
Abstract
Cancer therapy by endogenous or adoptively transferred anti-tumor T cells is considered complementary to conventional cancer treatment by surgery, radiotherapy or chemotherapy. However, the scope of promising immunotherapeutic protocols is currently limited because tumors can create a ‘hostile–immunosuppressive microenvironment that prevents their destruction by anti-tumor T cells. There is a possibility to develop better and more effective immunotherapies by inactivating mechanisms that inhibit anti-tumor T cells in the tumor microenvironment and thereby protect cancerous tissues from immune damage. This may be now possible because of the recent demonstration that genetic deletion of immunosuppressive A2A and A2B adenosine receptors (A2AR and A2BR) or their pharmacological inactivation can prevent the inhibition of anti-tumor T cells by the hypoxic tumor microenvironment and as a result facilitate full tumor rejection [Ohta A, Gorelik E, Prasad SJ et al (2006) Proc Natl Acad Sci USA 103(35):13132–3137]. This approach is based on in vivo genetic evidence that A2AR play a critical role in the protection of normal tissues from overactive immune cells in acutely inflamed and hypoxic areas. The observations of much improved T-cell-mediated rejection of tumors in mice with inactivated A2AR strongly suggest that A2AR also protects hypoxic cancerous tissues and that A2AR should be inactivated in order to improve tumor rejection by anti-tumor T cells.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Hypoxia-mediated protection of cancerous tissues from anti-tumor T cells
The important role of T cells in cancer immunosurveillance is now strongly supported by genetic studies in mice [1, 2] and human studies [3–5]. The presence of T cells in solid tumors is predictive of improved clinical outcome in some cases of human colorectal cancer [3], esophageal carcinoma [4] and ovarian cancer [5, 6]. Among recent advances in cancer immunology are reports of relatively successful adoptive T-cell therapy with selected forms of cancer [7] and better designs of cancer vaccines and treatments that improve the development of endogenous anti-tumor CD8+ T-cell effectors [8–12].
However, the potential of T-cell-based immunotherapy is limited by various immune escape mechanisms. The long sought after explanation of the co-existence of tumors and of anti-tumor immune cells in a patient (‘Hellstrom Paradox– [13–15] or in a mouse [16] has been a challenging problem to solve for more than 35 years. Why do anti-tumor T cells fail to completely and reliably destroy tumors in vivo even when the ability to recognize tumors is not the limiting factor and when very high numbers of highly lytic anti-tumor T cells are injected in a cancer patient [13, 15] or tumor-bearing mice [16]? What is it in the tumor microenvironment in vivo that prevents tumor destruction by the tumor-specific and highly lytic in vitro anti-tumor CD8+ T cells?
Some of the failures of adoptive therapy could be due to an inhibition of anti-tumor T cells by ‘passenger–T suppressor cells [17] and/or by cytokines, which inhibit the development of the anti-tumor T cells [18, 19]. In addition, convincing data from clinical and animal studies have shown that the tumor microenvironment itself can suppress anti-tumor T cells [13]. Although substantial progress has been made in studying tumor escape from anti-tumor immune response [19], the mechanism of inhibition of lethal anti-tumor T cells in a poorly understood ‘hostile–tumor microenvironment [13–16] is still perplexing. The importance of dealing with these issues and eliminating negative effects of the tumor microenvironment on cancer therapies is well recognized, as reflected in current extensive studies [20]. The puzzling inhibition of anti-tumor T cells in the tumor microenvironment provides yet another challenge [13, 15, 16].
Important clues to further understanding cancerous tissue protection from anti-tumor T cells have been provided by our recent insights into mechanisms of normal tissue protection from overactive immune cells [1]. We found that the still healthy normal tissues in acutely inflamed and therefore hypoxic areas under immune attack are protected from the continuing collateral immune damage by immunosuppressive signaling through extracellular A2 receptor [21–25] on the surface of immune cells.
