
Abstract
The existence of four dengue serotypes is associated with a phenomenon called “Antibody-Dependent Enhancement” that has been suggested to cause a severe form of dengue hemorrhagic fever and shock syndrome. To study the evolutionary event that drove the serotype separation, we employed the maximum likelihood approach by focusing on the Premembrane (prM) and Envelop (E) genes. We showed that the separation of dengue serotypes had been dominantly under purifying selection. In spite of the strong selective constraint, one codon of prM gene and twelve codons of E gene were detected to be under positive selection. This indicates that the E protein might have been under a stronger positive pressure than the PrM protein. The codons under positive selection were identified along the interserotypic branches, suggesting that changes at these sites were probably associated with the emergence of the four serotypes and/or adaptation to the new transmission environments.
Similar content being viewed by others


Introduction
Dengue (DEN) virus is a mosquito-borne virus of the Flavivirus genus, consisting of four distinct serotypes, designated DEN1, DEN2, DEN3, and DEN4. The 11-kb viral genome is composed of positive-stranded RNA, encoding 3 structural proteins (C, E, and PrM) and 7 non-structural proteins (NS1, 2A, 2B, 3, 4A, 4B, and NS5) that are involved in viral replication and pathogenesis [1]. Viral surface proteins, such as Premembrane (PrM) and Envelop (E) proteins, elicit antibodies that play an important role in viral neutralization and enhancement. Dejnirattisai et al. [2] showed that antibodies to PrM (anti-PrM) and antibodies to E (anti-E) were highly cross-reactive among four dengue serotypes. Relative to anti-Es, anti-PrMs appeared to enhance viral infection rather than neutralizing it, while anti-Es, especially those that responded to domain III, were important for viral neutralization.
Dengue virus causes a range of diseases, from the acute febrile illness dengue fever (DF) to life-threatening dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS). Several studies have suggested that serious complications of DHF is linked to a secondary infection by a heterologous dengue serotype, and this secondary infection has been proposed to be a cause of the “Antibody-Dependent Enhancement or ADE” phenomenon [3–5]. ADE has been described by which the antibody elicited by the first infection is not sufficient to neutralize a secondary infection that is often caused by a different virus serotype [6].
Origin of dengue virus has been suggested to be approximately 1,000 years ago [7]. The four distinct endemic/epidemic dengue serotypes are evolutionarily derived from at least three independent introductions into humans from wild primates between 125 and 320 years ago and probably occurred in Africa and Southeast Asia [7, 8]. Gubler [9] suggested that the endemic/epidemic forms of dengue probably evolved after urban human populations arose, by which the size of human population was sufficient to serve as reservoirs for the virus. An increase in genetic diversity within each serotype also coincides with an increased size and mobility of human population [10]. The influence of human host factors in dengue evolution has been shown in a few studies. Kawaguchi et al. [11] showed that ADE drives closely related strains to extinction, permitting only the coexistence of distantly related serotypes. Thus, the coexistence of the four dengue serotypes is maintained by ADE. Twiddy et al. [12, 13] analyzed the E sequences of different dengue serotypes separately and revealed a relatively weak positive selection in the DEN3, DEN4, and the Cosmopolitan and the Asian 2 genotypes of DEN2. They showed that the majority of the sites under such positive selection are located in or near potential T- and B-cell epitopes.
Despite a number of studies on dengue evolution, no study has so far looked at selection pressure and factors that drove the separation of the four dengue serotypes. Identifying this evolutionary process and genomic regions involved may provide a further insight into the correlation between dengue evolution and ADE, which is the major cause of death in dengue infection. Herein, we are interested in the natural selection pressure that played a role during the separation of dengue serotype and had acted to maintain the antigenic distance between different serotypes, allowing the occurrence of both neutralization and enhancement phenomenons. We focused on the Premembrane (prM) and Envelop (E) genes due to their involvement in neutralization and enhancement. A maximum likelihood approach was employed to identify natural selection pressure along four ancestral lineages of DEN1-4. Our analysis considered the prM or E genes of all four serotypes at the same time.
