
Abstract
Background
The dengue epidemic in Guangzhou has imposed a rising burden on society and health infrastructure. Here, we present the genotype data for dengue virus serotype 2 (DENV-2) to improve understanding of this dengue epidemic.
Methods
We sequenced the envelope gene of DENV-2 obtained from patient serum samples and subsequently performed maximum-likelihood phylogenetic analysis using PhyMLv3.1, maximum clade credibility analysis using BEAST v.1.10.4, and selection pressure analysis using Datamonkey 2.0.
Results
The prevalent DENV-2 strains identified in Guangzhou region are related to those in Southeast Asian countries. In particular, the Malaysia/Indian subcontinent genotype is prevailing in Guangzhou with no apparent genotype shift having occurred over the past 20 years. However, episodic positive selection was detected at one site.
Conclusions
Local control of the DENV-2 epidemic in Guangzhou requires effective measures to prevent and monitor imported cases. Moreover, the shift between the Malaysia/Indian subcontinent genotype lineages, which originated at different time points, may account for the rise in DENV-2 cases in Guangzhou. Meanwhile, the low rate of dengue haemorrhagic fever in Guangzhou may be explained by the dominance of the less virulent Malaysia/Indian subcontinent genotype.
Similar content being viewed by others


Background
Dengue is caused by infection with dengue virus (DENV), which is a member of the genus Flavivirus, family Flaviviridae. Approximately 96 million dengue infections were estimated globally in 2010, of which 70% were in Asia [1]. The clinical manifestations of dengue range from mild fever, known as dengue fever, to lethal forms of dengue haemorrhagic fever (DHF) and dengue shock syndrome (DSS). No effective vaccines or drugs against DENV have been developed to date, despite some vaccines available are important to decreases hospitalization for severe cases. Therefore, prevention strategies, such as avoiding additional mosquito bites, cleaning vector breeding sites, and use of pesticides, are the primary methods employed to control dengue infection.
There are four serotypes of DENV, namely, dengue virus serotype 1–4 (DENV-1–4). Each serotype can be farther divided into several genotypes based on phylogenetic analysis. The envelope of DENV plays a significant role during infection. DENV particles bind to host cell through envelope receptors. However, this attachment can be prevented via binding of antibodies specific for the envelope protein. Meanwhile, to avoiding antibody neutralisation, the env gene exhibits a high mutation rate. Indeed, the env gene is widely used to classify viruses into genetic groups (“genotypes”) within serotypes [2].
Guangzhou, located at 113°E, 23°N, is an important external city of China. With a population of 12 million, Guangzhou has borne a heavy burden of dengue for nearly 40 years since it was first recorded in 1978. In fact, 59,334 dengue cases were reported in mainland China from 2005 to 2015 [3], of which 39,325 cases were reported in Guangzhou (66.28%).
Over the past 2 decades, the dengue epidemic in Guangzhou was dominated by dengue virus serotype 1 (DENV-1) [4, 5]. However, DENV-2 has also been detected and associated with outbreaks in various communities. DENV-2 reportedly exhibits greater potential to cause DHF/DSS and is more readily transmitted than the other three serotypes. In fact, secondary infection with DENV-2 after the first heterotypic infection is more likely to result in DHF/DSS than secondary infection of the other three serotypes [6]. Therefore, with the prevalence of DENV-1in Guangzhou, the spread of DENV-2 may increase the risk of DHF/DSS outbreaks. Furthermore, a specific genotype may have the propensity to cause DHF/DSS and be transmitted efficiently by vectors [7, 8]. Therefore, it is necessary to closely monitor the genetic diversity and genotype variations of DENV-2 to better understand the dengue epidemic in Guangzhou.
To this end, we analysed serum samples of patients suspected of being infected with dengue that had been sent to the Guangzhou Center for Disease Control and Prevention from 2001 to 2020. We believe that phylogenetic analysis of the DENV-2 strains present throughout Guangzhou during this time period will provide critical insights to scientists and public health officials tasked with prevention and control of dengue.
Methods
Sample collection
Serum samples were obtained from suspected patients presenting with symptoms suggestive of dengue, such as sudden high fever with headache, arthralgia, and/or myalgia, according to diagnosis for dengue fever promulgated by the National Health Commission of the People’s Republic of China [9].
