Finally, P2X1, P2Y1, and P2Y12 receptor activation regulates platelet activity. These purinergic receptors mediate ED and participate in atherosclerosis. In short, P2X4, P2X7, P2Y1, and P2Y12 receptors are potential therapeutic targets for atherosclerosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Atherosclerosis is a chronic vascular disease and is the main pathological basis of cardiovascular disease (CVD) [1, 2]. Globally, CVD caused by atherosclerosis is spreading [3], leading to a heavy worldwide disease and economic burden [4]. Its pathogenesis involves lipid infiltration, endothelial injury, inflammation, and plaque formation [4], with endothelial injury and an abnormal inflammatory response being the keys to its occurrence [5]. As the physical barrier between the blood and blood vessel wall, the vascular endothelium is essential for maintaining blood vessel homeostasis [6]. Endothelial cells (ECs) bridge the blood and blood vessel walls and play a critical role in cardiovascular homeostasis by regulating shear stress, vascular tone, monocyte or leukocyte adhesion, and platelet aggregation [7]. Healthy ECs regulate the secretion and balance of vasodilating, vasoconstricting, anti-inflammatory, pro-inflammatory, oxidative, and antioxidant factors. A properly functioning vascular endothelium is regarded as the gatekeeper to cardiovascular health [8]. ECs are the first barrier in protecting blood vessels. Accordingly, atherosclerosis begins with their injury and dysfunction, which leads to plasma lipids invading the endothelium, monocyte or macrophage infiltration, vascular smooth muscle cell migration to the intima, and the formation of foam cells via the engulfment of lipids by macrophage. Moreover, ECs secrete a variety of pro-inflammatory factors, which eventually evolve into atherosclerosis [9].
A normally functioning endothelium regulates vascular tone by balancing the secretion of vasodilators and vasoconstrictors [10]. The decreased expression of vasodilatory factors such as nitric oxide (NO), prostacyclin I2 (PGI2), or endothelial-derived hyperpolarizing factor (EDHF) or the increased expression of vasoconstrictor factors such as endothelin 1 (ET-1), angiotensin II, or thromboxane A2 (TxA2) will imbalance vascular homeostasis and cause ED. As a semipermeable membrane, the vascular endothelium forms a biological barrier between the intravascular and extravascular spaces to regulate the transportation and exchange of molecules [11]. An incomplete endothelial barrier leads to vascular hyperpermeability, vascular swelling and edema, and eventually EC damage. ED is key in the occurrence and development of cardiovascular and cerebrovascular diseases. The main causes of ED are lipid metabolism disorders, inflammation, oxidative stress, and shear stress [12]. Under the action of these factors, ECs reduce the synthesis or activity of NO, PGI2, EDHF, and other vasodilators and increase the synthesis of vasoconstrictors such as endothelin (ET). Consequently, this perturbation of the balance between vasodilation and vasoconstriction induces the production of reactive oxygen species (ROS) and the release of the pro-inflammatory cytokines interleukin-1 (IL-1), interleukin-6 (IL-6), and tumor necrosis factor-α. Furthermore, the bioavailability of NO is reduced, which ultimately leads to ED [13].
Endothelial injury causes the abnormal expression of intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and platelet-endothelial cell adhesion molecule-1 (PECAM-1); it also promotes the adhesion of leukocytes to ECs. ICAM-1, VCAM-1, and PECAM-1 are specific indicators of vascular endothelial injury. Shear stress acts on ECs, causing leukocytes to migrate to adhesion molecules and chemokines in the arterial wall, leading to atherosclerosis [14]. The evaluation of EC function is an important tool for predicting the occurrence of CVD [15]. The improvement of EC function reduces the occurrence of CVD events, which include atherosclerosis [16, 17]. Interestingly, purinergic signaling is critical in regulating vascular tone and remodeling [18]. Increasing studies have reported that purinergic signaling mediates ED and participates in the formation of atherosclerosis [19].
In 1972, Burnstock introduced the concept of purinergic signaling, whereby he identified adenosine triphosphate (ATP) as an extracellular signaling molecule and called it a purinergic neurotransmitter [20]. In 1976, he suggested that extracellular purinergic signaling acts via purinergic receptors, thus first proposing the purinergic receptors [21]. The purinergic signaling system includes purinergic substances and their receptors. These purinergic substances include ATP, adenosine diphosphate (ADP), adenosine monophosphate (AMP), and adenosine. Almost all cells release ATP; it is the main universal energy currency in the cell, and it is released outside the cell as a signal molecule [22]. Intracellularly, ATP, ADP, and AMP are converted into adenosine via cytoplasmic 5′-nucleosidase activity. ATP can also be converted to cyclic adenosine monophosphate (cAMP) by adenylyl cyclase, which is then subsequently converted to AMP by phosphodiesterase. Cytoplasmic 5′-nucleotidase then converts AMP to adenosine. Additionally, adenosine is converted into inosine by adenosine deaminase, and adenosine can be converted back into AMP by adenosine kinase [23]. Extracellularly, ATP is released to the extracellular space via transporter-mediated or ATP-permeable ion channels, such as the pannexin 1 channel [24]. ATP is hydrolyzed to ADP and AMP mainly via the continuous action of ectonucleoside diphosphohydrolase triphosphate (CD39), while ecto-5′-nucleotidase (CD73) converts the AMP produced by the ATP and cAMP pathway into adenosine [25].