Delayed negative feedback mechanism of protection of normal tissues from overactive immune cells by extracellular adenosine A2 receptors on the surface of immune cells
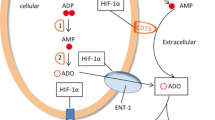
The A2 receptor-mediated mechanism of normal tissue protection from overactive immune cells in inflamed areas may be triggered by excessive collateral immune damage to endothelial cells and microcirculation with ensuing interruption of normal blood and oxygen supply [23]. In turn, this results in local tissue hypoxia [23, 24]. The hypoxia is associated with: (1) decrease in intracellular ATP [26]; (2) increase in intracellular AMP [26]; (3) inhibition of adenosine kinase [27]; (4) activation of 5–nucleotidase [26, 28]; (5) accumulation of intracellular adenosine [26, 27]; (6) subsequent transport or diffusion of adenosine from the cell into extracellular space [26].
The sufficiently high levels of extracellular adenosine trigger signaling by A2A receptors (A2AR) and/or A2B receptors (A2BR) on the surface of surrounding cells including activated T cells. This chain of events culminates in inhibition of overactive immune cells in a delayed negative feedback manner [22–25] due to the well-established immunosuppression by A2 adenosine receptors [22–34] (Fig. 1).

Model of hypoxia-mediated adenosine protection of tumors from T cells
Immunosuppression by extracellular A2AR and A2BR on immune cells
There are four different and widely distributed adenosine receptors: A1, A2A, A2B, and A3 [35, 36]. The high-affinity A1 receptor and low-affinity A3 receptor are Gi-protein coupled. The cAMP-elevating Gs-protein coupled A2 receptors are subdivided into high-affinity A2AR and low-affinity A2BR. Adenosine receptors are known to be immunosuppressive (reviewed in [23, 24, 36]). The CD8+ T cells, including anti-tumor CD8+ T cells and human T cells, predominantly express A2AR and A2BR and not the A3 receptor [29, 31, 37]. The cAMP-elevating signaling through A2AR or A2BR in T cells results in inhibition of TCR-triggered activation of T cells [32–34, 38] and of many effector functions, including proliferation, expansion and secretion by T cells of such important anti-tumor cytokines as IFN-γ [29, 39, 40] and TNF-α [41]. In contrast to our focus on A2AR and A2BR as the key immunosuppressive molecules in T cells, others have suggested instead the A3 adenosine receptors [42].
Tumor hypoxia and extracellular adenosine
Many solid tumors are characterized by an insufficient oxygen supply and transient or chronic hypoxia in some microenvironments [43, 44]. Tumor hypoxia may contribute to the propagation of oncogenic signals in the tumor microenvironment as was shown in demonstration of the switch to the angiogenic phenotype [45]. As a result, tumor hypoxia is associated with poor prognosis for the patient [46, 47].
Remarkable progress has been made in measuring and discriminating effects of moderate versus deep tumor hypoxia [46, 48]. New conceptual and methodological approaches in the area of cancer research offer novel opportunities for other studies far beyond cancer research. Indeed, an interrupted blood supply and transient or chronic hypoxia in some microenvironments are observed not only in cancerous tissues [43, 44, 46–48], but also in inflamed normal tissues [49, 53]. The hypoxia-associated accumulation of intracellular adenosine [26] in tumors and subsequent transport or diffusion of adenosine from the cell into extracellular space is one of the important mechanisms of extracellular adenosine accumulation in the tumor microenvironment [50].
It is important to emphasize that many normal tissue microenvironments are hypoxic to start with (reviewed in [23, 24]). Therefore, individual cancerous cells in newly-arising, small tumors in such anoxic ‘shelters–may be protected from T cells by the hypoxia-produced adenosine, but in this case it will not be the ‘tumor-hypoxia produced adenosine–but the ‘tissue microenvironment-produced adenosine– which then will be helped by contributions from adenosine produced by tumor tissues themselves.