Data and methods
Data
Sequences of prM and E genes of dengue were collected from NCBI database. We obtained 117 sequences of DEN1, 96 sequences of DEN2, 109 sequences of DEN3, and 21 sequences of DEN4 from Asia, North and South America, Europe and Pacific, from year 1963 to 2008. Sylvatic strains and sequences with 100% similarity were excluded from the analyses. The sequences were divided into 2 sets arbitrarily, and each set was analyzed separately to reduce the computing time required to analyze the sequences. The results obtained from each set of data would also be a good confirmation for one another. Set 1 and 2 contained 85 and 82 sequences, respectively. However, both data sets contained the same sequences of DEN4 due to limitation in DEN4 sequences around the world. Each data set composed of sequences representing 4 serotypes and all genotypes within each serotype. The accession numbers of all sequences used in this study are listed in the supplementary data 1.
Sequence alignment and phylogenetic tree construction
The sequences were aligned using MUSCLE and ClustalW implemented in program eBioX version 1.6b1 by Erik Lagercrantz. Gaps were deleted appropriately. Phylogenetic trees were constructed by Neighbor Joining method implemented in Phylogeny Inference Package (Phylip) version 3.69 by Joseph Felsenstein and by Maximum likelihood method implemented in PAUP version 4.0 by David Swofford. In all cases, we used GTR+G+I model of nucleotide substitution and undertook successive rounds of branch swapping. The resulting trees were further used as guided trees for estimating selection pressure in CodeML application program in Phylogenetic Analysis by Maximum Likelihood (PAML) package [14]. The branch lengths of the guided trees were calculated by CodeML application program, by which the branch length is defined as a number of nucleotide substitutions per codon. The trees are presented in the supplementary data 2 and 3.
Estimating natural selection pressure
The relative rates of non-synonymous (dN) and synonymous (dS) substitution across coding portions of the viral genes were examined by two codon-based likelihood models, the Branch-specific and the Branch-Site models. Positive selection is recognized when the dN/dS (ω) is greater than 1, while ω < 1 is considered to be under negative selection and ω = 1 is neutral selection. These models are implemented in the CodeML application program in Phylogenetic Analysis by Maximum Likelihood (PAML) package [14]. The Branch-specific model (M0, M1, and M2) was used to estimate the selection pressure along the lineage of interest [15]. In this study, the internal or interserotypic branches leading to individual serotypes are the branches of interest and will be referred as foreground branches. Different tests were performed to test the assumptions; One-ratio test (M0) assumes one ω for all branches in the phylogenetic tree, Several-ratio test (M2) assumes one ω (ω1) for foreground branches and one ω (ω0) for all the terminal branches (background branches) in the phylogenetic tree, Free-ratio test (M1) assumes that all branches in the tree have different ω. Different tests were compared using standard likelihood ratio tests (LRT), in which twice the log likelihood difference, 2Δl = 2(l 1 − l 0), was compared with X 2 distribution. For example, the likelihood values under the One-ratio (l 0) and Free-ratio (l 1) tests can be compared to test whether the ω values are different among lineages. If the P value is significant, then the One-ratio test is rejected and the result would suggest that different lineages are subject to different degrees of selective constrain. The Branch-Site model (M2NS2) was employed to detect sites or codons under positive selection along the lineage of interest [16]. A Bayesian approach was used to identify those individual codons most likely subject to positive selection [17]. The LRT was then employed to test the sensitivity of the results by comparing the model to the corresponding null hypothesis (ω = 1 fixed).
Results
Analysis of four dengue lineages
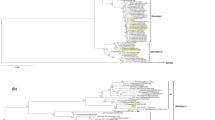
The Branch-specific models were used to examine natural selection pressure along the branches of interest (branches A–E), which were referred as foreground branches (Fig. 1). Branch A, B, C, and D were considered as ancestral lineages of DEN1, 2, 3, and 4, respectively. Branch E was the ancestral lineage of both DEN1 and DEN3. One-ratio test was performed to estimate average ω for all data sets. Both prM and E genes exhibited very small ω values of less than 1 (Table 1) and this result was consistent between the 2 data sets. Individual dN/dS ratios of the foreground (A–E) and background branches were then estimated using Free-ratio test. For both data sets 1 and 2, all foreground branches of both prM and E genes also showed ω < 1 (Table 1) and the majority of dN/dS ratios of the background branches were also less than 1.