Purinergic receptors can be subdivided into two categories: P1 and P2. P1 receptors are G protein-coupled and consist of the A1, A2A, A2B, and A3 receptor subtypes [26]; they are also known as adenosine receptors [27]. P2 receptors consist of two types, P2X and P2Y. P2X are ligand-gated cationic channel receptors and can be subdivided into the P2X1-7 receptor subtypes [28]. They mediate rapid responses to ATP [29]. By contrast, P2Y receptors are G protein-coupled and can be divided into the P2Y1, P2Y2, P2Y4, P2Y6, and P2Y11-14 receptor subtypes [28]; they respond to ATP and ADP [29]. Notably, the role of purinergic substances and their receptors in physiological and pathological processes has recently received more attention [30,31,32].
Purinergic receptors and endothelial dysfunction
Purinergic receptors are distributed throughout most tissues or cells and are the basis of endothelial-mediated vasodilation [33], inflammation [34], cell differentiation, migration, and proliferation [35]. Adenosine and ATP mediate the migration, growth, and proliferation of ECs [35]. During vascular remodeling, ATP and adenosine act on ECs via P1 and P2Y receptors to promote proliferation [19]. The purinergic activation of ECs leads to the release of NO, PGI2, and EDHF, thereby causing vasodilation [36, 37]. More specifically, endothelial P2Y receptors mediate vasodilation by releasing NO and EDHF [33].
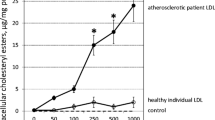
Reductions in NO bioavailability and increases in ROS release can cause ED [38]. Damage to the barrier function of the vascular endothelium promotes the deposition of low-density lipoprotein under the vascular intima, which is then modified into oxidized low-density lipoprotein (ox-LDL). As a key molecule in atherosclerosis, ox-LDL can bind to receptors and trigger a series of intracellular changes that cause vascular EC injury and dysfunction. The stimulation of monocyte macrophages to excessively engulf ox-LDL accelerates the transformation of macrophages into foam cells and forms plaques under the vascular endothelium, thereby promoting atherosclerosis development [39]. Notably, ox-LDL stimulates ECs to release ATP, thereby activating P2Y2 receptor. On one hand, it increases caspase-1 activity and interleukin-1β (IL-1β) secretion and activates the inflammasome [40]. On the other hand, it promotes ROS release, increases ICAM-1 and VCAM-1 content, promotes monocyte migration and EC adhesion, and participates in the pathogenesis of atherosclerosis [34]. P2Y2 receptors are an important medium for ox-LDL-mediated monocyte adhesion to ECs [34]. Table 1 summarizes the ED caused by purinergic receptors activation or inhibition, and Fig. 1 summarizes the role of purinergic receptors in atherosclerosis via their regulation of vascular tone, inflammation, and platelet aggregation.

Purinergic receptors mediate ED and participate in atherosclerosis. A2A, A2B, A3, P2X1, P2X4, P2X7, P2Y1, P2Y2, P2Y6, P2Y11, and P2Y12 receptors mediate endothelial cell dysfunction and participate in atherosclerosis. The activation of P2X4, P2X7, and P2Y2 receptors regulates vascular tone in response to shear stress. The activation of A2A, P2X4, P2X7, P2Y1, P2Y2, P2Y6, and P2Y12 receptors promotes the secretion of inflammatory cytokine interleukin (IL)-1β, 1L-6, IL-8, IL-1, intercellular adhesion molecule-1 and vascular cell adhesion molecule-1. P2X1, P2Y1, and P2Y12 receptor activation regulates platelet activity (platelet aggregation), mediates ED, and participates in atherosclerosis. R, receptors
P1 receptors in endothelial dysfunction
A1, A2A, A2B, and A3 receptors are expressed on ECs [41], among which the A2A and A2B receptors are the main purinergic receptors on ECs [42]. A2A receptor knockout in apolipoprotein E-deficient mice prevents the formation of atherosclerotic lesions [43]. Under hypoxic conditions, adenosine activates A3 and A2B receptors, increases vascular endothelial growth factor (VEGF) secretion, and promotes foam cell formation [44]. A2A and A2B receptors activation will increase cAMP levels, leading to vasodilation [45]. The subcutaneous injection of A2A receptor agonist in mice decreases VCAM-1, ICAM-1, and P-selectin expression. Adenosine inhibits early inflammation by activating the A2A receptors of ECs [46]. The knockout or inhibition of A1 receptor also reduces the concentration of pro-inflammatory cytokines, thereby reducing atherosclerotic lesions in apolipoprotein E-deficient mice [47]. In short, P1 receptors mediate ED and participate in atherosclerosis.
P2 receptors in endothelial dysfunction
P2X4, P2X7, P2Y1, P2Y2, P2Y6, and P2Y11 receptors are the most highly expressed P2 receptors in ECs and may be related to the release of ATP, NO, and EDHF [48]. P2X receptors are also expressed on ECs and are related to cell adhesion and permeability. The stimulation of human ECs via P2Y1 receptors activate VEGF-2 and leads to angiogenesis [49]. P2Y2 receptors in ECs regulate angiopoietin-2 and VEGFR-2 expression and play a crucial role in angiogenesis [50]. Endothelial P2Y2 receptor knockout will reduce endothelial nitric oxide synthase (eNOS) activity, resulting in reduced NO production, causing vasoconstriction, and ultimately leading to ED [51]. Ticagrelor, a P2Y12 inhibitor, improves endothelial function by activating endothelial eNOS in the vascular endothelium, thereby reducing circulating epidermal growth factor [52].