It was important to establish whether genetic deletion or pharmacological inactivation of A2AR and/or A2BR by drugs will make anti-tumor T cells more resistant to inhibition in the tumor microenvironment and thereby facilitate tumor destruction [1]. If that was the case, then the translation into clinical settings could be unusually immediate due to the well-established safety profiles and known limitations of available A2AR/A2BR antagonists, including 1,3,7-trimethylxanthine (caffeine).
Genetic and pharmacological in vitro and in vivo evidence shows that: (1) anti-tumor CD8+ T cells do express inhibitory A2AR and A2BR; (2) solid tumors are surrounded by extracellular adenosine; (3) A2 adenosine receptors are capable of inhibiting anti-tumor T cells in vitro; (4) most importantly, dramatic and complete rejection of tumors in mice is accomplished by genetic A2AR inactivation [1], providing the strongest evidence for the function of this mechanism in protection of cancerous tissues from anti-tumor CD8+ T cells in vivo; (5) T cells may be made much more resistant to inhibition in the tumor microenvironment by pharmacologically targeting the A2 receptors in vivo and this is reflected in observations of significant tumor growth retardation due to antagonism of A2AR and A2BR by drugs, including an A2 receptor antagonist caffeine; (6) better tumor rejection observed in A2 receptor antagonist-treated mice is at least partially explained by stronger inhibition of pro-tumor neo-vascularization due to an increased IFN-γ production by ‘de-inhibited–anti-tumor CD8+ T cells in the tumor microenvironment [1].
Importantly, the deficiency in A2AR did not prevent the establishment or the early growth of small inoculated tumors; rather, it improved the destruction of well-established tumors after the host has developed anti-CL8-1 or anti-RMA CD8+ T cells [1]. Observations of complete rejection of established melanoma and solid T lymphoma tumors in ~60% of A2AR-/- mice with no tumors rejected in A2AR+/+ controls indicated that A2AR do play an important role in the mechanism of tumor protection from immune cells [1].
Interestingly, the CD8+ T-cell-mediated anti-CL8-1 melanoma response in the A2AR-/- host was accompanied by a different appearance of tumors and of tumor-rejecting mice compared with the tumor-permissive A2AR-expressing wild-type control mice. While the solid, spherical, and well-defined tumors were continuously increasing in size in wild-type mice, the soft, flat, poorly defined tumors in A2AR-/- mice often showed central necrosis, and their disappearance and healing were, in some mice, accompanied by hair loss [1]. These signs of spontaneously resolved autoimmunity resemble autoimmunity in vaccinated melanoma-rejecting mice [51] and in melanoma patients undergoing immunotherapy with melanoma antigen-specific T cells [52]. These observations provide yet more strong evidence that inactivating the adenosine-A2AR/A2BR pathway did facilitate effector functions of anti-tumor T cells.
Also demonstrated was the feasibility of the pharmacological anti-adenosinergic strategy in several tumor rejection assays by endogenously developed and adoptively transferred T cells. The treatment with antagonists of A2 receptors significantly delayed the onset of rapid growth of CL8-1 melanoma, even if injections of antagonists ZM241385 or caffeine started after tumors reached 8 mm in diameter size [1].
Inhibition of tumor neovascularization by interruption of tumor-protection by the ‘hypoxia-adenosine-A2R–pathway
As it was shown, the dramatic inhibition of tumor neovascularization can be achieved by A2 receptor antagonists [1]. These data suggest that the improvement of tumor destruction by interruption of A2 receptor signaling could be due to the release of CD8+ T cells from A2R-mediated inhibition of IFN-γ production and subsequent IFN-γ -mediated inhibition of pro-tumor angiogenesis [1]. These observations are in agreement with recent findings that the angiogenesis-inhibiting properties of T-cell-produced IFN-γ are important for anti-tumor action of T cells in vivo [39].