Mid-root tree of the prM gene constructed by PAUP using GTR+G+I model. Similar tree topology was obtained with the E gene (supplementary data 3). The numbers on the foreground branches (branch A–E) represent Bootstrap values (at 1,000 replicates) calculated by Neighbor Joining method implemented in Phylogeny Inference Package (Phylip) version 3.69 by Joseph Felsenstein
To verify whether the selection pressure acting along the foreground branches A–E are significantly different to that of the background branches, the log likelihood value of the One-ratio test (test A) was compared to that of the Two-ratio test (test B) using the Likelihood ratio test (LRT). Test A is the null hypothesis, assuming the dN/dS ratios of all branches in the phylogenetic tree were equal. While test B is the test hypothesis, assuming the dN/dS ratios (ω1) of all foreground branches and dN/dS ratios (ω0) of the background branches were different. The Likelihood ratio test (LRT) compares the null hypothesis against the test hypothesis, in which the LRT will reject the null hypothesis if the P value is less than 0.05. The result showed that the dN/dS ratio of the foreground branches (branch A–E) of the prM gene is not significantly different to that of the background branches (P value > 0.05). This may imply a constant selective pressure throughout evolution of the prM gene. For E gene, the LRT comparing the log likelihood values of test A and test B suggested that the dN/dS ratio of the foreground branches (branch A–E) is significantly lower than the background branches (P value = 0.001). Therefore, further analysis was performed with the E gene. To examine which of the foreground branch contributes significantly to this difference, the log likelihood value of One-ratio test (test A) was compared with those of the Two-ratio tests (test C–G). In test C–G, each different foreground branch was assumed to have independent dN/dS ratio. The LRT showed that when an individual foreground branch was set independently, the dN/dS ratio of the foreground branch was not significantly different to that of the background branches (P value > 0.05, accepting the null hypothesis). This may imply that the degree of selection pressure acting on individual foreground branches is relatively similar. Eventhough the LRT of E gene suggested that the dN/dS ratios of the background branches are significantly lower than the foreground branches, both the background and foreground dN/dS ratios exhibited very small ω values and this indicated that all lineages were subject to a strong selective constraint.
Positive selection determined by the Branch-Site model
Adaptive evolution that occurred at a few time points and only affected a few amino acids may be missed using the Branch-specific models because the model assumes that there is no variation in ω among sites. In contrast, the Branch-Site model (M2NS2) allows the ω to vary among sites and lineages, thus the branches of interest were further analyzed using the Branch-Site model. The LRT was also employed to test the sensitivity of the results by comparing the model to the corresponding null hypothesis (ω = 1 fixed). Potential sites or codons under positive selection were selected using Bayes Empirical Bayes (BEB) analysis and the selected sites shown in Table 2 are those with posterior probability of >0.9.
Sites under positive selection were detected on branch A, B, C, and D of the phylogenetic tree constructed with prM gene. However, when the model was compared with the null hypothesis, only branch B rejected the null hypothesis in both data sets (Table 2). This indicated that only branch B exhibits ω that is significantly greater than 1. In data set 1, two sites were predicted to be under positive selection, while data set 2 only suggested 1 site under positive selection. Both data sets, however, are consistent with one another that amino acid position 82 was under positive selection.
For the E gene, sites under positive selection were detected along branch B, C, D, and E (Table 2). Most sites under positive selection were found along branch B, D, and E, while only one positive site was detected on branch C. The LRT of both data sets showed that branch B, D, and E rejected the null hypothesis, suggesting that the ω values along these branches are significantly greater than one (P value < 0.05). For branch C, the LRT of data set 2 suggested that the ω along branch C is significantly greater than 1. However, the LRT of data set 1 suggested otherwise. To avoid any ambiguity, only sites under positive selection along branches B, D, and E that are common in the 2 data sets were looked at further.
