
Abstract
For the strong expression of genes in plant tissue, the availability of specific gene regulatory sequences is desired. We cloned promoter and terminator sequences of an apple (Malus x domestica) ribulose biphosphate carboxylase small subunit gene (MdRbcS), which is known for its high expression and used gus reporter gene expression to test the regulatory activity of the isolated promoter and terminator sequences in transgenic tobacco. The MdRbcS promoter itself seemed to be less strong than the CaMV35S promoter when both used in combination with the nos terminator. However, the combination of the promoter and terminator of MdRbcS was able to drive gus to similar expression levels as the reference construct with CaMV35S promoter and nos terminator. This observation indicates the importance of the terminator sequence for gene expression. It is concluded that the combination of the MdRbcS promoter and terminator is a suitable regulatory sequence set for the expression of transgenes to a high level in plants and for intragenesis in apple specifically.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the production of genetically modified (GM) crops that are to be directly consumed by humans, there is a tendency to use preferentially genes and gene regulatory sequences that originate from plants themselves, rather than from, e.g., micro-organisms. Social studies aimed to elucidate factors determining consumer’s attitude toward GM crops indicate that such crops are likely to be accepted much more readily. Lusk and Sullivan (2002) found that consumers would prefer GM vegetables with genes coming from the same species. Taking this concept as a starting point, different scenarios can be envisaged.
Important for the introduction of a new trait by genetic modification are the function of the gene that is used and the expression of that gene. Expression is regulated by genomic DNA sequences that flank the coding region at its 5′- and 3′-ends. The 5′ upstream sequences or promoters determine the level of expression and the tissue specificity or timing, e.g., developmental stage or after induction by biotic or abiotic stress. For optimal desired performance, new combinations of coding regions and regulators might be made for genetic modification. Still, both types of DNA sequences can be taken from the recipient species. This type of genetic modification is referred to as intragenesis (Rommens et al. 2004). In case the gene or genes are used in their original configuration, which is with their own promoter and terminator regions, introns and in a sense orientation, this type of genetic modification has been called cisgenesis (Schouten et al. 2006; Schouten and Jacobsen 2008).
In our apple genetic modification program, a promoter was needed ensuring high level of transcription in many tissue types, comparable to the constitutive CaMV (cauliflower mosaic virus) 35S promoter, but obtained from apple. The promoter of the gene coding for the small subunit of the ribulose biphosphate carboxylase (RBC) has been studied extensively (Khoudi et al. 1997; Nomura et al. 2000; Gittins et al. 2000; Outchkourov et al. 2003) in both homologous and heterologous crops and was found to be able to provide strong transcriptional activity. RBC is one of the most abundant proteins in the plant kingdom and can constitute 40–60% of total leaf protein. The enzyme is composed of eight small and eight large subunits encoded respectively by the nuclear and the chloroplast genome (Jensen and Bahr 1977). The RBC small subunit (RbcS) genes constitute a gene family of 2–12 members that contribute to varying extents to the total transcriptional activity (Outchkourov et al. 2003). The expression of the genes is observed in photosynthetically active tissue and is regulated by light (Dean and Leech 1982). Expression levels of heterologous genes driven by RbcS promoter sequences in different crops range between half that obtained by the CaMV35S promoter to seven to eightfold higher than CaMV35S (Outchkourov et al. 2003; Khoudi et al. 1997). In apple, Gittins et al. (2000) tested heterologous RbcS promoters of tomato and soybean and concluded that they would be suitable for expression in green photosynthetic tissues. Also, the RbcS promoter of chrysanthemum was tested in our laboratory in apple, and strong transcriptional activity could be demonstrated (F.A. Krens, pers. communication). However, these heterologous promoters are not suitable for intragenesis in apple.
Here, we describe the isolation and cloning of the RbcS 5′ and 3′ regulatory sequences of apple, based on homology to RbcS sequences of other crops, and the preparation of an expression cassette. The expression cassette was combined with the gus reporter gene and used in transformation of tobacco and apple. Expression levels were quantified in tobacco and were compared to the levels obtained using the constitutive CaMV35S promoter.
Materials and methods
Isolation of genomic DNA
Genomic DNA was isolated from young leaves of the apple cultivar Elstar according to the method described by Doyle and Doyle (1987), including 1% (w/v) polyvinylpyrrolidone-10 in the DNA extraction buffer.