The genetic and pharmacological data described above are interpreted both as: (1) evidence for the role of a tumor hypoxia-adenosine-A2A/A2B receptor pathway in the physiological mechanism of cancerous tissue protection from anti-tumor T cells, and (2) promising proof of principle for future investigations of this strategy to enhance the immune-mediated tumor destruction by genetic targeting of A2AR and A2BR, or by combined inactivation of both A2 receptors.
The described retardation of tumor growth by A2A antagonists was observed even under conditions unfavorable for tumor rejection because the used drugs are ‘competitive–antagonists and at levels found in vivo they are expected to prevent inhibitory signaling of only ~30% of A2AR [1]. Moreover, such incomplete inhibition of A2AR by antagonists would be lasting no more than 30% of time due to short half-life of antagonists in vivo. This explains incomplete tumor rejection by drugs compared with genetic inactivation of A2AR, which accomplishes 100% deletion of receptors for 100% of the time [1]. In view of these considerations, the retardation of tumor growth by even inferior drugs bodes well for stronger anti-tumor effects of better and longer-lived antagonists.
Limitations of genetic targeting of A2AR alone
The described approach resulted so far in complete tumor rejection in only ~60% of mice with genetically targeted A2AR [1]. The tumor’s escape from CD8+ T cells that was observed in ~40% of A2AR-/- mice was not due to the loss of antigen-presenting molecules, but could be explained by the expression of inhibitory, lower affinity A2BR on A2AR-/- CD8+ T cells. While A2AR/A2BR antagonists ZM241385 and caffeine are capable to significantly delay tumor growth by relieving anti-tumor CD8+ T cells from inhibition, these drugs did not accomplish the complete rejection of immunogenic tumor [1]. Therefore, it is very important to further improve the therapeutic window. If the failure of tested antagonists to completely reject tumors are due to their short half-life, then the most straightforward alternative approach would be to use the longer-lived antagonists.
Conclusion
Despite strong evidence that the adenosine-A2A adenosine receptor pathway in T cells represent important novel target for improving cancer immunotherapy, the role of A2BR on T cells is not yet fully identified. It is up to future studies to find whether A2BR also may account for the failure of CD8+ T cells to destroy tumors in ~40% of A2AR-/- mice. Most importantly, the data described above strongly suggest the need to attempt the complete rejection of tumors by adoptively transferred or endogenous anti-tumor T cells by targeting both A2AR and A2BR.
References
Ohta A, Gorelik E, Prasad SJ et al (2006) A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci USA 103(35):13132–3137
Shankaran V, Ikeda H, Bruce AT et al (2001) IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 410:1107–111
Naito Y, Saito K, Shiiba K et al (1998) CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res 58:3491–494
Schumacher K, Haensch W, Roefzaad C et al (2001) Prognostic significance of activated CD8(+) T cell infiltrations within esophageal carcinomas. Cancer Res 61:3932–936
Zhang L, Conejo-Garcia JR, Katsaros D et al (2003) Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med 348:203–13
Nagorsen D, Scheibenbogen C, Marincola F et al (2003) Natural T cell immunity against cancer. Clin Cancer Res 9:4296–303
Rosenberg SA (2001) Progress in human tumour immunology and immunotherapy. Nature 411:380–84
Chambers CA, Kuhns MS, Egen JG et al (2001) CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol 19:565–94
Melief CJ, Van Der Burg SH, Toes RE et al (2002) Effective therapeutic anticancer vaccines based on precision guiding of cytolytic T lymphocytes. Immunol Rev 188:177–82