Sites under positive selection
The PrM protein is 166 amino acids long, composed of the Pr and M regions. The E protein consists of 3 domains designated domains I, II, and III. Domain I consists of amino acids 1–51, 132–192, and 280–295, forming 8-stranded center β barrel. Domain II consists of amino acid 52–131 and 193–279, forming 12 β-strands denoted a-l and two predicted α helix. Domain III is amino acid 296–393 that maintains the immunoglobulin constant region-like fold with 10 β-strands.
The Branch-Site model predicted 1 site (codon position 82) in PrM protein that was under positive selection. Amino acid position 82 is located in the premembrane region of the PrM protein, close to the furin cleavage site (position 87–91). This site was found along branch B that separated DEN2 from ancestor of DEN1 and DEN3. Twelve sites in E were predicted to be under positive selection, in which five sites were found on branch D separating DEN4 from the other serotypes. Site 148 and 186 are located in domain I, site 64 is on domain II, site 307 is on domain III, and site 406 is found on transmembrane domain. Predicted sites along branch E separated DEN2 from DEN1 to DEN3 were also found in all domains of E. Two sites along branch B were located in domain I and transmembrane domain.
Sites predicted to be under positive selection were mapped on the crystal structure of DEN2 recombinant PrM-E heterodimer (PDB ID: 3C6E) obtained from NCBI 3D structure database [18, 19]. The structure represents 81 and 394 amino acids of PrM and E proteins, respectively. The transmembrane domains of both PrM and E are absent in the structure. Most of the sites, except amino acid positions 59 and 186, appeared to be exposed on the surface of the protein (Fig. 2).

Crystal structure of Dengue 2 recombinant PrM-E heterodimer (PDB ID: 3C6E) obtained from NCBI 3D structure database [19]. The E protein is shown in yellow and the Pr protein is shown in blue with attached glycan. Red color represents predicted residues under positive selection by the Branch-Site mode
Discussion
The Branch-specific model tests estimated the dN/dS ratios along the interserotypic branches leading to the four serotypes to be less than one, suggesting that purifying selection played a dominant role during serotype separation. The dN/dS ratios estimated by the Free-ratio test were very small, ranging from 0.03 to 0.05, which indicated a very strong selective constraint throughout dengue evolution. These results are found to be consistent in both the prM and E gene data sets. The evidence of a strong selective constraint has previously been described in other studies, for example Twiddy et al. [13] and Bennett et al. [20] showed that the majority of dN/dS ratios along intraserotypic branches of different DEN2 genotypes are less than one. Reduced positive selection and significantly lower rates of nucleotide substitution have also been observed in vector-borne RNA viruses, compared with those transmitted by other routes [21–23]. In addition, the adaptive evolution in viral epidemic was the result of genetic turn over within a focal population, by which the most common genotype at a particular sampling time often arose from a previously rare lineage rather than by new introduction of genetic mutations [20, 23]. Therefore, it seems that evolution of dengue has not been driven by rapid mutations, like that seen with influenza virus. It has been suggested that the selective constraint observed in vector-borne RNA viruses may be imposed by the two-host factor, such that mutations that enhance viral fitness in insect cells may have a deleterious effect on mammalian cells, or by the absence of/weak exposure to immune selection [24].
Twiddy et al. [7] suggested that the separation of the four dengue serotypes occurred before the transition from sylvatic to endemic/epidemic strains, indicating that the influence of human host factors occurred after the serotype separation. This also implies that ADE is not the cause of serotype separation but acts to maintain the antigenic distance between serotypes. Therefore, positive selection sites detected along the four ancestral lineages of dengue virus are ancient positively selected sites, involving in the emergence of each individual serotypes. These sites may be responsible for an increase in the viral fitness either to sylvatic mosquitos and non-human primates, or adaptation to new hosts such as humans and peridomestic mosquitos.
