
Abstract
Obesity has reached epidemic proportion worldwide and in all ages. Available evidence points to a multifactorial pathogenesis involving gene predisposition and environmental factors. Gut microbiota plays a critical role as a major interface between external factors, i.e., diet, lifestyle, toxic chemicals, and internal mechanisms regulating energy and metabolic homeostasis, fat production and storage. A shift in microbiota composition is linked with overweight and obesity, with pathogenic mechanisms involving bacterial products and metabolites (mainly endocannabinoid-related mediators, short-chain fatty acids, bile acids, catabolites of tryptophan, lipopolysaccharides) and subsequent alterations in gut barrier, altered metabolic homeostasis, insulin resistance and chronic, low-grade inflammation. Although animal studies point to the links between an “obesogenic” microbiota and the development of different obesity phenotypes, the translational value of these results in humans is still limited by the heterogeneity among studies, the high variation of gut microbiota over time and the lack of robust longitudinal studies adequately considering inter-individual confounders. Nevertheless, available evidence underscores the existence of several genera predisposing to obesity or, conversely, to lean and metabolically health phenotype (e.g., Akkermansia muciniphila, species from genera Faecalibacterium, Alistipes, Roseburia). Further longitudinal studies using metagenomics, transcriptomics, proteomics, and metabolomics with exact characterization of confounders are needed in this field. Results must confirm that distinct genera and specific microbial-derived metabolites represent effective and precision interventions against overweight and obesity in the long-term.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Obesity, a chronic complex disease defined by excessive adiposity that can impair health [1], has nearly tripled since 1975. In 2016, more than 1.9 billion adults 18 years and older (39%), were overweight, with over 650 million (13%) who were obese [2]. The epidemic proportions of obesity worldwide are a huge social, economic and health burden [3, 4]. With obesity, public health is negatively exposed to the increasing incidence of several comorbidities and related complications by cardiovascular diseases, metabolic syndrome, type 2 diabetes mellitus (T2D), liver steatosis, cholesterol cholelithiasis, several tumours [5] and increased risk of severe COVID-19 [6]. Mean body mass index (BMI) is increasing globally in both women and men [3] and has become a marker of chronic positive energy balance, i.e., when energy intake is more than energy expenditure. Major causes of obesity are excess caloric intake [7], behavioural changes with decreased energy expenditure [8], and socio-economic status as environmental factors driving epigenetic modifications throughout gene-environment interaction [9,10,11].
The human gut hosts a huge collection of diverse microbial species (microbiome) which contribute to human health [12]. The gut microbiome contributes to several functions, including body metabolism, adiposity, homoeostasis, and energy balance. Gut microbes play a role also in signals linked to central appetite and food reward while bacterial strains and/or their metabolites can target the brain directly via vagal stimulation or indirectly through immune-neuroendocrine mechanisms. The microbiome is the site hosting the production of a wide variety of metabolites which interact with receptors on host cells. From this interaction made of short-chain fatty acids (SCFAs), bile acids (BA), peptidoglycan, and lipopolysaccharides (LPS) modulated by diet and environment [13,14,15], signalling pathways are activated or inhibited with effects on the host and health. It is becoming increasingly clear that a shift in microbiome diversity and composition can contribute to obesity by altering the host’s capacity to harvest and use energy. Consequences include insulin resistance, metabolic chronic inflammation, leaky gut, and fat deposition [16].
The aim of this review is to discuss key aspects relating onset and progression of obesity and metabolic abnormalities with gut barrier, microbiome, and key microbial products.
2 The gut barrier
The human gut consists of a luminal surface of 200–300 m2 [17,18,19] which is part of the complex morpho-functional machinery named the intestinal barrier [20] (Table 1).
Several layers are components of the intestinal barrier. The microbial layer populates the mucin layer [18, 19, 21]. In the healthy status, the microbes do not come in close contact with the epithelial enterocytes, but rather grow in the central mucin layer. The next layer is the functional combination of gastrointestinal secretions and motility which contributes to the oro-aboral transit of gut content in the fasting state and postprandially. The mucosal level is the combination of a monolayer of several intestinal epithelial cells (IECs) which show distinct histological and functional features. The lamina propria hosts the immunological cluster of distinct cells and their receptors. The muscularis mucosae is the layer below the lamina propria. The gut-vascular layer consists of endothelium, pericytes, enteric glial cells and their junctions. A last barrier is represented by the liver with the collection of immunological cells. At the gut barrier, a continuous crosstalk occurs at the interface between external and internal body environments [22]. Key events include the interaction between nutrients, microbiome, and host: during digestion most of fat (> 95%) will be emulsified and absorbed, and a similar pattern occurs for simple carbohydrates (absorbed as sugars) and proteins (broken into aminoacids). Complex carbohydrates in non-digestible dietary fibres will escape the small intestinal digestion (because of lack of specific enzymes) and will enter the colon as energetic substrates to the resident microbiome. Additional interactions occur with toxins and toxic chemicals vehiculated by ingested food, water supply, and host gastric, biliary, pancreatic secretes.
