
Abstract
Five palladium(II) complexes with pyridine derivative ligands have been synthesized. The molecular structures of the complexes were determined by X-ray crystallography, and their spectroscopic properties were studied. Based on the crystal structures, computational investigations were carried out in order to determine the electronic structures of the complexes. The electronic spectra were calculated with the use of time-dependent DFT methods, and the electronic spectra of the transitions were correlated with the molecular orbitals of the complexes. The emission properties of the complexes have been examined.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Palladium(II) complexes currently attract a considerable interest because of their potentially beneficial pharmacological properties. Palladium complexes with aromatic N-containing ligands, e.g., derivatives of pyridine, quinoline, pyrazole, and 1,10-phenanthroline, have shown very promising antitumor characteristics [1–4]. Some of these complexes, especially the trans analogs with nonplanar heterocyclic amine ligands, have been found to overcome multifactorial cisplatin resistance in human ovarian cell lines [5, 6]. On the other hand, Pd(II) complexes have been widely explored due to their catalytic efficiency, e.g., for various carbon–carbon and carbon–nitrogen bond-forming reactions [7–11]. While most studies of palladium have concentrated on the reactivity of its complexes, little information about their electronic structures has been obtained.
Complexes with pyridine derivative ligands and such metals as ruthenium(II), osmium(II), and rhenium(I) are by far the most studied due to their luminescent properties. In contrast, d 8 metal complexes have been much less studied. The luminescent properties of palladium(II) complexes strongly depend on their electronic structure. For this reason, study of the electronic structures of such complexes is valuable as a means to the prediction of their properties [12].
In this paper, we present the synthesis, crystal, molecular, electronic structures, and the spectroscopic characterization of five palladium(II) complexes with pyridine-type ligands. The electronic structures of the complexes have been determined by density functional theory (DFT) and employed for the discussion of bonding properties.
Experimental
All reagents used for the synthesis of the complexes were commercial products and have been used without further purification.
Synthesis of [PdCl2(L)2] complexes
A typical procedure for the preparation of the complexes is as follows: a solution of PdCl2 (0.177 g, 0.1 mmol) and 2-hydroxy-6-methylpyridine (1) (0.22 g, 0.2 mmol), 2-aminomethylpyridine (2) (0.2 cm−3, 0.2 mmol), 2-hydroxypropylpyridine (3) (0.28 cm−3, 0.2 mmol), 2-benzoilpyridine (4) (0.36 g, 0.2 mmol), or 4-(3-phenylpropyl)pyridine (5) (0.4 cm−3, 0.2 mmol) in acetonitrile (50 cm3) was refluxed for about 2 h and than filtered. Crystals suitable for X-ray analysis were obtained by slow evaporation of solvent.
1: IR (KBr) [cm−1]: 3,400 νOH; 3,163 νArH; 2,917 νCH; 1,614 νC=N; 1,578 νC=C; 1,452, 1,306 δ(C–CH in the plane); 1,434 νPh; 1,099, 1,037 δOH; 788 δ(C–C out of the plane); 742 δ(C–C out of the plane). 1H NMR [δ, ppm]: (DMSO-d6): 7.743, 7.303 (py), 6.095 (OH), 2.152 (CH3). UV–VIS [λ nm, (ε)] (acetonitrile): 417 (1.21), 305 (2.14), 280 (2.21), 236 (4.87), 202 (4.37).
2: IR (KBr) [cm−1]: 3,330 νNH; 3,060, 3,022 νArH; 2,942, 2,883 νCH; 1,605 νC=N; 1,583 νC=C; 1,480, 1,247 δ(C–CH in the plane); 1,444 νPh; 1,060, 1,022 δNH; 922 δ(C–H out of the plane); 804 δ(C–C out of the plane); 702 δ(C–C out of the plane). 1H NMR [δ, ppm]: (DMSO–d6): 9.012, 7.743, 7.723 (py), 4.169 (NH), 1.597 (CH2). UV–VIS [λ nm (ε)] (acetonitrile): 370 (1.98), 319 (2.17), 255 (4.75), 211 (4.88).