Virus isolation and sequencing
The serum samples were analysed by reverse transcription-quantitative polymerase chain reaction (RT-qPCR) using a dengue virus RT-qPCR kit (Jiangsu BioPerfectus Technologies Co., Ltd., China).
Samples from 2001to 2018 that tested positive by RT-qPCR were diluted 1:50 in RPMI-1640 medium (Life Technologies Corporation, Grand Island, NY, USA). Aedes albopictus cloneC6/36(ATCC CRL-1660) cell monolayers (Cell Bank of the Chinese Academy of Sciences, Shanghai, China) were inoculated with the diluted samples and incubated at 28 °C. Supernatants of cultures with cytopathic effects (CPE) observed within 7 days were harvested for further sequencing. Supernatants of cell cultures with no CPE were added to new C6/36 monolayers. Inoculated cells without CPE after three generations were considered negative for DENV isolation.
In 2019 and 2020, to improve sequence quality and fidelity, RNA extracted from serum samples was analysed via RT-qPCR and direct sequencing of the envelope gene without virus isolation, as recommended by Leitmeyer et al. [10]. Therefore, more sequences were acquired in 2019 than in other years.
Viral RNA was extracted from the supernatants of cell cultures (2001–2018) or sera of patients (2019–2020) using the QIAamp Viral RNA Mini kit (Qiagen, Germany) according to the manufacturer’s instructions. Nucleotides from positions 937 to 2376 of the DENV-2 coding envelope gene were amplified using the SuperScript III One-Step RT–PCR System with Platinum Taq DNA polymerase (Invitrogen, USA). The sense primer was 5ʹ-CCAGGCTTTACCATAATGGC-3ʹ and the anti-sense primer was 5ʹ-CCAGCTGCACAACGCAACCAC-3ʹ. The reaction was initiated at 50 °C for 30 min, followed by denaturation at 94 °C for 2 min; 40 cycles of denaturation at 94 °C for 30 s, primer annealing at 52 °C for 30 s, primer extension at 72 °C for 2 min, and a final extension step at 72 °C for 7 min. The product was sequenced using Sanger sequencing.
Phylogenetic and molecular clock analysis
The maximum-likelihood phylogenetic tree of the obtained envelope sequences acquired in Guangzhou, along with the reference sequences retrieved from GenBank, was constructed using PhyML v3.1 [11]. The substitution model was determined by SMART model selection of the Bayesian information criterion [12]. The fast likelihood-based method of eBayes was applied owing to the large number of sequences. Genotypes were grouped according to the criteria defined by Rico-Hesse [2].
A maximum clade credibility (MCC) tree was constructed for the same sequences using BEAST v.1.10.4. The detected date for each sequence was used as the calibration date. The tree was visualised using Bayesian Evolutionary Analysis Utility (BEAUti) with the following settings: substitution model, GTR; base frequencies, estimated; site heterogeneity model, gamma + invariant sites; number of gamma categories, 4; clock type, uncorrelated relaxed clock; tree model, random starting tree; length of chain, 100,000,000; echo state to screen every 10,000; and log parameters every 10,000. The results of both the maximum-likelihood and MCC phylogenetic trees were visualised and edited using FigTree.
Selection pressure analysis
Episodic adaptive selection was evaluated using the Mixed Effects Model of Evolution (MEME) algorithm implemented by Datamonkey 2.0 [13]. A positive selection signature for each site was determined when the β+ parameter, representing the rate of nonsynonymous substitutions (dN), was greater than α, representing the rate of synonymous substitutions (dS).
Results
A total of 37,431 serum samples from suspected patients from 2001 to 2020 were sent to the lab at the Guangzhou Center for Disease Control and Prevention for RT-qPCR screening. A total of 2942 positive samples were identified. From these samples, 1003 sequences were obtained, comprising 754 DENV-1, 148 DENV-2, 63 DENV-3, and 38 DENV-4. Table 1 summarises the DENV-2 cases detected by RT-qPCR in the serum samples of patients suspected to have dengue from 2001 to 2020. There were 416 cases of DENV-2 infection, comprising 373 domestic cases (89.66%) and 43 (10.34%) imported cases. Cases from Southeast Asian countries constituted 83.72% (n = 36) of imported cases. Before 2010, only one DENV-2 infection was detected in 2005. Since 2010, DENV-2 infections have been detected each year.