Genome walking
Isolation of 5′-end (promoter) and 3′-end (terminator) flanking genomic sequences of the apple RbcS gene (MdRbcS) was performed following a genome walking protocol. In short, eight different in vitro genomic DNA libraries of the apple cultivar Elstar were constructed by digesting genomic DNA with eight different blunt-end cutting restriction enzymes followed by a ligation of an adapter to the ends of the restriction fragments. The adapter is described by van der Linden et al. (2004). The libraries were used as templates in a genome-walking PCR in which, for the amplification of promoter fragments, two primers specific for the 5′-end of the MdRbcS cDNA sequence (GenBank accession number L24497), pGSP1 (5′-GTGAATGTGTGATGGTGGTTSGCCTCT-3′) and pGSP2 (5′-AGGTTATCTATTAGGGTGCGTTTGTTG-3′), and for the amplification of terminator fragments, two primers specific for the 3′-end of the MdRbcS cDNA sequence, tGSP1 (5′-AGGGTCCCGGTGGTCTGTTTAAGTTTG-3′) and tGSP2 (5′-TCGGATTCAAGATGGTGTTGGTTGAGA-3′), were used together with adapter-specific primers (van der Linden et al. 2004). Maximase™ Polymerase (Transgenomic) was used for amplification. Following a primary and nested PCR, different PCR fragments were obtained, and two putative promoter fragments of 0.9 and 1.6 kb, amplified from an HpaI- and Hinc-digested genomic DNA library, respectively, and a putative terminator fragment of 1.1 kb, amplified from a Hinc-digested genomic DNA library, were cloned in a PCR cloning vector pGEM-T Easy (Promega) and completely sequenced.
Construction of transformation vectors
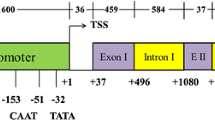
For the construction of expression vectors, two MdRbcS genomic fragments of 1,232 and 1,679 bp located directly upstream of the translational start and a single MdRbcS genomic fragment of 582 bp located directly downstream of the translational stop of MdRbcS were amplified by PCR using genomic apple DNA as template (primers: 1.6-kb 5′-end fragment: pMdRbcS1.6-FW, 5′-TTAATTAAGTGTTGGACCATCTCAGAGT-3′ + pMdRbcS-REV, GGCGCGCCATGGCATTGCTCTCTCTCTCTGTTTTCTCTTC; 1.2 kb 5′-end fragment: pMdRbcS-1.2-FW, TTAATTAAGACGGTGATAAATCATTTTTGTCT + pMdRbcS-REV; 0.5 kb 3′-end fragment: tMdRbcS-FW, 5′-CCATGGTACCGTTGTTCTACAATTTTCATAAATAATGTTGT-3′ + tMdRbcS-REV, 5′-GGCGCGCCGTTTGGTTACACTACCCTAAGGTC-3′). The promoter and terminator fragments included the MdRbcS 3′-UTR and 5′-UTR, respectively. For cloning of the promoter sequences, PacI and AscI + NcoI restriction sites were attached to the forward and reverse primers sequences, respectively (restriction site sequences underlined), and for cloning of the terminator, NcoI + KpnI and AscI restriction sites were attached to the forward and reverse primers, respectively. The amplified MdRbcS promoter and terminator sequences were cloned in pGEM-Teasy, and the sequences were verified by sequencing. For promoter study, two lengths of 1,200 and 1,600 bp of the MdRbcS promoter (p1.2MdRbcS and p1.6MdRbcS) were combined with the terminator (tMdRbcS) sequence by introducing the terminator as NcoI-AscI fragment behind the promoter sequences. Both resulting vectors (p1.2 + tMdRbcS and p1.6 + tMdRbcS) contain the MdRbcS promoter and terminator sequences separated by an NcoI and a KpnI site. As reporter gene, an intron containing β-glucuronidase (GUS) gene (gus) was amplified using the vector pCambia1301 (GenBank accession number AF234297) as template. For