3: IR (KBr) [cm−1]: 3,485 νOH; 3,070 νArH; 2,919, 2,874, 2,788 νCH; 1,607 νC=N; 1,570 νC=C; 1,482, 1,260 δ(C–CH in the plane); 1,453 νPh; 1,073, 1,052 δOH; 917 δ(C–H out of the plane); 768 δ(C–C out of the plane); 749 δ(C–C out of the plane). 1H NMR [δ, ppm]: (DMSO–d6): 8.322, 8.031, 7.369 (py), 5.172 (OH), 4.171 (t), 3.812, 2.029 (CH2). UV–VIS [λ nm, (ε)] (acetonitrile): 392 (1.83), 319 (2.06), 265 (3.96), 232 (4.13), 212 (4.97).
4: IR (KBr) [cm−1]: 3,082, 3,050 νArH; 1,676 νC=O; 1,595 νC=N; 1,566 νC=C; 1,446 νPh; 1,318, 1,283, 1,160 δC=O; 943, 928; 917 δ(C–H out of the plane); 776 δ(C–C out of the plane); 706 δ(C–C out of the plane). 1H NMR [δ, ppm]: (DMSO–d6): 9.027, 8.049, 7.880, 7.719, 7.616, 7.469 (py, Ph). UV–VIS [λ nm, (ε)] (acetonitrile): 370 (1.62), 262 (3.97), 237 (4.17), 211 (5.06).
5: IR (KBr) [cm−1]: 3,070 νArH; 2,918, 2,858 νCH; 1,618 νC=N; 1,578 νC=C; 1,490, 1,267 δ(C–CH in the plane); 1,452 νPh; 918 δ(C–H out of the plane); 756 δ(C–C out of the plane); 706 δ(C–C out of the plane). 1H NMR [δ, ppm]: (DMSO–d6): 8.823, 7.102 (py), 7.461–6.952 (Ph), 2.628, 1.973 (CH2). UV–VIS [λ nm, (ε)] (acetonitrile): 377 (1.23), 285 (2.96), 212 (4.95).
Physical measurements and DFT calculations
Infrared spectra were recorded on a Nicolet Magna 560 spectrophotometer in the range of 4,000–450 cm−1 using KBr pellets. Electronic spectra were measured on a Lab Alliance UV–VIS 8,500 spectrophotometer in the range of 1,100–180 nm in methanol solution. 1H NMR spectra were obtained at room temperature in CDCl3 using a Bruker 400 spectrometer.
The calculations were carried out using the Gaussian09 [13] program. The DFT/B3LYP [14, 15] method was used for the geometry optimization and electronic structure determination. The electronic spectra were calculated by the TD-DFT [16] method with use of the CAM-B3LYP functional [17]. The calculations were performed using the DZVP basis set [18] with f functions with exponents 1.94722036 and 0.748930908 on palladium and polarization functions for all other atoms: 6–31G** for chlorine, carbon, nitrogen and 6–31G for hydrogen. The PCM solvent model was used in the Gaussian calculations with acetonitrile as the solvent. Natural bond orbital (NBO) calculations were performed with the NBO code [19] included in Gaussian09. GaussSum 2.2 [20] was used to calculate group contributions to the molecular orbitals and to prepare the partial density of states (DOS) and overlap population density of states (OPDOS) spectra. The contribution of a group to a molecular orbital was calculated using Mulliken population analysis. The PDOS and OPDOS spectra were created by convoluting the molecular orbital information with Gaussian curves of unit height and FWHM of 0.3 eV.
Crystal structures determination and refinement
Yellow crystals of [PdCl2(CH3pyOH)2] (1), [PdCl2(pyCH2NH2)2] (2), [PdCl2(py(CH2)3OH)2] (3), [PdCl2(bopy)2] (4), and [PdCl2(pyCHPh)2] (5) were mounted in turn on an Xcalibur, Atlas, Gemini ultra Oxford Diffraction automatic diffractometer equipped with a CCD detector, and used for data collection. X-ray intensity data were collected with graphite monochromated MoKα radiation (λ = 0.71073 Å) at a temperature of 295.0(2) K, with ω scan mode. Ewald sphere reflections were collected up to 2θ = 50.10°. The unit cell parameters were determined from least-squares refinement of the setting angles of 2,839, 4,094, 6,556, 6,894, and 6,541 strongest reflections. The details concerning crystal data and refinement are gathered in Table 1. During the data reduction, the decay correction coefficients were taken into account. Lorentz, polarization, and numerical absorption corrections were applied. The structures were solved by Patterson methods. All the nonhydrogen atoms were refined anisotropically using full-matrix, least-squares techniques on F 2. All the hydrogen atoms were found from difference Fourier synthesis after four cycles of anisotropic refinement and refined as “riding” on the adjacent atoms with individual isotropic temperature factor equal 1.2 times the value of equivalent temperature factor of the parent atom, with geometry idealization after each cycle. The Olex2 [21] and SHELXS97 [22], SHELXL97 [23] programs were used for all the calculations. Atomic scattering factors were those incorporated in the computer programs.