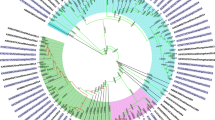
Over the 20 years, 148 DENV-2 envelope gene sequences (1440 bp) were obtained from isolated strains or sera of patients in Guangzhou. All sequences were deposited in GenBank. The phylogenetic tree shown in Fig. 1 was constructed based on these 148 sequences detected in Guangzhou and 56 sequences downloaded from GenBank. Most of the sequences (n = 142, 95.95%) identified from 2001 to 2020 in Guangzhou belonged to the Malaysia/Indian subcontinent genotype, while only six sequences (4.05%) clustered in the Southeast Asia genotype. No American genotype or West African genotype was detected throughout the study period.

Maximum-likelihood midpoint rooted phylogenetic tree of DENV-2 envelope sequences from Guangzhou and reference sequences. Guangzhou sequences (n = 148) are identified according to the accession number, isolated year, and lab number. Reference sequences (n = 56) from GenBank (marked with a triangle) are identified using the accession number, country, and year. Bootstrap support, with > 75%, is shown next to the branches. Different colours present different genotypes
The Malaysia/Indian subcontinent genotype can be further divided into several lineages: GZ1, GZ2, GZ3, GZ4, and GZ5. The sequences acquired in 2005, 2010, and 2013 all belonged to lineage GZ4; thereafter, a shift was observed to lineage GZ5 in 2014. Moreover, nine subspecies were detected in 2015, three of which (33.33%) clustered in lineage GZ2, while the remaining six (66.67%) clustered in lineage GZ5. Thereafter, most sequences belonged to lineage GZ5; however, a few were scattered among the GZ1, GZ2, GZ3, and GZ4 lineages.
When several identical sequences were detected in the same year, one was retained as a representative for constructing the MCC tree (Fig. 2). This tree was based on 80 sequences obtained in Guangzhou along with 53 sequences retrieved from GenBank. All of the Malaysia/Indian subcontinent genotype strains shared an ancestor in 1955. However, the various lineages of this genotype manifested different introduction times. Lineage GZ5, which was the most prevalent, emerged in 1995 and comprised sequences from 2014 to 2019.

Maximum clade credibility tree of envelope gene nucleotide sequences collected in Guangzhou and from GenBank. Guangzhou sequences, n = 80; GenBank sequences (triangle), n = 53. Posterior probabilities higher than 0.80 are shown at each node. tMRCA = time to the most recent common ancestor
Positive selection of the envelope gene for lineage GZ5was analysed using MEME. The sequences of Guangzhou were compared with those of the reference sequence DQ518635 isolated from Malaysia in 2003. Episodic positive selection was detected at one site, codon position 364, with β+ = 2160.36, α = 0.00, P = 0.06. Additionally, molecular characterisation identified two amino acid differences (E329D and I439V) in all sequences from the Guangzhou isolates in comparison with the DQ518635 sequence from Malaysia in 2003.
Discussion
Over the last century, DENV-2 epidemics were reported in 1986, 1987, 1988, and 1993 in Guangzhou [14, 15], thereafter, they have subsided. However, our findings indicate that DENV-2 has been spreading throughout Guangzhou from 2001 to 2020. Although no DENV-2 infection was detected prior to 2005, the number of annual DENV-2 cases continued to increase between 2010 and 2018. We also determined that the percentage of DENV-2 domestic cases increased from 80.95 to 95.31% between 2015 and 2018, reaching a peak in both number and percentage in 2018. Thereafter, the number and percentage of domestic cases declined slightly in 2019 but remained the second highest compared to those in previous years. Moreover, a sharp decrease in DENV-2 cases occurred in 2020, which might be related to the quarantine imposed due to the coronavirus disease 2019 outbreak [16]. That is, limiting imported cases with the quarantine may have controlled the local dengue epidemic, highlighting the relevance of monitoring imported cases for local control.
The DENV-2 epidemic in Guangzhou is greatly impacted by Southeast Asian countries. In fact, our epidemiological investigation revealed that 83.72% of DENV-2 imported cases, most of which were returning travellers, originated from Southeast Asian countries. The results of a BLAST search in GenBank and the phylogenetic tree (Fig. 1) further revealed that the DENV-2 strains sequenced in Guangzhou were closely related to those in Southeast Asian countries, which was similar to the characteristic of dengue epidemics involving the other three serotypes in Guangzhou [4, 17, 18]. Indeed, the World Health Organization statistics revealed that infections in Southeast Asian countries account for half of the global dengue burden. Meanwhile, from 2015 to 2019, dengue cases in Southeast Asia increased by 46% (from 451,442 to 658,301) [19]. China not only contiguous with Southeast Asian countries, but also is farther connected by economic ties. Moreover, with the opening of private travel abroad, the number of travellers to Southeast Asian countries has steadily increased [20,21,22]. This situation is not unique to China, as the spread of dengue viruses by travellers has become a global issue [23]. Therefore, better preparation is needed, with strict regulations, to prevent the spread of infection when travelling in endemic areas. For example, a convenient and rapid method for screening viremia that can be applied at customs may help curb the import and spread of DENV.