Shear stress is the frictional resistance acting on the vascular cavity surface [53]. It can regulate the structure and function of the endothelium [54, 55] and is closely related to the development, physiology, and pathology of the blood vessel. ECs transduce fluid shear stress into biochemical signals to regulate endothelial function [56]. That is, in response to shear stress or hypoxia, ECs release ATP, which then act on P2X and P2Y receptors on the endothelium to release NO, PGI2, and EDHF, causing vasodilation [19, 57]. When the endothelium is injured, it promotes the release of vasoconstrictors, resulting in vasospasm [58]. Ca2+ plays an important role in the EC response to shear stress. As the P2X4 receptors of ECs participate in shear stress-mediated Ca2+ transmission, they may have shear sensor properties [59]. The ECs of mice with knockout P2X4 receptors respond to shear stress, and as the amount of released NO decreases, vascular tone increases [60]. Under the action of shear stress, ATP acts on ECs via P2X7 receptors, which induces E-selectin and IL-1β secretion and promotes endothelial inflammation in atherosclerotic sites [61]. In response to shear stress, ECs also release ATP, which acts on the P2Y2 receptor of ECs to induce NO release, and regulate vascular tone [62]. Correspondingly, deletion of the P2Y2 gene reduces shear stress-induced vasodilation [63].
Purinergic signaling mediating ED is involved in atherosclerosis, mainly via P2 receptor-mediated EC inflammation. ATP is released into the extracellular space and binds to P2 receptors to mediate inflammation [64]. During inflammation, ECs release ATP via the pannexin 1 channels to promote leukocyte adhesion and migration [24]. Notably, the release of ATP also involves the connexin 43 channels [65]. High glucose and palmitate upregulated the expression of P2X4 and P2X7 in human umbilical vein ECs, as well as increased the release of the inflammatory factors IL-1β, 1L-6, IL-8, ICAM-1, and VCAM-1. These results indicate that P2X4 and P2X7 receptors regulate high-glycemic inflammation in ECs. Hyperglycemia and hyperlipidemia cause ED, leading to oxidative stress and inflammation [66]. ATP acts on the P2X7 receptor, activates the inflammasome, and induces the release of inflammatory cytokines IL-1β and IL-18 as well as the production of ROS [67]. It promotes inflammation and ED and ultimately leads to atherosclerosis. Vascular traction injury has been reported to promote ATP release, which inhibits the production of NO via EC P2X7 receptors, leading to ED [68]. P2X7 receptors are highly expressed in mice with atherosclerotic lesions, and the realization of clinical patients is also consistent. P2X7-deficient mice exhibit reduced inflammasome activation and reduced leukocyte rolling and adhesion. These results indicate that P2X7 underactivity reduces atherosclerotic lesions by inhibiting the inflammatory response [69]. P2X7 receptor antagonists reduce IL-1β levels in atherosclerotic blood vessels [70]. As a ligand-gated ion channel, P2X7 receptor binds to extracellular ATP and promotes IL-1β release via the pannexin-1 channel [71].
P2Y1 receptor knockout in apolipoprotein E-deficient mice decreases macrophage infiltration and VCAM-1 levels, indicating that atherosclerosis is related to the P2Y1 receptors on ECs [72]. P2Y2 receptor causes vascular inflammation by increasing VCAM-1 expression in ECs, which eventually causes atherosclerosis [51]. In P2Y2 receptor-deficient mice, ATP-induced leukocyte adhesion is reduced. In addition, VCAM-1 and ICAM-1 RNA levels are decreased, indicating that extracellular ATP induces vascular inflammation and atherosclerosis by activating P2Y2 receptor [73]. In P2Y6-deficient mice, leukocyte adhesion to the vessel wall is reduced, as are ox-LDL and lipid content and RNA expression of ICAM-1 and IL-6. These reductions indicate that P2Y6 deficiency reduces inflammation and inhibits atherosclerosis [74]. During lipopolysaccharide-induced vascular inflammation, P2Y6 receptor knockout or injection of P2Y6 antagonists weakens the inflammatory response, indicating that P2Y6 receptor are closely related to vascular inflammation [75]. Inhibition P2Y12 receptor expression protects EC barrier function by increasing the concentration of cAMP in ECs [76]. Notably, P2Y11 receptor is also closely related to inflammation, as their stimulation reduces oxidative stress and improves ED [77].
Interestingly, purinergic signaling is also involved in atherosclerosis by regulating platelet activity. Platelets adhere and fuse into the vascular endothelium, thereby maintaining its integrity. P2X1, P2Y1, and P2Y12 receptors are expressed on platelets and mediate their aggregation [58]. ADP is a platelet agonist; it causes platelet shape change and aggregation via the P2X1, P2Y1, and P2Y12 receptors and generates TxA2 [78]. ATP diphosphohydrolase hydrolyzes extracellular ATP and ADP to AMP, and ADP inhibits platelet aggregation via ATP diphosphohydrolase. ATP diphosphohydrolase has the same functionality as CD39 [79]. CD39 is expressed on the surface of ECs, inhibits ADP-induced platelet aggregation, and is the main regulator of platelet activation [80]. Moreover, it protects against atherosclerosis [81]. Aspirin or clopidogrel, P2Y12 receptor antagonists that mediate platelet aggregation, is widely used to treat stroke and thrombosis [82, 83].
Conclusion
Although a review of purinergic signaling in the cardiovascular system has been previously published [58], this review focuses on purinergic signaling mediating ED caused by atherosclerosis. Atherosclerosis begins with vascular ED, and ED involves purinergic signaling. ATP and its degradation products ADP, AMP, adenosine, uridine diphosphate, and cAMP stimulate a series of receptors and affect endothelial function. So far, the A2A, A2B, A3, P2X1, P2X4, P2X7, P2Y1, P2Y2, P2Y6, P2Y11, and P2Y12 receptors have been demonstrated to mediate EC dysfunction and participate in atherosclerosis.