this, a forward primer starting at the gus start codon (Gus-start, 5′-CCATGGTCCGTCCTGTAGAAACCCCAAC-3′) and a reverse primer located at the gus stop codon (Gus-stop, 5′-GGTACCTCATTGTTTGCCTCCCTGCTGCGGTT-3′) were used. The forward and reverse primers were extended with an NcoI and KpnI site, respectively, to facilitate cloning. As a control, the promoter of the cauliflower mosaic virus 35S gene (pCaMV35S) and the terminator of the Agrobacterium tumefaciens nopaline synthase gene (tnos) were used, and these were also amplified from pCambia1301, together with the gus reporter gene. For amplification of pCaMV35S + gus, a primer designed at pCaMV35S (p35S-FW, 5′-TTAATTAACATGGAGTCAAAGATTCAAATAGAG-3′), which was extended with a PacI site, was used together with primer Gus-stop, and for amplification of gus + tnos, a primer designed at tnos (tnos-REV, 5′-GGCGCGCCCGATCTAGTAACATAGATGACAC-3′), which was extended with a AscI site, was used together with primer Gus-start. Finally, a fragment with pCaMV35S + gus + tnos was amplified using primers p35S-FW and tnos-REV. The amplified gus, pCaMV35S-gus, gus-tnos, and pCaMV35S + gus + tnos fragments were cloned in pGEM-Teasy, and the sequences were verified by sequencing. The gus fragment was introduced as NcoI-KpnI fragment into both vectors p1.2 + tMdRbcS and p1.6 + tMdRbcS, which were opened by digestion with NcoI-KpnI. The pCaMV35S-gus fragment was combined with the MdRbcS terminator by introducing it as PacI-KpnI fragment in vector p1.2 + tMdRbcS, which was opened by digestion with PacI and KpnI, thereby removing the MdRbcS promoter, and the gus-tnos fragment was combined with both MdRbcS promoters by introducing it as NcoI-AscI fragment in vector p1.2 + tMdRbcS or p1.6 + tMdRbcS, which were opened by digestion with NcoI and AscI, thereby removing the MdRbcS terminator. In total six different combinations of promoter and terminator sequences were constructed (p1.2MdRbcS-gus-tMdRbcS; p1.2MdRbcS-gus-tnos; p1.6MdRbcS-gus-tMdRbcS; p1.6MdRbcS-gus-tnos; pCaMV35S-gus-tMdRbcS, and CaMV35S-gus-tnos (see Fig. 1)), and these were introduced as AscI-PacI fragments in the multiple cloning site of the binary vector pBINplus (van Engelen et al. 1995). The ultimate constructs were transferred to the supervirulent A. tumefaciens strain Agl0 (Lazo et al. 1991).

Schematic representation of the different promoter–terminator combinations used for plant transformation
Plant transformation
For expression analysis of the cloned promoter fragments, transgenic tobacco (Nicotiana tabacum cv. Samsung) plants containing T-DNA with the different promoter-gus terminator constructs were produced according to Horsch et al. (1985). For each construct, 15 independent kanamycin-resistant transgenic tobacco lines were selected, and per construct two to four transgenic lines showing intense GUS staining of leaf tissue were selected for further analysis.
Apple cv. Gala was transformed with the p1.6MdRbcS-gus-tMdRbcS constructs only. Transformation was performed according to Puite and Schaart (1996).
Histochemical GUS assay
Histochemical GUS staining of plant material was performed as described by Jefferson (1987) using a modified staining solution containing 1 mM 5-bromo-4-chloro-3-indolyl β-glucuronide (X-gluc) in 50 mM sodium phosphate buffer (pH 7.5), 10 mM EDTA, 0.1% (v/v) Triton X-100, 0.5 mM potassium ferricyanide, and 5% (w/v) polyvinylpyrrolidone-40.