The Malaysia/Indian subcontinent genotype was responsible for the epidemic in Guangzhou. As displayed in the phylogenetic tree in Fig. 1, this genotype, shown in yellow, constitutes 95.95% (142 sequences) of the total 148 DENV-2 sequences. Meanwhile only six sequences of the Southeast Asia genotype were detected. Moreover, no genotype shift in the Malaysia/Indian subcontinent genotype was observed over the 20-year study period. The most recent common ancestor of all Malaysia/Indian subcontinent strains was estimated to have appeared in 1955. However, when dividing the Malaysia/Indian subcontinent genotype into its different lineages, a shift from lineage GZ4 to GZ5, was observed between 2013 and 2014, which coincided with a rising number of DENV-2 cases since 2014. Although there is no clear relationship between lineage and virulence in DENV, outbreaks, limited circulation, and spreading related to shifts in lineages have been reported [24,25,26,27]. Substitutions in the envelope gene that may result in maturation and activation of macrophages, with consequent enhancement of the immune response characterised by increased production of cytokines, are considered to be likely causes of the prevailing differences among lineages. Meanwhile, positive selection analysis of the GZ5 lineage by MEME exhibited signs of directional selection, which may contribute to the prevalence of GZ5. However, further research is needed to confirm whether this lineage shift is responsible for the rise in cases.
With the prevalence of the Malaysia/Indian subcontinent genotype in Guangzhou, the relatively low rate of DHF/DSS suggests that this genotype may be less virulent. Secondary infection with DENV-2 after infection with heterotypic DENV is believed to be associated with an increased risk of DHF/DSS [6]. However, with the DENV-1epidemic in Guangzhou persisting for more than 20 years and the rising number of DENV-2 cases [4, 5], the incidence of DHF/DSS remained relatively low compared with the global incidence and that of Southeast Asian countries [28,29,30,31]. Another study revealed that secondary infection with the American genotype of DENV-2 failed to cause DHF/DSS [32], whereas other extensive studies indicated that the Southeast Asian genotype was more efficient at infection and was also more likely to cause DHF/DSS [7, 8, 32, 33]. Of the 148 sequences detected in Guangzhou, only six (4.05%) belonged to the Southeast Asia genotype, four of which were identified from imported cases. This revealed that the Southeast Asia genotype was rare in Guangzhou, which may explain the low incidence of DHF/DSS. Meanwhile, the prevailing Malaysia/Indian subcontinent genotype has a limited capacity for leading to DHF/DSS. However, further studies are needed to determine whether the incidence of DHF/DSS is low in other areas, with an epidemic dominated by the Malaysia/Indian subcontinent genotype. Determining the critical differences between genotypes and host immune mechanisms that may enhance the pathogenesis of genotypes might provide new insights to advance the current understanding regarding the mechanism of DHF/DSS.
The DENV-2 epidemic in Guangzhou was complex. Specifically, the MCC tree in Fig. 2 revealed that the Guangzhou strains originated from different time periods, indicating various evolution and dissemination paths. Strains that clustered into lineage GZ5 shared the eldest ancestor in 1995, whereas strains belonging to lineage GZ1 emerged in 2016. Meanwhile, the domestic and foreign strains of lineage GZ2 detected in 2014 and 2016 shared the same ancestor in 2012. The strain then evolved during 2014 and 2016 in Guangzhou and spread not only in China but also throughout Thailand and Vietnam. However, within the same year, the strains may have also be distributed in different lineages; for example, the sequences from isolates obtained in 2018 were distributed among lineages GZ1, GZ3, GZ4 and GZ5. This co-epidemic of different lineages showing different origins has complicated the epidemic situation in Guangzhou.