This review reports that purinergic signaling mediates EC dysfunction and participates in atherosclerosis in different ways. Purinergic signaling plays an important role in regulating endothelial structure and function in response to shear stress. ECs release ATP in response to shear stress, which acts on P2X4 [60], P2X7 [61], and P2Y2 receptors [62] to regulate vascular tone. Disturbances in vascular tone will result in ED. Purinergic signaling also plays an important role in the inflammatory response, particularly by activating the A2A [46], P2X4 [66], P2X7 [69], and P2Y1 receptors [73] to induce the release of inflammatory factors and to promote the adhesion of leukocytes to ECs. Purinergic signaling also activates the P2X1, P2Y1, and P2Y12 receptors [78], causing platelet shape changes and aggregation and mediating ED to contribute to atherosclerosis.
In combination with the current understanding of the pathogenesis underlying atherosclerosis caused by ED and the in-depth understanding of purinergic signaling, further study is needed to determine the specific P2 receptors that mediate ED. Since different purinergic receptor subtypes can mediate similar functional effects, it is challenging to selectively target specific receptor subtypes. From this review, P2X4, P2X7, P2Y1, and P2Y12 receptors may be potential therapeutic targets for atherosclerosis. While few studies have been published on the involvement of P1 receptor-mediated ED in atherosclerosis, we will pay more attention to the relationship between P1 receptors and ED in the future. Therefore, purinergic receptors may become potential therapeutic targets for atherosclerosis.
Code availability
Not applicable.
Abbreviations
- CVD:
-
Cardiovascular disease
- ECs:
-
Endothelial cells
- ED:
-
Endothelial dysfunction
- NO:
-
Nitric oxide
- PGI2 :
-
Prostacyclin I2
- EDHF:
-
Endothelium-derived hyperpolarizing factor
- ET-1:
-
Endothelin 1
- TXA2 :
-
Thromboxane A2
- ET:
-
Endothelin
- ROS:
-
Reactive oxygen species
- IL-1:
-
Interleukin-1
- IL-6:
-
Interleukin-6
- ICAM-1:
-
Intercellular adhesion molecule-1
- VCAM-1:
-
Vascular cell adhesion molecule-1
- PECAM-1:
-
Platelet-endothelial cell adhesion molecule-1
- ATP:
-
Adenosine triphosphate
- ADP:
-
Adenosine diphosphate
- AMP:
-
Adenosine monophosphate
- cAMP:
-
Cyclic adenosine monophosphate
- CD39:
-
Ectonucleoside diphosphohydrolase triphosphate
- CD73:
-
Ecto-5′-nucleotidase
- ox-LDL:
-
Oxidized low-density lipoprotein
- IL-1β:
-
Interleukin-1β
- VEGF:
-
Vascular endothelial growth factor
- eNOS:
-
Endothelial nitric oxide synthase
References
Xu S, Kamato D, Little PJ, Nakagawa S, Pelisek J, Jin ZG (2019) Targeting epigenetics and non-coding RNAs in atherosclerosis: from mechanisms to therapeutics. Pharmacol Ther 196:15–43. https://doi.org/10.1016/j.pharmthera.2018.11.003
Zhu Y, Xian X, Wang Z, Bi Y, Chen Q, Han X, Tang D, Chen R (2018) Research progress on the relationship between atherosclerosis and inflammation. Biomolecules 8(3):80. https://doi.org/10.3390/biom8030080
Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Delling FN, Djousse L, Elkind MSV, Ferguson JF, Fornage M, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, Lichtman JH, Longenecker CT, Loop MS, Lutsey PL, Martin SS, Matsushita K, Moran AE, Mussolino ME, Perak AM, Rosamond WD, Roth GA, Sampson UKA, Satou GM, Schroeder EB, Shah SH, Shay CM, Spartano NL, Stokes A, Tirschwell DL, VanWagner LB, Tsao CW (2020) Heart disease and stroke statistics-2020 update: a report from the American Heart Association. Circulation 141(9):e139–e596. https://doi.org/10.1161/cir.0000000000000757
Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, Tokgözoğlu L, Lewis EF (2019) Atherosclerosis nature reviews. Disease primers 5(1):56. https://doi.org/10.1038/s41572-019-0106-z
Zmysłowski A, Szterk A (2017) Current knowledge on the mechanism of atherosclerosis and pro-atherosclerotic properties of oxysterols. Lipids Health Dis 16(1):188. https://doi.org/10.1186/s12944-017-0579-2
Godo S, Shimokawa H (2017) Endothelial functions. Arterioscler Thromb Vasc Biol 37(9):e108–e114. https://doi.org/10.1161/atvbaha.117.309813
Sturtzel C (2017) Endothelial Cells. Adv Exp Med Biol 1003:71–91. https://doi.org/10.1007/978-3-319-57613-8_4
Sun HJ, Wu ZY, Nie XW, Bian JS (2020) Role of endothelial dysfunction in cardiovascular diseases: the link between inflammation and hydrogen sulfide. Front Pharmacol 10:1568. https://doi.org/10.3389/fphar.2019.01568
Wu MY, Li CJ, Hou MF, Chu PY (2017) New insights into the role of inflammation in the pathogenesis of atherosclerosis. Int J Mol Sci 18(10):2034. https://doi.org/10.3390/ijms18102034