Quantitative (RT)-PCR
For quantification of gus, expression levels and of gus gene copy number quantitative (RT)-PCR was performed. For quantification of gus expression levels, total RNA was isolated from in vitro leaves of N. tabacum transgenic plants basically as described by Asif et al. (2000). For each transgenic line, two independent RNA isolations were performed. cDNA was synthesized using the IScript first-strand cDNA synthesis system for RT-PCR (Biorad) according to the instruction manual. For quantification of gus gene copy number, genomic DNA was isolated using the CTAB method of Doyle and Doyle (1987). Quantitative (RT)-PCR was performed as described by Schaart et al. (2002) using the MyiQ Single-Color Real-Time Detection System (BioRad, USA), but instead of the fluorogenic TaqMan probes, SYBR Green was used for detection of PCR products. For amplification of gus, the forward and reverse gus-primers (5′-CGGAAGCAACGCGTAAACTC-3′ and 5′-TGAGCGTCGCAGAACATTACAT-3′) were used (product size 80 bp). As endogenous control, the elongation factor 1α gene (EF1α) from N. tabacum (GenBank accession number AF120093) was selected as a reference gene (forward EF1α primer 5′-GATTGGTGGAATTGGTACTGTC-3′, reverse EF1α primer 5′-AGCTTCGTGGTGCATCTC-3′, product size 130 bp). All PCR reactions were performed in duplo. For the real-time analysis, PCR products were detected directly by monitoring the increase in fluorescence from the incorporated SYBR green dye. The amplification was plotted as the normalized reporter signal, ΔR n (the reporter dye was normalized to the internal reference dye and corrected for the baseline value), toward the number of PCR cycles. For each reaction, the threshold cycle, C T, which is defined as the PCR cycle at which a statistically significant increase of ΔR n is first detected, was determined. The relative quantification was done using the comparative C T-method in which the differences in the C T for the gus-amplicon and the C T for the endogenous control EF1α, called ΔC T, were calculated to normalize for the differences in the total amount of cDNA or gDNA copies present in each reaction and for cDNA, the efficiency of the RT step. For comparison of two samples, the ΔC T values were subtracted from each other, giving a ΔΔC T value, and finally, the relative amount of gus mRNA or gDNA copies was calculated by \( {2^{ - \Delta \Delta {C_{\rm{T}}}}} \).
Results
Isolation of MdRbcS promoter and terminator fragments
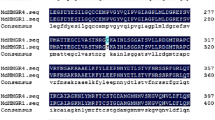
Using a genome walking protocol, several DNA fragments were amplified from the 5′- and 3′-flanking genomic regions of an MdRbcS gene. For this reverse primers located around the start-codon of the MdRbcS gene (together with adaptor primers) were designed for amplification of putative promoter sequences, and primers located around the stop-codon of the MdRbcS gene were designed for amplification of a putative terminator sequence. Two putative promoter fragments of 0.9 and 1.6 kb were cloned and sequenced (GenBank accession numbers HM222640 and HM222639, respectively), and a single putative terminator fragment was also cloned and sequenced (GenBank accession number HM222641). The first 260 bp of both promoter sequences near the transcription start showed a high level of similarity (91% identity) but more distal from transcription (from start similarity was low (46% identity)). Blasting of the promoter sequences showed that they were sharing partial homology to promoter sequences of RbcS of different other plant species. Both putative MdRbcS promoter sequences contain a TATA-box and several other putative cis-acting elements like two GATA-boxes (Fig. 2), which are known for their light responsive activity (Castresana et al. 1988). Also an element showing similarity to a GT-1 binding box II, which was identified in the promoter of pea RbcS-3A (Green et al. 1987), is present in the MdRbcS promoter. This element is associated with high expression levels of RbcS-3A in pea. Polymorphisms that discriminate both promoter fragments were all located outside the predicted cis-acting elements (Fig. 2). The terminator sequence contained two polyadenylation signals, 70 and 116 bp downstream of the translational stop codon.

The consensus sequence of the first 270 bp of the MdRbcS promoter, based on both isolated fragments. The consensus contains several putative cis-acting elements. Polymorphisms are given as IUPAC ambiguity codes, and lowercase-denoted nucleotides are indel polymorphisms. Light-gray shaded and not boxed, GATA-box; dark-gray shaded and not boxed, G-box; non-shaded box, Pea RbcS GT-1-like box; light-gray and boxed, TATA-box
Construction of expression vectors
For the cloning of the promoter, two sizes of 1.2 and 1.6 kb were taken. The promoter fragments were amplified from genomic DNA using forward primers that were based on one of the cloned 5′-flanking sequences of the MdRbcS gene. The reverse primer was located directly upstream of the start-codon of the MdRbcS gene, so the amplified sequence included the 5′-UTR. Both promoter sequences differed only in a few single nucleotide polymorphisms. For cloning of the MdRbcS terminator, a similar approach was followed using a forward primer just downstream of the stop-codon of MdRbcS (so, including the 3′-UTR) and a reverse primer that was based on the cloned 3′-flanking sequence of the MdRbcS gene. For convenient cloning, appropriate restriction sites were included into the primers. For comparison of the regulatory function of the isolated promoter and terminator sequences, the CaMV35S promoter and the nos terminator were used as reference. The six possible combinations of the three promoters (p1.2MdRbcS, p1.6MdRbc, or pCaMV35S) with the two terminators (tMdRbcS or tnos) were made and combined with the gus reporter gene and cloned into the binary vector pBinplus.