Results and discussion
Palladium(II) complexes with pyridine derivative ligands were obtained by reactions of PdCl2 with 2-hydroxyl-6-methylpyridine (1), 2-aminomethylpyridine (2), 2-hydroxypropylpyridine (3), 2-benzoilpyridine (4), or 4-(3-phenylpropyl)pyridine (5) in acetonitrile solutions. In the IR spectra of the complexes, the ring C=N and C=C stretching modes of the pyridine-type ligands are present at 1,595–1,618 cm−1 and 1,583–1,566 cm−1, respectively. The stretching and bending modes of the OH and NH2 group in complexes 1, 2, and 3 have maxima at 3,330–3,400 cm−1, 1,099–1,037, and 1,060–1,022 cm−1 in complexes 1, 3, and 2, respectively. The C=O stretching mode of 2-benzoilpyridine in complex 4 is present at 1,676 cm−1 which confirms coordination number 4 in the complex. The 1H NMR spectra of the complexes display the expected signals from the ligands. The signals of the hydroxyl and ammine protons are present at 6.095, 5.172, and 4.169 ppm in complexes (1), (3), and (2), respectively.
Crystal structures
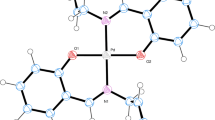
The complexes crystallize in the space groups given in Table 1. The molecular structures of the complexes are shown in Fig. 1. Selected bond lengths and angles are listed in Table 2. The palladium(II) atoms in these complexes have square planar environments with the ligands bonded to the metal centre through the pyridine nitrogen atoms. The Pd–N(py) and Pd–Cl distances are similar and comparable with other palladium(II) complexes. The conformations of molecules of 2 and 3 are stabilized by inter- and intramolecular hydrogen bonds as collected in Table 3. In Fig. 2, the crystal packing of complex 2 viewing down the a axis with hydrogen bonds indicated by dotted lines is shown; the network formed by hydrogen bonds and the π-stacking interactions are visible.

ORTEP drawing of [PdCl2(CH3pyOH)2] (1), [PdCl2(pyCH2NH2)2] (2), [PdCl2(py(CH2)3OH)2] (3), [PdCl2(bopy)2] (4), and [PdCl2(pyCHPh)2] (5) complexes with 50% probability displacement ellipsoids

Crystal packing of complex 2 viewing down the a axis with hydrogen bonds indicated by dotted lines
Electronic structure
To gain insight into the electronic structures and bonding properties of these complexes, DFT calculations were carried out. Before the calculations of electronic structures of the complexes, their geometries were optimized in triplet states using the B3LYP functional. From the data collected in Table 2, one may see that the bond lengths are maximally elongated by ~ 0.11 Ǻ in the calculated gas-phase structures, while the bond angles almost unchanged. Figure 3 shows that the calculated and experimental IR spectra of complex 3 are in good agreement.

The calculated frequencies and experimental IR spectrum of complex (3)
The formal charge of palladium is +2 in the complexes. The calculated charges on the palladium atoms, obtained from natural population analysis, are 0.04 in 1, 0.30 in 2, 0.19 in 3, 0.24 in complex 4, and 0.21 in 5. This is a result of charge donation from the ligands to the metal center and also shows that 2-hydroxy-6-methylpyridine is the strongest σ-donor among the ligands used.
The calculations utilize both α and β occupied molecular orbitals. Among the occupied MOs of the complexes, the largest numbers constitute orbitals of the pyridine-type ligands that are not relevant for the discussion. The d yz , \( d_{{z^{2} }} \), d zx orbitals of palladium(II) contribute to the HOMOs and in the range from HOMO–1 to HOMO–7 with α spin and in the HOMO–1 (2, 3, 4), HOMO–2/–3/–4 and HOMO–7/–8/–9 with β spin. These molecular orbitals are mixed with π orbitals of the pyridine derivatives and chloride ligands. The d xy and \( d_{{x^{2} - y^{2} }} \) orbitals are visible in the LUMO and LUMO + 1 orbitals with β spin, with an antibonding admixture of pyridine and chloride orbitals.