Xu S, Ilyas I, Little PJ, Li H, Kamato D, Zheng X, Luo S, Li Z, Liu P, Han J, Harding IC, Ebong EE, Cameron SJ, Stewart AG, Weng J (2021) Endothelial dysfunction in atherosclerotic cardiovascular diseases and beyond: from mechanism to pharmacotherapies. Pharmacol Rev 73(3):924–967. https://doi.org/10.1124/pharmrev.120.000096
Cahill PA, Redmond EM (2016) Vascular endothelium - gatekeeper of vessel health. Atherosclerosis 248:97–109. https://doi.org/10.1016/j.atherosclerosis.2016.03.007
Abraham D, Distler O (2007) How does endothelial cell injury start? The role of endothelin in systemic sclerosis. Arthritis research & therapy 9 Suppl 2(Suppl 2):S2. https://doi.org/10.1186/ar2186
Ghosh A, Gao L, Thakur A, Siu PM, Lai CWK (2017) Role of free fatty acids in endothelial dysfunction. J Biomed Sci 24(1):50. https://doi.org/10.1186/s12929-017-0357-5
Brown AJ, Teng Z, Evans PC, Gillard JH, Samady H, Bennett MR (2016) Role of biomechanical forces in the natural history of coronary atherosclerosis. Nat Rev Cardiol 13(4):210–220. https://doi.org/10.1038/nrcardio.2015.203
Guazzi M, Phillips SA, Arena R, Lavie CJ (2015) Endothelial dysfunction and lung capillary injury in cardiovascular diseases. Prog Cardiovasc Dis 57(5):454–462. https://doi.org/10.1016/j.pcad.2014.11.003
Haybar H, Shahrabi S, Rezaeeyan H, Shirzad R, Saki N (2019) Endothelial cells: from dysfunction mechanism to pharmacological effect in cardiovascular disease. Cardiovasc Toxicol 19(1):13–22. https://doi.org/10.1007/s12012-018-9493-8
Yamagata K (2017) Docosahexaenoic acid regulates vascular endothelial cell function and prevents cardiovascular disease. Lipids Health Dis 16(1):118. https://doi.org/10.1186/s12944-017-0514-6
Burnstock G, Ralevic V (2014) Purinergic signaling and blood vessels in health and disease. Pharmacol Rev 66(1):102–192. https://doi.org/10.1124/pr.113.008029
Burnstock G (2016) Purinergic signalling and endothelium. Curr Vasc Pharmacol 14(2):130–145. https://doi.org/10.2174/1570161114666151202204948
Burnstock G (1972) Purinergic nerves. Pharmacol Rev 24(3):509–581
Burnstock G (1976) Purinergic receptors. J Theor Biol 62(2):491–503. https://doi.org/10.1016/0022-5193(76)90133-8
Lazarowski ER (2012) Vesicular and conductive mechanisms of nucleotide release. Purinergic Signalling 8(3):359–373. https://doi.org/10.1007/s11302-012-9304-9
Reiss AB, Grossfeld D, Kasselman LJ, Renna HA, Vernice NA, Drewes W, Konig J, Carsons SE, DeLeon J (2019) Adenosine and the cardiovascular system. American journal of cardiovascular drugs: drugs, devices, and other interventions 19(5):449–464. https://doi.org/10.1007/s40256-019-00345-5
Lohman AW, Leskov IL, Butcher JT, Johnstone SR, Stokes TA, Begandt D, DeLalio LJ, Best AK, Penuela S, Leitinger N, Ravichandran KS, Stokes KY, Isakson BE (2015) Pannexin 1 channels regulate leukocyte emigration through the venous endothelium during acute inflammation. Nat Commun 6:7965. https://doi.org/10.1038/ncomms8965
Zhou R, Dang X, Sprague RS, Mustafa SJ, Zhou Z (2020) Alteration of purinergic signaling in diabetes: focus on vascular function. J Mol Cell Cardiol 140:1–9. https://doi.org/10.1016/j.yjmcc.2020.02.004
Nell PG, Albrecht-Küpper B (2009) The adenosine A1 receptor and its ligands. Prog Med Chem 47:163–201. https://doi.org/10.1016/s0079-6468(08)00204-x
Burnstock G, Pelleg A (2015) Cardiac purinergic signalling in health and disease. Purinergic Signalling 11(1):1–46. https://doi.org/10.1007/s11302-014-9436-1
Abbracchio MP, Burnstock G, Verkhratsky A, Zimmermann H (2009) Purinergic signalling in the nervous system: an overview. Trends Neurosci 32(1):19–29. https://doi.org/10.1016/j.tins.2008.10.001
Fountain SJ (2013) Primitive ATP-activated P2X receptors: discovery, function and pharmacology. Front Cell Neurosci 7:247. https://doi.org/10.3389/fncel.2013.00247
Inoue K (2021) Nociceptive signaling of P2X receptors in chronic pain states. Purinergic Signalling 17(1):41–47. https://doi.org/10.1007/s11302-020-09743-w
Tam TH, Salter MW (2021) Purinergic signalling in spinal pain processing. Purinergic Signalling 17(1):49–54. https://doi.org/10.1007/s11302-020-09748-5
Zhang WJ (2021) Effect of P2X purinergic receptors in tumor progression and as a potential target for anti-tumor therapy. Purinergic Signalling 17(1):151–162. https://doi.org/10.1007/s11302-020-09761-8
Wihlborg AK, Malmsjö M, Eyjolfsson A, Gustafsson R, Jacobson K, Erlinge D (2003) Extracellular nucleotides induce vasodilatation in human arteries via prostaglandins, nitric oxide and endothelium-derived hyperpolarising factor. Br J Pharmacol 138(8):1451–1458. https://doi.org/10.1038/sj.bjp.0705186