Expression study in tobacco and apple
The different promoter-gus terminator constructs were transformed to tobacco, and for each construct, approximately 15 independent kanamycin-resistant lines were selected. In vitro leaves from all transgenic lines were checked for gus-expression by a histochemical GUS-assay. For each construct, the transgenic lines showed variation in staining intensity from low to high level of blue staining (results not shown), indicating that all promoter–terminator combinations were able to drive gus expression to high levels. No clear difference in staining intensity was found between plants transformed with the different constructs. For each construct, a number of independent transformed tobacco lines, which showed a clear uniform blue staining of in vitro leaf tissue plant, was selected for quantitative gene expression analysis. Using quantitative RT-PCR, it was determined that on average, lines with the gus reporter gene under regulation of the MdRbcS promoter showed a lower gus expression level than lines transformed with pCaMV35S-gus (Fig. 3; Table 1). Both MdRbcS promoters did not differ significantly in their activity when combined with the MdRbcS terminator, but when combined with the nos terminator, gus expression driven by p1.6MdRbcS was significantly lower (unpaired Student’s t test; P < 0.05) than for p1.2 MdRbcS. For all three promoters used, the presence of the terminator sequence of MdRbcS resulted in significantly higher expression levels when compared to the use of the nos terminator (Fig. 3; Table 1). The use of the MdRbcS promoters combined with the MdRbcS terminator gave comparable gus expression levels as the CaMV35S promoter combined with tnos did.

Relative gus expression levels in leaves of transgenic tobacco plants. Numbers on the x-axis correspond to independent transgenic lines. For each transgenic line, RNA was isolated from young leaves pooled from three different in vitro grown plants, and RNA isolation was done in duplicate. Error bars represent ± standard error
To investigate the effect of transgene copy number on the transgene expression level for the different plant lines, the relative transgene copy number was estimated using Q-PCR (Table 2). The average ΔC T value of a group of plant lines with a corresponding large ΔC T value, and thus a similar low copy number, was used as calibrator. Half of the transgenic tobacco plants contained a relatively low number of transgene copies, but in some plant lines, high transgene copy number was found. No clear correlation was observed between transgene copy number and transgene expression level (Table 2).
In order to test the functionality of the apple rubisco promoters in transgenic apples, the p1.6MdRbcS-gus-tMdRbcS construct was transformed to apple cv. Gala. Several calli that produced shoots on kanamycin containing shoot regeneration medium were stained for the presence of GUS activity. Most regenerated shoots showed a strong blue staining (Fig. 4).

Composition of four photographs showing regenerating shoots (white arrows) of apple cv. Gala that was transformed with a binary vector with p1.6MdRbcS-gus-tMdRbcS. Blue staining indicates presence of GUS activity
Discussion
For the intragenic expression of genes in apple, the availability of suitable apple regulatory sequences is desirable. Our goal was to isolate gene regulatory sequences which drive gene expression in leaf tissue to a high level comparable to that of the CaMV35S promoter. The RbcS family is a photosynthesis-associated nuclear gene which is known to be highly expressed in green photosynthetic tissues and was therefore a suitable candidate to isolate regulatory sequences from. Gittins et al. (2000) tested RbcS promoters of tomato and soybean in apple and concluded that they would be suitable for expression in green photosynthetic tissues. They found that RbcS promoters of the different origins were able to drive gus expression to half that of plants containing the CaMV35S promoter.