In the frontier region, neighboring orbitals may show a quasi-degeneracy of the energy levels. In such cases, consideration of only the HOMO and LUMO may not yield a realistic description of the frontier orbitals. For this reason, partial density of states (DOS) and overlap population density of states (OPDOS) in terms of Mulliken population analysis were calculated using the GaussSum program. They provide a pictorial representation of the MO compositions and their contributions to chemical bonding. The DOS diagram (β spin) for complex 3 is shown in Fig. 4 as an example. The DOS plot mainly presents the composition of the fragment orbitals contributing to the molecular orbitals. As may be seen from the DOS plots, the HOMO has d Pd and p Cl character and the LUMO is composed of d palladium and π* orbitals of the pyridine ligands. The OPDOS gives an indication of the bonding, nonbonding, and antibonding characteristics with respect to the particular fragments. A positive value in the OPDOS plot means a bonding interaction, while a negative value represents an antibonding interaction, and a near zero value indicates a nonbonding interaction. At the same time, an analysis of the OPDOS diagram allows us to determine the donor–acceptor properties of the ligands. From Fig. 5, it may be concluded that the 2-hydroxy-6-methyl-pyridine and 4-(3-phenylpropyl)pyridine ligands have comparable and strongest σ-donor and π-acceptor properties which is consistent with the charges on the palladium atoms given above. It is connected with an increase in energy of the HOMO associated with strong donor properties and strong π-acceptor ability of the ligand.

The density of states (β spin DOS) diagram for [PdCl2(CH3pyOH)2] (1) complex

Interaction between Pd(II) central ions and pyridine derivative ligands on the overlap partial density of states (OPDOS) diagrams for the complexes
Electronic spectra
The UV–VIS spectra of the complexes are very similar. The nature of the transitions observed in the UV–Vis spectra has been analyzed using time-dependent DFT (TDDFT) based on the optimized triplet state geometry. The electronic transitions were calculated by use of the long range corrected version of the B3LYP functional—CAM-B3LYP which employs the Coulomb-attenuating method. This functional provides a better estimation of excitation energies and oscillators strengths, especially for transitions with Charge-Transfer character. Figure 6 presents the experimental spectra with the calculated transitions of complexes 3 and 5.

UV–Vis spectra with calculated transitions for complexes (1) and (3)
The calculations show that the longest wavelength experimental bands have a Charge-Transfer character connected with transitions from occupied orbitals localized on the pyridine derivatives ligands to β-spin LUMO, LUMO + 1 orbitals localized on the d xy and \( d_{{x^{2} - y^{2} }} \) palladium orbitals. In these bands, contributions of d → d transitions (LF) are also visible. For example, in the case of H-8(β) → L + 1(β) (94%) transition in complex 2, both occupied and virtual molecular orbitals are mainly composed of d Pd orbitals. From the data collected in Table 4, one can see that the bands with maxima in the range 319–250 nm originate from transitions between lower occupied orbitals and the LUMO + 1. Therefore, these bands have CT character. The experimental bands with maxima near 200 nm are attributed to intraligand (IL) (π(py) → π*(py)) and ligand–ligand charge transfer (π(Cl) → π*(py)) transitions.
The emission properties of the complexes have been examined in methanol solutions (concentration of 5 × 10−4 mol/dm3) at room temperature. Figure 7 presents the emission spectra of complexes (2), (3), and (4). The excitations wavelengths were 390 nm for (3) and 370 nm for (2) and (4) which are the absorption maxima which may be assigned to a combination of d → d, MLCT (dπ(Pd) → π*py) and 1ClLCT (n(Cl) → π*) transitions. From the data collected in Table 4 and in the DOS diagrams, one may see that the pyridine ligands play similar role in the occupied and virtual frontier orbitals in all four complexes. Therefore, the emission originating from the lowest energy metal to ligand charge transfer (MLCT) state derived from the excitation involving a dπ → π*ligand transition is observed. This assignment is also supported by analysis of the frontier orbitals of the corresponding complexes that show a contribution from the ligands. The complicated structure of the luminescence spectra suggests that more than one state is involved in luminescence processes.