Eun SY, Park SW, Lee JH, Chang KC, Kim HJ (2014) P2Y(2)R activation by nucleotides released from oxLDL-treated endothelial cells (ECs) mediates the interaction between ECs and immune cells through RAGE expression and reactive oxygen species production. Free Radical Biol Med 69:157–166. https://doi.org/10.1016/j.freeradbiomed.2014.01.022
Burnstock G (2006) Pathophysiology and therapeutic potential of purinergic signaling. Pharmacol Rev 58(1):58–86. https://doi.org/10.1124/pr.58.1.5
Bogle RG, Coade SB, Moncada S, Pearson JD, Mann GE (1991) Bradykinin and ATP stimulate L-arginine uptake and nitric oxide release in vascular endothelial cells. Biochem Biophys Res Commun 180(2):926–932. https://doi.org/10.1016/s0006-291x(05)81154-4
Van Coevorden A, Boeynaems JM (1984) Physiological concentrations of ADP stimulate the release of prostacyclin from bovine aortic endothelial cells. Prostaglandins 27(4):615–626. https://doi.org/10.1016/0090-6980(84)90097-2
Lee NT, Ong LK, Gyawali P, Nassir C, Mustapha M, Nandurkar HH, Sashindranath M (2021) Role of purinergic signalling in endothelial dysfunction and thrombo-inflammation in ischaemic stroke and cerebral small vessel disease. Biomolecules 11(7):994. https://doi.org/10.3390/biom11070994
Di Pietro N, Formoso G, Pandolfi A (2016) Physiology and pathophysiology of oxLDL uptake by vascular wall cells in atherosclerosis. Vascul Pharmacol 84:1–7. https://doi.org/10.1016/j.vph.2016.05.013
Jin H, Ko YS, Park SW, Kim HJ (2019) P2Y(2)R activation by ATP induces oxLDL-mediated inflammasome activation through modulation of mitochondrial damage in human endothelial cells. Free Radical Biol Med 136:109–117. https://doi.org/10.1016/j.freeradbiomed.2019.04.004
Borea PA, Gessi S, Merighi S, Vincenzi F, Varani K (2018) Pharmacology of adenosine receptors: the state of the art. Physiol Rev 98(3):1591–1625. https://doi.org/10.1152/physrev.00049.2017
Burnstock G (2009) Purinergic regulation of vascular tone and remodelling. Auton Autacoid Pharmacol 29(3):63–72. https://doi.org/10.1111/j.1474-8673.2009.00435.x
Wang H, Zhang W, Zhu C, Bucher C, Blazar BR, Zhang C, Chen JF, Linden J, Wu C, Huo Y (2009) Inactivation of the adenosine A2A receptor protects apolipoprotein E-deficient mice from atherosclerosis. Arterioscler Thromb Vasc Biol 29(7):1046–1052. https://doi.org/10.1161/atvbaha.109.188839
Gessi S, Fogli E, Sacchetto V, Merighi S, Varani K, Preti D, Leung E, Maclennan S, Borea PA (2010) Adenosine modulates HIF-1{alpha}, VEGF, IL-8, and foam cell formation in a human model of hypoxic foam cells. Arterioscler Thromb Vasc Biol 30(1):90–97. https://doi.org/10.1161/atvbaha.109.194902
Rekik M, Mustafa JS (2003) Modulation of A2A adenosine receptors and associated G alphas proteins by ZM 241385 treatment of porcine coronary artery. J Cardiovasc Pharmacol 42(6):736–744. https://doi.org/10.1097/00005344-200312000-00007
McPherson JA, Barringhaus KG, Bishop GG, Sanders JM, Rieger JM, Hesselbacher SE, Gimple LW, Powers ER, Macdonald T, Sullivan G, Linden J, Sarembock IJ (2001) Adenosine A(2A) receptor stimulation reduces inflammation and neointimal growth in a murine carotid ligation model. Arterioscler Thromb Vasc Biol 21(5):791–796. https://doi.org/10.1161/01.atv.21.5.791
Teng B, Smith JD, Rosenfeld ME, Robinet P, Davis ME, Morrison RR, Mustafa SJ (2014) A1 adenosine receptor deficiency or inhibition reduces atherosclerotic lesions in apolipoprotein E deficient mice. Cardiovasc Res 102(1):157–165. https://doi.org/10.1093/cvr/cvu033
Wang L, Karlsson L, Moses S, Hultgårdh-Nilsson A, Andersson M, Borna C, Gudbjartsson T, Jern S, Erlinge D (2002) P2 receptor expression profiles in human vascular smooth muscle and endothelial cells. J Cardiovasc Pharmacol 40(6):841–853. https://doi.org/10.1097/00005344-200212000-00005
Rumjahn SM, Yokdang N, Baldwin KA, Thai J, Buxton IL (2009) Purinergic regulation of vascular endothelial growth factor signaling in angiogenesis. Br J Cancer 100(9):1465–1470. https://doi.org/10.1038/sj.bjc.6604998
Mühleder S, Fuchs C, Basílio J, Szwarc D, Pill K, Labuda K, Slezak P, Siehs C, Pröll J, Priglinger E, Hoffmann C, Junger WG, Redl H, Holnthoner W (2020) Purinergic P2Y(2) receptors modulate endothelial sprouting. Cellular and molecular life sciences : CMLS 77(5):885–901. https://doi.org/10.1007/s00018-019-03213-2
Chen X, Qian S, Hoggatt A, Tang H, Hacker TA, Obukhov AG, Herring PB, Seye CI (2017) Endothelial cell-specific deletion of P2Y2 receptor promotes plaque stability in atherosclerosis-susceptible ApoE-null mice. Arterioscler Thromb Vasc Biol 37(1):75–83. https://doi.org/10.1161/atvbaha.116.308561