Although the coding regions of different RbcS gene family members are highly similar, the level and pattern of expression can vary considerably (Anisimov et al. 2007; Sawchuk et al. 2008). The apple RbcS gene we used in this study to isolate regulatory sequences from shows homology to the RbcS1A gene subfamily from Arabidopsis thaliana, which was maximally expressed in differentiating and mature leaf mesophyll cells of Arabidopsis (Sawchuk et al. 2008). Following a genome walking approach, 5′- and 3′-flanking genomic DNA sequences of MdRbcS were amplified, cloned, and characterized. In order to study the two isolated putative MdRbcS promoters and the putative MdRbcS terminator sequence independently, the CaMV35S promoter and the nos terminator, which are generally used in genetic modification studies, were used as reference, and different combinations of promoters and terminators with the gus reporter gene were made and tested in tobacco. Based on Q-RT-PCR analysis of transgenic tobacco leaf material, we could conclude that the MdRbcS promoter was able to drive gus expression to a relatively high level, but the promoter was less active than the CaMV35S promoter. Furthermore, Q-RT-PCR data revealed that the presence of the terminator of MdRbcS had a positive effect on gus gene expression. Dean et al. (1989) reported for the first time in plants that sequences 3′ to the coding region (of petunia RbcS) had a significant effect on transcription rate. This finding was confirmed by Ingelbrecht et al. (1989) who showed that terminator sequences of different plant genes modulated the level of gene expression in plant cells in a different way. In their study, the presence of a terminator sequence from an Arabidopsis RbcS gene enhances transcription of the nptII gene threefold, when compared to the presence of the terminator of the A. tumefaciens octopine synthase (ocs) gene. They also showed by deletion analysis of the 3′-flanking region of the ocs gene that sequences downstream of the polyadenylation signal are required for optimal gene expression. These observations indicate the importance of the terminator sequence for (trans)gene transcription. Although the MdRbcS promoter combined with the nos terminator was inferior to the reference construct in which the CaMV35S promoter and the nos terminator were combined, the MdRbcS promoter combined with the MdRbcS terminator gave similar high gus expression as the reference construct. Therefore, the combination of both the MdRbcS promoter and terminator is a suitable regulatory sequence set for the expression of transgenes to a high level in plants and for intragenesis in apple specifically. A preliminary test of the functionality of the p1.6MdRbcS-gus-tMdRbcS construct in apple resulted in an intense blue staining in a histochemical GUS staining of regeneration transgenic apple shoots, suggesting the MdRbcS promoter and terminator combination to be strongly active in apple plant tissue. Whether the MdRbcS promoter and terminator combination drives gene expression also to a high level in mature leaves and if the combination is also active in other tissues, for example in the fruit skin green photosynthetic active tissue, has still to be investigated. In apple, the MdRbcS promoter–terminator combination would be suitable to confine the expression of, for example resistance genes, to the leaves which is an important site of infection for microbial diseases and insect pests. Currently, we are testing the suitability of the MdRbcS promoter and terminator for overexpression of the apple HcrVf scab resistance genes (Belfanti et al. 2004) in GM apples.
References
Anisimov A, Koivu K, Kanerva A, Kaijalainen S, Juntunen K, Kuvshinov V (2007) Cloning of new rubisco promoters from Brassica rapa and determination of their activity in stably transformed Brassica napus and Nicotiana tabacum plants. Mol Breed 19:241–253
Asif MH, Dhawan P, Nath P (2000) A simple procedure for the isolation of high quality RNA from ripening banana fruit. Plant Mol Bio Rep 18:109–115
Belfanti E, Silfverberg-Dilworth E, Tartarini S, Patocchi A, Barbieri M, Zhu J, Vinatzer BA, Gianfranceschi L, Gessler C, Sansavini S (2004) The HcrVf2 gene from a wild apple confers scab resistance to a transgenic cultivated variety. Proc Natl Acad Sci USA 101:886–890