Several factors can disrupt the gut barrier integrity, and include bacterial overgrowth of pathogens such as E. Coli, a diet enriched in fat, microbial fragments such as LPS, drugs such as proton pump inhibitors (PPIs) and nonsteroidal anti-inflammatory drugs (NSAIDs), food allergens, and gluten component such as gliadin [23, 24] (Fig. 1).

Components of the intestinal barrier in health. The extracellular barrier consists of microbes populating the mucing in its central layer. The functional barrier is the combination of motility and secretions of gastric acid, pancreatic juice, and liver bile. The mucosa layer consists of epithelium with several cell lines, the lamina propria, the immunological cells, the Pattern Recognition Receptors (PRRs), and the muscularis mucosae. The gut vascular layer prevents the translocation of bacteria and/or microbial components across the extracellular and the intestinal epithelial barrier. The liver barrier keeps the liver free of bacteria. For details see Table 1. Adapted from Di Ciaula et al. [20]
3 The microbiota
The gut microbiome consists of a polymicrobial community of about 100 trillion of microorganisms with over 1,000 bacterial species [25], viruses, fungi, archaea, and eukaryotes, and includes their genes and products as well. Overall, the microbiome has key functions which include the maturation of the host immune system, digestion and absorption of nutrients, protection from pathogenic infection, maintenance of intestinal barrier function, and maintenance of selective gut permeability.
Microbes start colonizing the human gut shortly after birth and will influence the microbiome composition throughout life. Important factors are type of infant delivery i.e., vaginal vs. caesarean, type of feeding i.e., breastfed vs. bottle-fed, and the type of microbes present in the mother’s mouth and skin. Environmental factors which influence the type of colonization include the aging process, lifestyles, geographical locations, drugs, and stress [26]. At the age of three the microbiome community becomes stable with Firmicutes, Bacteroidetes, and Clostridium cluster IV/XIV phyla [27, 28]. This step contributes to the development of the natural and adaptive immunity within intestinal lymphoid tissues [29, 30], tolerance towards the microbiota determinants, and prevention of overgrowth by intestinal pathogens [31, 32]. After colonization, the microbial community will grow within the most superficial (luminal) mucin layer as essential component of the gut barrier [20, 29, 30].
The total abundance of bacteria with respect to pH and partial pressure of O2 (pO2) is variable according to the different body sites, and the human gut hosts the highest density (Table 2). This is a huge number meaning about three bacterial cells for every single human body cell.
This huge number of microbes and the gradient across the stomach-colon tract, is under the control of general and local factors which include gastric acid, hepato-intestinal bile acids, pancreatic juice, gastrointestinal motility, digestive enzymes and antimicrobial proteins, all factors governing the pH, oxygen concentrations and redox potential.
The two major phyla in the human gut are the Firmicutes which are enriched in Gram-positive bacteria with two main classes of Bacilli (obligate or facultative aerobes) and Clostridia (anaerobic), and the Bacteroidetes which consist of Gram-negative anaerobic bacteria. There are minor quantities of Actinobacteria, Proteobacteria, and Verrucomicrobia (Akkermansia) [34]. Actinobacteria like Bifidobacterium are populated by Gram-positive anaerobic bacteria while Proteobacteria like Escherichia, Klebsiella, Enterobacter, Helicobacter consist of Gram-negative aerobic or facultative anaerobic bacteria. Looking at the gut distribution, anaerobic microbes represent the vast majority in the colon with thousands of species, millions of genes and the two phyla of Firmicutes mainly Ruminococcaceae and Lachnospiraceae, and Bacteroidetes, plus Actinobacteria, Proteobacteria and Verrucomicrobia [12]. Microbes populating the distal intestine such as Bacteroides, Bifidobacterium, Enterococcus can synthesize vitamins B12 and K, a function contributing to the daily requirement for vitamins [35]. The microbiota contributes to the host gut homeostasis by production of short-chain fatty acids (SCFAs) [21, 36,37,38]. In the small intestine, studies have shown that colonising Streptococcus and Lactobacillus spp express highly effective transport systems which compete with the host for sugar uptake and use. Microbes generate lactate and acetate which, in turn, are substrates for Veillonella spp which produce propionate [39]
At this level, the microbe–microbe interaction implies mechanisms like: i) direct competition between the resident microbiota and pathobionts i.e., C. difficile to gain nutrients and space [40]; ii) production of metabolic products inhibiting the growth of other microbes, such as SCFAs inhibition of Enterobacteriaceae [41], Salmonella enterica and Escherichia coli; iii) consumption of luminal oxygen which inhibits the growth of pathogenic bacteria [42]; iv) production of superoxide anion by Enterococcus faecalis inhibiting Staphylococcus aureus [43]; v) the type VI secretion system (T6SS) of Gram- negative bacteria secretes toxins against pathogenic bacteria [44]; vi) the gut microbiota produces bacteriocins as antimicrobial peptides (AMPs) which kill or inhibit pathogenic bacteria [45] (Fig. 2).