Vieceli Dalla Sega F, Fortini F, Aquila G, Pavasini R, Biscaglia S, Bernucci D, Del Franco A, Tonet E, Rizzo P, Ferrari R, Campo G (2018) Ticagrelor improves endothelial function by decreasing circulating epidermal growth factor (EGF). Front Physiol 9:337. https://doi.org/10.3389/fphys.2018.00337
Baeyens N, Bandyopadhyay C, Coon BG, Yun S, Schwartz MA (2016) Endothelial fluid shear stress sensing in vascular health and disease. J Clin Investig 126(3):821–828. https://doi.org/10.1172/jci83083
Russo TA, Banuth AMM, Nader HB, Dreyfuss JL (2020) Altered shear stress on endothelial cells leads to remodeling of extracellular matrix and induction of angiogenesis. PLoS ONE 15(11):e0241040. https://doi.org/10.1371/journal.pone.0241040
Siasos G, Sara JD, Zaromytidou M, Park KH, Coskun AU, Lerman LO, Oikonomou E, Maynard CC, Fotiadis D, Stefanou K, Papafaklis M, Michalis L, Feldman C, Lerman A, Stone PH (2018) Local low shear stress and endothelial dysfunction in patients with nonobstructive coronary atherosclerosis. J Am Coll Cardiol 71(19):2092–2102. https://doi.org/10.1016/j.jacc.2018.02.073
Chatterjee S (2018) Endothelial mechanotransduction, redox signaling and the regulation of vascular inflammatory pathways. Front Physiol 9:524. https://doi.org/10.3389/fphys.2018.00524
Sathanoori R, Rosi F, Gu BJ, Wiley JS, Müller CE, Olde B, Erlinge D (2015) Shear stress modulates endothelial KLF2 through activation of P2X4. Purinergic Signalling 11(1):139–153. https://doi.org/10.1007/s11302-014-9442-3
Burnstock G (2017) Purinergic signaling in the cardiovascular system. Circ Res 120(1):207–228. https://doi.org/10.1161/circresaha.116.309726
Yamamoto K, Korenaga R, Kamiya A, Ando J (2000) Fluid shear stress activates Ca(2+) influx into human endothelial cells via P2X4 purinoceptors. Circ Res 87(5):385–391. https://doi.org/10.1161/01.res.87.5.385
Yamamoto K, Sokabe T, Matsumoto T, Yoshimura K, Shibata M, Ohura N, Fukuda T, Sato T, Sekine K, Kato S, Isshiki M, Fujita T, Kobayashi M, Kawamura K, Masuda H, Kamiya A, Ando J (2006) Impaired flow-dependent control of vascular tone and remodeling in P2X4-deficient mice. Nat Med 12(1):133–137. https://doi.org/10.1038/nm1338
Green JP, Souilhol C, Xanthis I, Martinez-Campesino L, Bowden NP, Evans PC, Wilson HL (2018) Atheroprone flow activates inflammation via endothelial ATP-dependent P2X7-p38 signalling. Cardiovasc Res 114(2):324–335. https://doi.org/10.1093/cvr/cvx213
Sathanoori R, Bryl-Gorecka P, Müller CE, Erb L, Weisman GA, Olde B, Erlinge D (2017) P2Y(2) receptor modulates shear stress-induced cell alignment and actin stress fibers in human umbilical vein endothelial cells. Cellular and molecular life sciences : CMLS 74(4):731–746. https://doi.org/10.1007/s00018-016-2365-0
Wang S, Iring A, Strilic B, Albarrán Juárez J, Kaur H, Troidl K, Tonack S, Burbiel JC, Müller CE, Fleming I, Lundberg JO, Wettschureck N, Offermanns S (2015) P2Y2 and Gq/G11 control blood pressure by mediating endothelial mechanotransduction. The Journal of clinical investigation 125(8):3077-3086. https://doi.org/10.1172/jci81067
Idzko M, Ferrari D, Eltzschig HK (2014) Nucleotide signalling during inflammation. Nature 509(7500):310–317. https://doi.org/10.1038/nature13085
Eltzschig HK, Eckle T, Mager A, Küper N, Karcher C, Weissmüller T, Boengler K, Schulz R, Robson SC, Colgan SP (2006) ATP release from activated neutrophils occurs via connexin 43 and modulates adenosine-dependent endothelial cell function. Circ Res 99(10):1100–1108. https://doi.org/10.1161/01.Res.0000250174.31269.70
Sathanoori R, Swärd K, Olde B, Erlinge D (2015) The ATP receptors P2X7 and P2X4 modulate high glucose and palmitate-induced inflammatory responses in endothelial cells. PLoS ONE 10(5):e0125111. https://doi.org/10.1371/journal.pone.0125111
Savio LEB, de Andrade MP, da Silva CG, Coutinho-Silva R (2018) The P2X7 receptor in inflammatory diseases: angel or demon? Front Pharmacol 9:52. https://doi.org/10.3389/fphar.2018.00052
Komalavilas P, Luo W, Guth CM, Jolayemi O, Bartelson RI, Cheung-Flynn J, Brophy CM (2017) Vascular surgical stretch injury leads to activation of P2X7 receptors and impaired endothelial function. PLoS ONE 12(11):e0188069. https://doi.org/10.1371/journal.pone.0188069
Stachon P, Heidenreich A, Merz J, Hilgendorf I, Wolf D, Willecke F, von Garlen S, Albrecht P, Härdtner C, Ehrat N, Hoppe N, Reinöhl J, von Zur MC, Bode C, Idzko M, Zirlik A (2017) P2X(7) Deficiency blocks lesional inflammasome activity and ameliorates atherosclerosis in mice. Circulation 135(25):2524–2533. https://doi.org/10.1161/circulationaha.117.027400
Lombardi M, Mantione ME, Baccellieri D, Ferrara D, Castellano R, Chiesa R, Alfieri O, Foglieni C (2017) P2X7 receptor antagonism modulates IL-1β and MMP9 in human atherosclerotic vessels. Sci Rep 7(1):4872. https://doi.org/10.1038/s41598-017-05137-y