Conclusions
Our study has investigated the epidemiology of DENV-2 and shed light on epidemiologic impact of the Malaysia/Indian subcontinent genotype and its different lineages, in Guangzhou, an important dengue circulation city in China. In particular, the increase in DENV-2 cases may be due to an observed lineage shift. Taken together, our study findings suggest that global cooperation is required to curb the spread of dengue.
Availability of data and materials
The sequences analysed during the current study are available in the GenBank repository, https://www.ncbi.nlm.nih.gov/genbank/. The data that support the findings of this study are available from the corresponding author.
Abbreviations
- BEAUti:
-
Bayesian Evolutionary Analysis Utility
- CPE:
-
Cytopathic effects
- DENV:
-
Dengue virus
- DENV1-4:
-
Dengue virus serotype 1–4
- DHF:
-
Dengue haemorrhagic fever
- DSS:
-
Dengue shock syndrome
- MCC:
-
Maximum clade credibility
- MEME:
-
Mixed Effects Model of Evolution
References
Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, et al. The global distribution and burden of dengue. Nature. 2013;496:504–7.
Rico-Hesse RH. Microevolution and virulence of dengue viruses. Adv Virus Res. 2003;59:315–41.
Sun J, Lu L, Wu H, Yang J, Xu L, Sang S, et al. Epidemiological trends of dengue in mainland China, 2005–2015. Int J Infect Dis. 2017;57:86–91.
Jiang L, Wu X, Wu Y, Bai Z, Jing Q, Luo L, et al. Molecular epidemiological and virological study of dengue virus infections in Guangzhou, China, during 2001–2010. Virol J. 2013;10:4.
Bai Z, Cheng Liu L, Jiang L, Luo L, Feng H, Lin P, et al. Evolutionary and phylodynamic analyses of dengue virus serotype I in Guangdong Province, China, between 1985 and 2015. Virus Res. 2018;256:201–8.
Sangkawibha N, Rojanasuphot S, Ahandrik S, Viriyapongse S, Jatanasen S, Salitul V, et al. Risk factors in dengue shock syndrome: a prospective epidemiologic study in Rayong, Thailand. I. The 1980 outbreak. AmJ Epidemiol. 1984;120:653–69.
Cologna R, Rico-Hesse R. American genotype structures decrease dengue virus output from human monocytes and dendritic cells. J Virol. 2003;77:3929–38.
Anderson JR, Rico-Hesse R. Aedes aegypti vectorial capacity is determined by the infecting genotype of dengue virus. Am J Trop Med Hyg. 2006;75:886–92.
China National Health Commission of the People’s Republic of China. Diagnosis for Dengue Fever. 2018. http://www.nhc.gov.cn/wjw/s9491/201803/d524df26df28453eada8371dc3565818.shtml. Accessed 4 Nov 2021.
Leitmeyer KC, Vaughn DW, Watts DM, Salas R, Villalobos I, de Chacon, et al. Dengue virus structural differences that correlate with pathogenesis. J Virol. 1999;73:4738–47.
Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. New Algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21.
Lefort V, Longueville JE, Gascuel O. SMS: smart model selection in PhyML. Mol Biol Evol. 2017;34:2422–4.
Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond S. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012;8:e1002764.
Pan ZM, Qiu J, Chen X, Liao Y, Yan Z. Epidemiology of dengue in Guangzhou, during 1990s. South China J Prev Med. 2000;26:33–5.
Luo L, Yang Z-C, Wang Y-L, Liu Y-F. Analysis of the epidemiologic features of dengue fever from 1978 to 2006 in Guangzhou. China Chin J Infect Dis. 2008;26:490–3.
Jiang L, Liu Y, Su W, Liu W, Yang Z. Decreased dengue cases attributable to the effect of COVID-19 in Guangzhou in 2020. PLoS Negl Trop Dis. 2021;15:e0009441.
Liu Y, Jiang LY, Luo L, Cao YM, Jing QL, Yang ZC. Phylogenetic analysis of envelope gene of dengue virus serotype 2 in Guangzhou, 2001–2015. Chin J Epidemiol. 2017;38:90–5.
Liu Y, Jiang LY, Jing QL, Su WZ, Cai WF, Di B, et al. Molecular characterization and phylogenetic analysis of the envelope gene of dengue virus type 3 in Guangzhou, China 2001–2016. Chin J Zoonoses. 2018;3:487–90.
WHO. Dengue and severe dengue. 2021. https://www.who.int/southeastasia/health-topics/dengue-and-severe-dengue. Accessed 28 Oct 2021.