We detected one site (codon position 82) under positive selection along ancestral lineage (branch B) of DEN2 prM gene using the Branch-Site model. The site locates in the premembrane (Pr) region of the PrM protein. The PrM protein associates with domain II of the E protein in 1:1 fashion, and is believed to act as a chaperone for the folding of E and to prevent the premature fusion of virus to membrane inside the producing cell [19]. The PrM cleavage site will be cleaved by the host furin into a C-terminal M portion containing a transmembrane domain that remains associated with the viral particle, and a pr peptide dissociates upon the release of the virus. The site detected in this study is proximal to the furin cleavage site at position P10. The cleavage site contains basic amino acids at position P1, P2, and P4, often with Arg(P4)-X(P3)-X(P2)-Arg(P1) (where X is any amino acids). Positions P5–P13 are additional basic residues that are found at the location, and these residues are quite conserved in dengue virus [25]. Replacement of these 13 amino acids (from P1 to P13) with those of tick-borne encephalitis virus (TBEV), yellow fever virus (YEV), and Japanese encephalitis virus (JEV) showed changes in the level of PrM cleavage and viral export [25]. Multiple point mutations in the prM cleavage site, including residue P10, confirmed that these amino acids affect the cleavage level as well as virus replication [24]. The prM cleavage of dengue is associated with viral maturation and has found to be incomplete in many cell lines. Comparing to complete cleavage of PrM in the other flaviviruses, this partial cleavage in dengue has raised many questions, such as the association between the partial cleavage and enhancement property of PrM [26, 27]. The ability of PrM to enhance viral infection was demonstrated in the study by Dejnirattisai et al. (2010) showing that antibodies to PrM (anti-PrMs) appeared to enhance viral infection rather than neutralizing it, while antibodies to Es (anti-Es), especially those that responded to domain III, were important for viral neutralization [2]. They also showed that both anti-PrMs and anti-Es were highly cross-reactive among four dengue serotypes, but when compare the cross-reactivity to JE, the majority of anti-Es tested cross-react with JE, while most of the anti-PrMs do not. This implies a more structural conservation of PrM and thus, a more conservative function of the protein. Taken together, the evidence of codon at position 82, which is referred to as position P10 in the cleavage site, being under positive selection suggests that the PrM cleavage plays an important role in dengue evolution, perhaps involving in modulation of the levels of viral release or infectivity. Whether this site is correlated with the evidence suggesting that DEN2 has been associated more often with DHF/DSS requires further investigation.
The Branch-Site model detected more sites under positive selection along the ancestral lineages of E gene, which may indicate that the E gene was subject to a stronger positive selection than the prM gene. This result is consistent with the result obtained from the Branch-specific model. The viral E protein carries the main antigenic determinants of the virus as well as involves in receptor recognition and fusion between viral and cellular membrane [28, 29]. Comparing to a more conservative and protective functions of PrM as a chaperon, it is not surprising if the E protein would be under a stronger positive selection pressure than the PrM protein, due to its direct interaction with the host. The potential positively selected sites were detected in all domains of E and the majority appears to be located on the surface of the protein. This may imply that these sites may have a direct contact with the host cellular membrane, especially those that are located in the domain III. However, we cannot rule out the possibility that these amino acids may have an important role in the structure stabilization and the interaction with PrM. We have detected two potential positively selected sites along ancestral lineage of DEN4, codon positions 64 and 148, which appear to be on the surface of the E protein (Fig. 2) and are found to locate in a functionally important site. These two sites are close to the two N-linked glycosylation sites, at Asn-67 and Asn-153. The Asn-153 site is conserved in most flaviviruses, while the Asn-67 is unique for dengue (12). The Asn-67 and Asn-153 glycosylation sites were found to interact with DC-SIGN [30]. In mammalian cells, a number of proteins have been proposed as dengue receptor protein, including DC-SIGN, which is a tetrameric C-type lectin [31]. DC-SIGN uses its C-terminal carbohydrate recognition domain (CRD) to bind high-mannose N-linked glycan on the surfaces of dengue viral particles. The Asn-67 and Asn-153 are associated with viral morphogenesis, infectivity, and tropism. Removing of Asn-67 site from the viral particles abolishes the ability to produce new infectious particles, while removing of Asn-153 reduces the viral infectivity [32] A single substitution of Isoleucine for threonine-155 that diminishes one of these two glycosylation sites in DEN4 yielded a virus that was neurovirulence [33]. Moreover, a change in amino acid 148 during the emergence of DEN4 has also been observed in the study by Wang et al. [8]. The study has shown that substitution of this amino acid may have accompanied the adaptation of ancestral sylvatic dengue to human transmission. Therefore, it is possible that amino acids changes at position 64 and 148 that occur throughout evolution especially that of DEN4 may have an effect on the glycosylation sites that consequently results in a more efficient virus-host interaction.