Pelegrin P, Surprenant A (2006) Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J 25(21):5071–5082. https://doi.org/10.1038/sj.emboj.7601378
Hechler B, Freund M, Ravanat C, Magnenat S, Cazenave JP, Gachet C (2008) Reduced atherosclerotic lesions in P2Y1/apolipoprotein E double-knockout mice: the contribution of non-hematopoietic-derived P2Y1 receptors. Circulation 118(7):754–763. https://doi.org/10.1161/circulationaha.108.788927
Stachon P, Geis S, Peikert A, Heidenreich A, Michel NA, Ünal F, Hoppe N, Dufner B, Schulte L, Marchini T, Cicko S, Ayata K, Zech A, Wolf D, Hilgendorf I, Willecke F, Reinöhl J, von Zur MC, Bode C, Idzko M, Zirlik A (2016) Extracellular ATP induces vascular inflammation and atherosclerosis via purinergic receptor Y2 in mice. Arterioscler Thromb Vasc Biol 36(8):1577–1586. https://doi.org/10.1161/atvbaha.115.307397
Stachon P, Peikert A, Michel NA, Hergeth S, Marchini T, Wolf D, Dufner B, Hoppe N, Ayata CK, Grimm M, Cicko S, Schulte L, Reinöhl J, von zur Muhlen C, Bode C, Idzko M, Zirlik A, (2014) P2Y6 deficiency limits vascular inflammation and atherosclerosis in mice. Arterioscler Thromb Vasc Biol 34(10):2237–2245. https://doi.org/10.1161/atvbaha.114.303585
Riegel AK, Faigle M, Zug S, Rosenberger P, Robaye B, Boeynaems JM, Idzko M, Eltzschig HK (2011) Selective induction of endothelial P2Y6 nucleotide receptor promotes vascular inflammation. Blood 117(8):2548–2555. https://doi.org/10.1182/blood-2010-10-313957
Gündüz D, Tanislav C, Schlüter KD, Schulz R, Hamm C, Aslam M (2017) Effect of ticagrelor on endothelial calcium signalling and barrier function. Thromb Haemost 117(2):371–381. https://doi.org/10.1160/th16-04-0273
Dănilă MD, Privistirescu A, Duicu OM, Rațiu CD, Angoulvant D, Muntean DM, Sturza A (2017) The effect of purinergic signaling via the P(2)Y(11) receptor on vascular function in a rat model of acute inflammation. Mol Cell Biochem 431(1–2):37–44. https://doi.org/10.1007/s11010-017-2973-5
Jin J, Quinton TM, Zhang J, Rittenhouse SE, Kunapuli SP (2002) Adenosine diphosphate (ADP)-induced thromboxane A(2) generation in human platelets requires coordinated signaling through integrin alpha(IIb)beta(3) and ADP receptors. Blood 99(1):193–198. https://doi.org/10.1182/blood.v99.1.193
Kaczmarek E, Koziak K, Sévigny J, Siegel JB, Anrather J, Beaudoin AR, Bach FH, Robson SC (1996) Identification and characterization of CD39/vascular ATP diphosphohydrolase. J Biol Chem 271(51):33116–33122. https://doi.org/10.1074/jbc.271.51.33116
Marcus AJ, Broekman MJ, Drosopoulos JH, Islam N, Alyonycheva TN, Safier LB, Hajjar KA, Posnett DN, Schoenborn MA, Schooley KA, Gayle RB, Maliszewski CR (1997) The endothelial cell ecto-ADPase responsible for inhibition of platelet function is CD39. J Clin Investig 99(6):1351–1360. https://doi.org/10.1172/jci119294
Kanthi Y, Hyman MC, Liao H, Baek AE, Visovatti SH, Sutton NR, Goonewardena SN, Neral MK, Jo H, Pinsky DJ (2015) Flow-dependent expression of ectonucleotide tri(di)phosphohydrolase-1 and suppression of atherosclerosis. J Clin Investig 125(8):3027–3036. https://doi.org/10.1172/jci79514
Gimbel M, Qaderdan K, Willemsen L, Hermanides R, Bergmeijer T, de Vrey E, Heestermans T, Tjon Joe Gin M, Waalewijn R, Hofma S, den Hartog F, Jukema W, von Birgelen C, Voskuil M, Kelder J, Deneer V, Ten Berg J (2020) Clopidogrel versus ticagrelor or prasugrel in patients aged 70 years or older with non-ST-elevation acute coronary syndrome (POPular AGE): the randomised, open-label, non-inferiority trial. Lancet (London, England) 395(10233):1374–1381. https://doi.org/10.1016/s0140-6736(20)30325-1
Johnston SC, Amarenco P, Denison H, Evans SR, Himmelmann A, James S, Knutsson M, Ladenvall P, Molina CA, Wang Y (2020) Ticagrelor and aspirin or aspirin alone in acute ischemic stroke or TIA. N Engl J Med 383(3):207–217. https://doi.org/10.1056/NEJMoa1916870
Funding
This work was supported by the National Natural Science Foundation of China (82160941, 82160937).
Author information
Authors and Affiliations
Contributions
Xian-Ming Wu, Ning Zhang, and Xiao-Fang Yang drafted the manuscript. All authors contributed to manuscript revision and have read and approved the submitted version.
Corresponding author
Ethics declarations
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflicts of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, XM., Zhang, N., Li, JS. et al. Purinergic receptors mediate endothelial dysfunction and participate in atherosclerosis. Purinergic Signalling 19, 265–272 (2023). https://doi.org/10.1007/s11302-021-09839-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-021-09839-x