Bao FH. Disparity of size and rank for the outbound tourism of mainland China. Area Res Dev. 2018;37:92–7.
Bao FH, Chen Y. Research on the spatial-temporal distribution characteristics of China’s outbound tourism in the past 10 years. World Reg Stud. 2017;26:127–39.
Ding J, Li LF. A study on tourist behavior of outbound travel for Guangzhou residents. Geogr Res. 2004;2:705–14.
Wilder-Smith A, Ooi E-E, Horstick O, Wills B. Dengue. Lancet. 2019;393:350–63.
Oliveira MF, Araujo JMG, Ferreira C Jr, Ferreira DF, Lima DB, Santos FB, et al. Two lineages of dengue virus type 2. Brazil Emerg Infect Dis. 2010;16:576–8.
Faria NR, Nogueira RMR, de Filippis AMB, Simões JBS, de Bruycker Nogueira F, da Rocha Queiroz Lima M, et al. Twenty years of DENV-2 activity in Brazil: molecular characterization and phylogeny of strains isolated from 1990 to 2010. PLoS Negl Trop Dis. 2013;7:e2095.
McElroy KL, Santiago GA, Lennon NJ, Birren BW, Henn MR, Muñoz-Jordán JL. Endurance, refuge, and reemergence of dengue virus type 2, Puerto Rico, 1986–2007. Emerg Infect Dis. 2011;17:64–71.
Mir D, Romero H, de Carvalho LMF, Bello G. Spatiotemporal dynamics of DENV-2 Asian–American genotype lineages in the Americas. PLoS ONE. 2014;9:e98519.
Lu JY, Chen ZQ, Ma MM, Cai RB, Su WZ, Li YL, et al. Epidemic trend of dengue fever in Guangzhou City from 2008 to 2017. J Trop Med. 2018;18:973–6.
Karyanti MR, Uiterwaal CSPM, Kusriastuti R, Hadinegoro SR, Rovers MM, Heesterbeek H, et al. The changing incidence of dengue haemorrhagic fever in Indonesia: a 45-year registry-based analysis. BMC Infect Dis. 2014;14:412.
Guo C, Zhou Z, Wen Z, Liu Y, Zeng C, Xiao D, et al. Global epidemiology of dengue outbreaks in 1990–2015: a systematic review and meta-analysis. Front Cell Infect Microbiol. 2017;7:317.
Cummings DA, Iamsirithaworn S, Lessler JT, McDermott A, Prasanthong R, Nisalak A, et al. The impact of the demographic transition on dengue in Thailand: insights from a statistical analysis and mathematical modeling. PLoS Med. 2009;6:e1000139.
Watts DM, Porter KR, Putvatana P, Vasquez B, Calampa C, Hayes CG, et al. Failure of secondary infection with American Genotype Dengue 2 to cause dengue haemorrhagic fever. Lancet. 1999;354:1431–4.
Rico-Hesse R, Harrison LM, Salas RA, Tovar D, Nisalak A, Ramos C, et al. Origins of dengue type 2 viruses associated with increased pathogenicity in the Americas. Virology. 1999;230:244–51.
Acknowledgements
We thank our colleagues in Virology Department.
Funding
This work was funded by grants from the Guangzhou Municipal Science and Technology Project [Grant No. 202002030117], the Key Project of Medicine Discipline of Guangzhou [Grant No. 2021-2023-11], and the Basic Research Project of Key Laboratory of Guangzhou [Grant No. 2021BRP004].
Author information
Authors and Affiliations
Contributions
LJ conducted most of the experiments and drafted the manuscript. YL designed the experiments and analysed data. QJ collected the epidemiological information. WS and YC cultured the virus and sequenced the genes. XW and ZY participated in the detection of dengue virus in the samples. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Ethics Committee of the Guangzhou Centre for Disease Control and Prevention approved the present study. Written informed consent was obtained from all the participants in the study or from the parent/guardian when the participant was a minor (younger than 18 years old). All procedures involved in this work complied with the ethical standards of the relevant national and institutional committees on human experimentation and with the Declaration of Helsinki (2008 amendment).
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jiang, L., Liu, Y., Su, W. et al. Circulation of genotypes of dengue virus serotype 2 in Guangzhou over a period of 20 years. Virol J 19, 47 (2022). https://doi.org/10.1186/s12985-022-01773-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-022-01773-7