Hurwitz AA, Yanover P, Markowitz M et al (2003) Prostate cancer: advances in immunotherapy. BioDrugs 17:131–38
Ribas A, Butterfield LH, Glaspy JA et al (2003) Current developments in cancer vaccines and cellular immunotherapy. J Clin Oncol 21:2415–432
Wang XY, Kazim L, Repasky EA et al (2003) Immunization with tumor-derived ER chaperone grp170 elicits tumor-specific CD8+ T-cell responses and reduces pulmonary metastatic disease. Int J Cancer 105:226–31
Marincola FM, Jaffee EM, Hicklin DJ et al (2000) Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv Immunol 74:181–73
Hellstrom I, Hellstrom KE, Pierce GE et al (1968) Cellular and humoral immunity to different types of human neoplasms. Nature 220:1352–354
Rosenberg SA (2001) Progress in the development of immunotherapy for the treatment of patients with cancer. J Intern Med 250:462–75
Hanson HL, Donermeyer DL, Ikeda H et al (2000) Eradication of established tumors by CD8+ T cell adoptive immunotherapy. Immunity 13:265–76
Peng L, Kjaergaard J, Plautz GE et al (2002) Tumor-induced L-selectinhigh suppressor T cells mediate potent effector T cell blockade and cause failure of otherwise curative adoptive immunotherapy. J Immunol 169:4811–821
Halak BK, Maguire HC Jr, Lattime EC (1999) Tumor-induced interleukin-10 inhibits type 1 immune responses directed at a tumor antigen as well as a non-tumor antigen present at the tumor site. Cancer Res 59:911–17
Kobie JJ, Wu RS, Kurt RA et al (2003) Transforming growth factor beta inhibits the antigen-presenting functions and antitumor activity of dendritic cell vaccines. Cancer Res 63:1860–864
Jung YD, Ahmad SA, Akagi Y et al (2000) Role of the tumor microenvironment in mediating response to anti-angiogenic therapy. Cancer Metastasis Rev 19:147–57
Ohta A, Sitkovsky M (2001) Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 414:916–20
Sitkovsky MV (2003) Use of the A(2A) adenosine receptor as a physiological immunosuppressor and to engineer inflammation in vivo. Biochem Pharmacol 65:493–01
Sitkovsky MV, Lukashev D, Apasov S et al (2004) Physiological control of immune response and inflammatory tissue damage by hypoxia inducible factors and adenosine A2A receptors. Annu Rev Immunol 22:2101–126
Sitkovsky M, Lukashev D (2005) Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat Rev Immunol 5:712–21
Lukashev D, Ohta A, Apasov S et al (2004) Cutting edge: Physiologic attenuation of proinflammatory transcription by the Gs protein-coupled A2A adenosine receptor in vivo. J Immunol 173:21–4
Kobayashi S, Zimmermann H, Millhorn DE (2000) Chronic hypoxia enhances adenosine release in rat PC12 cells by altering adenosine metabolism and membrane transport. J Neurochem 74:621–32
Decking UK, Schlieper G, Kroll K et al (1997) Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circ Res 81:154–64
Synnestvedt K, Furuta GT, Comerford KM et al (2002) Ecto-5–nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest 110:993–002
Koshiba M, Kojima H, Huang S et al (1997) Memory of extracellular adenosine/A2a purinergic receptor-mediated signalling in murine T cells. J Biol Chem 272:25881–5889
Huang S, Koshiba M, Apasov S et al (1997) Role of A2a adenosine receptor-mediated signaling in inhibition of T cell activation and expansion. Blood 90:1600–610
Koshiba M, Rosin DL, Hayashi N et al (1999) Patterns of A2A extracellular adenosine receptor expression in different functional subsets of human peripheral T cells. Flow cytometry studies with anti-A2A receptor monoclonal antibodies. Molec Pharmacol 55:614–24
Sitkovsky MV, Trenn G, Takayama H (1988) Cyclic AMP-dependent protein kinase as a part of the possible down-regulating pathway in the antigen receptor-regulated cytotoxic T lymphocyte conjugate formation and granule exocytosis. Ann NY Acad Sci 532:350–58