Overall, we have shown that purifying selection played an important role during the separation of four dengue serotypes. It appears that the E protein was subject to a stronger positive pressure than PrM protein, probably due to its direct interaction with the hosts. We have also identified twelve ancient positively selected sites along the interserotypic branches of four dengue serotypes that have yet been identified in other studies. We predict that these sites are associated with the emergence of the four serotypes as well as the adaptation to new hosts. Future mutagenesis study on these positively selected sites will bring a deeper understanding of the emergence of the four serotypes and may shed some light to problem, such as the antibody-dependent enhancement that has been a big obstacle for dengue vaccine development.
References
B.D. Lindenbach, C.M. Rice, Adv. Virus Res. 59, 23–61 (2003)
W. Dejnirattisai, A. Jumnainsong, N. Onsirisakul, P. Fitton, S. Vasanawathana, W. Limpitikul, C. Puttikhunt, C. Edwards, T. Duangchinda, S. Supasa, K. Chawansuntati, P. Malasit, J. Mongkolsapaya, G. Screaton, Science 328, 745–748 (2010)
S.B. Halstead, E.J. O’Rourke, Nature 265, 739–741 (1977)
N. Sangkawibha, S. Rojanasuphot, S. Ahandrik, S. Viryaponse, S. Japanasen, V. Salitul, B. Phantumchinda, S.B. Halstead, Am. J. Epidemiol. 120, 653–669 (1984)
M.G. Guzmán, G. Kouri, L. Valdes, J. Bravo, M. Alvarez, S. Vazques, I. Delgado, S.B. Halstead, Am. J. Epidemiol. 152, 793–799 (2000)
M.S. Diamond, T.C. Pierson, D.H. Fremont, Immunol. Rev. 225, 212–225 (2008)
S.S. Twiddy, E.C. Holmes, A. Rambaut, Mol. Biol. Evol. 20, 122–129 (2003)
E. Wang, H. Ni, R. Xu, A.D. Barrett, S.J. Watowich, D.J. Gubler, S.C. Weaver, J. Virol. 74, 3227–3234 (2000)
D.J. Gubler, Dengue and dengue hemorrhagic fever: its history and resurgence as a global public health problem, in Dengue and dengue hemorrhagic fever, ed. by D.J. Gubler, G. Kuno (CAB International, New York, NY, 1997), pp. p1–p22
P.M. Zanotto, E.A. Gould, G.F. Gao, P.H. Harvey, E.C. Holmes, Proc. Natl. Acad. Sci. USA 93, 548–553 (1996)
I. Kawaguchi, A. Sasaki, M. Boots, Proc. Biol Sci. 270, 2241–2247 (2003)
S.S. Twiddy, C.H. Woelk, E.C. Holmes, J. Gen. Virol. 83, 1679–1689 (2002)
S.S. Twiddy, J.J. Farrar, N.V. Chau, B. Wills, E.A. Gould, T. Gritsun, G. Lloyd, E.C. Holmes. Virology 298, 63–72 (2002)
Z. Yang, Mol. Biol. Evol. 24, 1586–1591 (2007)
Z. Yang, Mol. Biol. Evol. 15, 568–573 (1998)
Z. Yang, R. Nielsen, Mol. Biol. Evol. 19, 908–917 (2002)
Z. Yang, W.S.W. Wong, R. Nielsen, Mol. Biol. Evol. 22, 1107–1118 (2005)
I.M. Yu, W. Zhang, H.A. Holdaway, L. Li, V.A. Kostyuchenko, P.R. Chipman, R.J. Kuhn, M.G. Rossmann, J. Chen. Sci. 319, 1834–1837 (2008)