Emission spectra of complexes (2), (3), and (4) in acetonitrile solutions (c = 5.10−4 mol/dm3)
Conclusion
Summarizing, new palladium(II) complexes with pyridine derivative ligands were synthesized. Molecular structures of the complexes were determined by X-ray, and the spectroscopic properties were studied. The crystal structures were used for DFT calculations in order to determine the electronic structures of the complexes. The results were used to compare π-donor/acceptor properties of the pyridine-type ligands. The electronic spectra were calculated with use of TD-DFT, and the transition characters were assigned in connection with the molecular orbitals of the complexes. The emission properties of the complexes were examined. The emissions originating from the lowest energy MLCT state derived from the excitation involving a dπ → π*ligand transition were observed. This assignment was supported by the analysis of the frontier orbitals of the corresponding complexes, showing a partial contribution from the ligands.
Supplementary data
CCDC 793379, CCDC 782336, CCDC 782033, CCDC 782241, and CCDC 794606 contain the supplementary crystallographic data for complexes [PdCl2(CH3pyOH)2] (1), [PdCl2(pyCH2NH2)2] (2), [PdCl2(py(CH2)3OH)2] (3), [PdCl2(bopy)2] (4), and [PdCl2(pyCHPh)2] (5), respectively. These data can be obtained free of charge from http://www.ccdc.cam.ac.uk/conts/retrieving.html or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223-336-033; or e-mail: deposit@ccdc.cam.ac.uk.
References
Kovala-Demertzi D, Demertzis MA, Filiou E, Pantazaki AA, Yadav PN, Miller JR, Zheng Y, Kyriakidis DA (2003) Biometals 16:411
Budizisz E, Krajewska U, Rozalski M, Szulawska A, Czyż M, Nawrot B (2004) Eur J Pharmacol 502:59
Kuduk-Jaworska J, Puszko A, Kibiak M, Pelczynska M (2004) J Inorg Biochem 98:1447
Budzisz E, Miernicka M, Lorenz I-P, Mayer P, Krajewska U, Rozalki M (2009) Polyhedron 28:637
Najajreh Y, Perez JM, Navarro-Ranninger C, Gibson D (2002) J Med Chem 45:5189
Khazanov E, Barenholtz Y, Gibson D, Najajreh Y (2002) J Med Chem 45:5196
Andrade-Lopez N, Alvarado-Rodrıguez JG, Gonzalez-Montiel S, Rodrıguez-Mendez MG, Paez-Hernandez ME, Galan-Vidal CA (2007) Polyhedron 26:4825
Ojwach SO, Guzei IA, Darkwa J, Mapolie SF (2007) Polyhedron 26:851
Liang H, Liu J, Li X, Li Y (2004) Polyhedron 23:1619
Piro NA, Owen JS, Bercaw JE (2004) Polyhedron 23:2797
Chen R, Bacsa J, Mapolie SF (2003) Polyhedron 22:2855
Anbalagan V, Srivastava TS (2004) Polyhedron 23:3173
Gaussian 09, Revision A.1, Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr., Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian, Inc., Wallingford CT
Becke AD (1993) J Chem Phys 98:5648
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785
Casida ME (1996) In: Seminario JM (ed) Recent developments and applications of modern density functional theory, theoretical and computational chemistry, vol 4. Elsevier, Amsterdam, p 391
Yanai T, Tew D, Handy N (2004) Chem Phys Lett 393:51
Eichkorn K, Weigend F, Treutler O, Ahlrichs R (1997) Theor Chim Acc 97:119
Glendening ED, Reed AE, Carpenter JE, Weinhold F NBO (version 3.1)
O’Boyle NM, Tenderholt AL, Langner KM (2008) J Comp Chem 29:839
Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H (2009) J Appl Cryst 42:339
Sheldrick GM (1990) Acta Cryst A46:467
Sheldrick GM (1997) SHELXL97. Program for the solution and refinement of crystal structures. University of Göttingen, Germany
Acknowledgments
The GAUSSIAN09 calculations were carried out in the Wrocław Centre for Networking and Supercomputing, WCSS, Wrocław, Poland (http://www.wcss.wroc.pl grant number 18).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
The Gaussian09 calculations were carried out in Wrocław Centre for Networking and Supercomputing, WCSS, Wrocław, Poland.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Małecki, J.G., Maroń, A. Spectroscopic, structure and DFT studies of palladium(II) complexes with pyridine-type ligands. Transition Met Chem 36, 297–305 (2011). https://doi.org/10.1007/s11243-011-9469-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-011-9469-z