Takayama H, Sitkovsky MV (1988) Potential use of antagonists of cAMP-dependent protein kinase to block inhibition and modulate T-cell receptor-triggered activation of cytotoxic T-lymphocytes. J Pharm Sci 78:8–0
Sugiyama H, Chen P, Hunter M et al (1992) The dual role of the cAMP-dependent protein kinase C alpha subunit in T- cell receptor-triggered T-lymphocytes effector functions. J Biol Chem 267:25256–5263
Fredholm BB, Ijzerman AP, Jacobson KA et al (2001) International union of pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev 53:527–52
Linden J (2001) Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annu Rev Pharmacol Toxicol 41:775–87
Lukashev DE, Smith PT, Caldwell CC et al (2003) Analysis of A2a receptor-deficient mice reveals no significant compensatory increases in the expression of A2b, A1, and A3 adenosine receptors in lymphoid organs. Biochem Pharmacol 65:2081–090
Torgersen KM, Vang T, Abrahamsen H et al (2002) Molecular mechanisms for protein kinase A-mediated modulation of immune function. Cell Signal 14:1–
Qin Z, Blankenstein T (2000) CD4+ T cell-mediated tumor rejection involves inhibition of angiogenesis that is dependent on IFN gamma receptor expression by nonhematopoietic cells. Immunity 12:677–86
Sun H, Gutierrez P, Jackson MJ et al (2000) Essential role of nitric oxide and interferon-gamma for tumor immunotherapy with interleukin-10. J Immunother 23:208–14
Poehlein CH, Hu HM, Yamada J et al (2003) TNFalpha plays an essential role in tumor regression after adoptive transfer of perforin/IFN-gamma double knockout effector T cells. J Immunol 170:2004–013
Hoskin DW, Butler JJ, Drapeau D et al (2002) Adenosine acts through an A3 receptor to prevent the induction of murine anti-CD3-activated killer T cells. Int J Cancer 99:386–95
Harris AL (2002) Hypoxia a key regulatory factor in tumour growth. Nat Rev Cancer 2:38–7
Vaupel P, Thews O, Hoeckel M (2001) Treatment resistance of solid tumors: role of hypoxia and anemia. Med Oncol 18:243–59
Laderoute KR, Alarcon RM, Brody MD et al (2000) Opposing effects of hypoxia on expression of the angiogenic inhibitor thrombospondin 1 and the angiogenic inducer vascular endothelial growth factor. Clin Cancer Res 6:2941–950
Evans SM, Koch CJ (2003) Prognostic significance of tumor oxygenation in humans. Cancer Lett 195:1–6
Giatromanolaki A, Sivridis E, Kouskoukis C et al (2003) Hypoxia-inducible factors 1alpha and 2alpha are related to vascular endothelial growth factor expression and a poorer prognosis in nodular malignant melanomas of the skin. Melanoma Res 13:493–01
Ziemer LS, Evans SM, Kachur AV et al (2003) Noninvasive imaging of tumor hypoxia in rats using the 2-nitroimidazole 18F–EF5. Eur J Nucl Med Mol Imaging 30:259–66
Thiel M, Chouker A, Ohta A et al (2005) Oxygenation inhibits the physiological tissue-protecting mechanism and thereby exacerbates acute inflammatory lung injury. PLoS Biol 3:e174
Blay J, White TD, Hoskin DW (1997) The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res 57:2602–605
Overwijk WW, Lee DS, Surman DR et al (1999) Vaccination with a recombinant vaccinia virus encoding a ‘self–antigen induces autoimmune vitiligo and tumor cell destruction in mice: requirement for CD4(+) T lymphocytes. Proc Natl Acad Sci USA 96:2982–987
Phan GQ, Yang JC, Sherry RM et al (2003) Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci USA 100:8372–377
Lahat N, Rahat MA, Ballan M et al (2003) Hypoxia reduces CD80 expression on monocytes but enhnaces their LPS-stimulated TNF-a secretion. Leukoc Biol 74:197–05
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Lukashev, D., Sitkovsky, M. & Ohta, A. From ‘Hellstrom Paradox–to anti-adenosinergic cancer immunotherapy. Purinergic Signalling 3, 129–134 (2007). https://doi.org/10.1007/s11302-006-9044-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-006-9044-9