L. Long, S.M. Lok, I.M. Yu, Y. Zhang, R.J. Kuhn, J. Chen, M.G. Rossmann, Science 319, 1830–1834 (2008)
S.N. Bennett, E.C. Holmes, M. Chirivella, D.M. Rodriguez, M. Beltran, V. Vorndam, D.J. Gubler, W.O. McMillan, J. Gen. Virol. 87, 885–893 (2006)
G.M. Jenkins, A. Rambaut, O.G. Pybus, E.C. Holmes, J. Mol. Evol. 54, 156–165 (2002)
C.H. Woelk, E.C. Holmes, Mol. Biol. Evol. 19, 2333–2336 (2002)
E.C. Holmes, C.H. Woelk, R. Kassis, H. Bourhy, Virology 292, 247–257 (2002)
S.N. Bennett, E.C. Holmes, M. Chirivella, D.M. Rodriguez, M. Beltran, V. Vorndam, D.J. Gubler, W.O. McMillan, Mol. Biol. Evol. 20, 1650–1658 (2003)
P. Keelapang, R. Sriburi, S. Supasa, N. Panyadee, A. Songjaeng, A. Jairungsri, C. Puttikhunt, W. Kasinrerk, P. Malasit, N. Sittisombut, J. Virol. 78, 2367–2381 (2004)
J. Junjhon, M. Lausumpao, S. Supasa, S. Noisakran, A. Songjaeng, P. Saraithong, K. Chaichoun, U. Utaipat, P. Keelapang, A. Kanjanahaluethai, C. Puttikhunt, W. Kasinrerk, P. Malasit, N. Sittisombut, J. Virol. 82, 10776–10791 (2008)
J. Junjhon, T.J. Edwards, U. Utaipat, V.D. Bowman, H.A. Holdaway, W. Zhang, P. Keelapang, C. Puttikhunt, R. Perera, P.R. Chipman, W. Kasinrerk, P. Malasit, R.J. Kuhn, N. Sittisombut, J. Virol. 84, 8353–8358 (2010)
W.D. Crill, J.T. Roehrig, J. Virol. 75, 7769–7773 (2001)
S.L. Allison, J. Schalich, K. Stiasny, C.W. Mandl, F.X. Heinz, J. Virol. 75, 4268–4275 (2001)
E. Pokidysheva, Y. Zhang, A.J. Battisti, C.M. Bator-Kelly, P.R. Chipman, C. Xiao, G.G. Gregorio, W.A. Hendrickson, R.J. Kuhn, M.G. Rossmann, Cell 124, 485–493 (2006)
B. Tassaneetrithep, T.H. Burgess, A. Granelli-Piperno, C. Trumpfheller, J. Finke, W. Sun, M.A. Eller, K. Pattanapanyasat, S. Sarasombath, D.L. Birx, R.M. Steinman, S. Schlesinger, M.A. Marovich. J. Exp. Med. 197, 823–829
J.A. Mondotte, P.Y. Lozach, A. Amara, A.V. Gamarnik, J. Virol. 81, 7136–7148 (2007)
H. Kawano, V. Rostapshov, L. Rosen, C.J. Lai, J. Virol. 67, 6567–6575 (1993)
Acknowledgments
This work was supported by the Office of the Higher Education Commission and Mahidol University under the National Research Universities Initiative. P. Rodpothong thanks Mahidol University for a Postdoctoral scholarship.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.

{kind=link}

{kind=link}

{kind=link}

{kind=link}
Rights and permissions
About this article
Cite this article
Rodpothong, P., Auewarakul, P. Positive selection sites in the surface genes of dengue virus: phylogenetic analysis of the interserotypic branches of the four serotypes. Virus Genes 44, 408–414 (2012). https://doi.org/10.1007/s11262-011-0709-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-011-0709-2