Castresana C, Garcia-Luque I, Alonso E, Malik VS, Cashmore AR (1988) Both positive and negative regulatory elements mediate expression of a photoregulated CAB gene from Nicotiana plumbaginifolia. EMBO J 7:1929–1936
Dean C, Leech RM (1982) Genome expression during normal leaf development. Plant Physiol 69:904–910
Dean C, Favreau M, Bond-Nutter D, Bedbrook J, Dunsmuir P (1989) Sequences downstream of translation start regulate quantitative expression of two petunia rbcS genes. Plant Cell 1:201–208
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure from small quantities of fresh leaf tissues. Phytochem Bull 19:11–15
Gittins JR, Pellny TK, Hiles ER, Rosa C, Biricolti S, James DJ (2000) Transgene expression driven by heterologous ribulose-1, 5-bisphosphate carboxylase/oxygenase small-subunit gene promoters in the vegetative tissues of apple (Malus pumila Mill.). Planta 210:232–240
Green PJ, Kay SA, Chua N-H (1987) Sequence-specific interactions of pea nuclear factor with light-responsive elements upstream of the rbcs-3A gene. EMBO J 6:2543–2549
Horsch RB, Fry JE, Hoffman NL, Eichholtz D, Rogers SG, Fraley RT (1985) A simple and general method for transferring genes into plants. Science 227:1229–1231
Ingelbrecht LW, Herman LMF, Dekeyser RA, Van Mountagu MC, Depicker AG (1989) Different 3′ end regions strongly influence the level of gene expression in plant cells. Plant Cell 1:671–680
Jefferson RA (1987) Assaying chimeric genes in plants: the GUS gene fusion system. Plant Mol Biol Rep 5:387–405
Jensen RG, Bahr JT (1977) Ribulose 1,5-bisphosphate carboxylase-oxygenase. Ann Rev Plant Physiol 28:379–400
Khoudi H, Vezina LP, Mercier J, Castonguay Y, Allard G, Laberge S (1997) An alfalfa rubisco small subunit homologue shares cis-acting elements with the regulatory sequences of the RbcS-3A gene from pea. Gene 15:343–351
Lazo GR, Stein PA, Ludwig RA (1991) A DNA transformation competent Arabidopsis genomic library in Agrobacterium. Biotechnol 9:963–967
Lusk JL, Sullivan P (2002) Consumersacceptance of genetically modified foods. Food Technol 56:32–37
Nomura M, Katayama K, Nishimura A, Ishida Y, Ohta S, Komari T, Miyao-Tokutomi M, Tajima S, Matsuoka M (2000) The promoter of rbcS in a C3 plant (rice) directs organ-specific, light-dependent expression in a C4 plant (maize), but does not confer bundle sheath cell-specific expression. Plant Mol Biol 44:99–106
Outchkourov NS, Peters J, de Jong J (2003) The promoter–terminator of chrysanthemum rbcS1 directs very high expression levels in plants. Planta 216:1003–1012
Puite KJ, Schaart JG (1996) Genetic modification of the commercial apple cultivars Gala, Golden Delicious, and Elstar via an Agrobacterium tumefaciens-mediated transformation method. Plant Sci 119:125–133
Rommens CM, Humara JM, Ye J, Yan H, Richael C, Zhang L, Perry R, Swords K (2004) Crop improvement through modification of the plant's own genome. Plant Physiol 135:421–431
Sawchuk MG, Donner TJ, Head P, Scarpella E (2008) Unique and overlapping expression patterns among members of photosynthesis-associated nuclear gene families in Arabidopsis. Plant Physiol 148:1908–1924
Schaart JG, Salentijn EJM, Krens FA (2002) Tissue-specific expression of the β-glucuronidase reporter gene in transgenic strawberry (Fragaria × ananassa) plants. Plant Cell Rep 21:313–319
Schouten HJ, Jacobsen E (2008) Cisgenesis and intragenesis, sisters in innovative plant breeding. Trends Plant Sci 13:260–261
Schouten HJ, Krens FA, Jacobsen E (2006) Cisgenic plants are similar to traditionally bred plants. EMBO Rep 7:750–753
van der Linden CG, Wouters DCAE, Mihalka V, Kochieva EZ, Smulders MJM, Vosman B (2004) Efficient targeting of plant disease resistance loci using NBS profiling. Theor Appl Genet 109:384–393
van Engelen FA, Molthoff JW, Conner AJ, Nap J-P, Pereira A, Stiekema WJ (1995) pBINPLUS: an improved plant transformation vector based on pBIN19. Transgenic Res 4:288–290
Acknowledgments
This study was supported by the EU-ISAFRUIT Project (FP6-FOOD-016279). We thank Thomas Sevestre for performing the Q-(RT)-PCR experiments.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by R. Velasco
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Schaart, J.G., Tinnenbroek-Capel, I.E.M. & Krens, F.A. Isolation and characterization of strong gene regulatory sequences from apple, Malus × domestica . Tree Genetics & Genomes 7, 135–142 (2011). https://doi.org/10.1007/s11295-010-0320-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11295-010-0320-z