Factors contributing to microbe-microbe interaction. The microbe-microbe interaction confers a stable intestinal environment. Direct and indirect interactions exist due to complex mechanisms of action and microbial factors in health (eubiosis) or disease (dysbiosis). Mechanisms potentially involved in microbe–microbe interaction in the gut include: 1) direct competition between resident and pathogenic bacteria; 2) production of growth inhibitory metabolites such as short chain fatty acids (SCFAs); 3) consumption of luminal oxygen which inhibits the growth of pathogenic bacteria; 4) production of superoxide anion (i.e., by Enterococcus faecalis inhibiting Staphylococcus aureus); 5) secretion of toxins against pathogenic bacteria mediating the type VI secretion system (T6SS) of Gram- negative bacteria; 6) production of antimicrobial peptides (AMPs)
3.1 Lipopolysaccharides/Pathogen-associated molecular patterns
The first gut barrier layers, i.e., the resident microbiome, the intestinal epithelial cells (IECs) mucin, and the innate immune cells of the host oppose to the inappropriate immune response and mucosal damage caused by other pathogenic bacteria.
The innate immune cells of the host express pathogen recognition receptors (PRRs) which are sensors of microorganisms to activate the appropriate host’s response. The four major PRRs subfamilies are the toll-like receptors (TLRs), the nucleotide-binding oligomerisation domain-containing proteins (NODs), the retinoic acid-inducible gene 1 (RIG-1)-like receptors (aka RIG-1-like helicases), and the C-type lectin receptors [46]. Such receptors recognize conserved microbial sequences, the pathogen (microbial)-associated molecular patterns (P(M)AMPs), and the metabolism-associated molecular patterns (MAMPs) [47]. The bacterial lipopolysaccharides (LPS) belong to PAMPs, and are a collection of endotoxins found on the cell membranes of Gram-negative pathogens and potent pro-inflammatory structures which can have systemic effects [48].
The TLRs comprise 10 receptor members in humans and 13 in mice which are sensors for both external stimuli (PAMPs), MAMPs [49] and internal signals from cellular damage, i.e., Damage-associated molecular patterns (DAMPs) which are component of the innate immune response released from damaged or dying cells due to trauma or an infection by a pathogen. TLRs are expressed on the cell surface or in the endosomal membranes of IECs, in immune cells such as macrophages, neutrophils, dendritic cells, natural killer cells, mast cells, basophils and eosinophils [50], on hepatocytes, and adipocytes [51]. Receptors respond to LPS, peptidoglycans, nucleotides, proteins, lipoproteins, small and large molecules from distinct microbial pathogens. In particular, TLR2 senses bacterial lipoproteins [52] and TLR4 senses bacterial LPS [53]. TLRs activation implies the antigen-presenting step and cell activation and connection between innate and adaptive immune responses. The resulting signalling cascades creates a defense mechanism against microbes or paves the way to tissue repair. There is a precise crosstalk between the host and the microbiome since TLRs and NOD signalling control the production of antimicrobial peptides (AMPs) by the host epithelial cells and the microbiota also controls TLRs and NOD signalling.
The AMP RegIIIγ is produced from the ileum and mucus layer in the colon and is regulated by TLR stimulation and has antibacterial properties. Mice deficient in the TLR signalling adaptor MyD88 produced less RegIIIγ, while mice lacking RegIIIγ developed intestinal bacterial overgrowth in the intestine. Such mechanisms protect against enteric infection and diseases such as inflammatory bowel diseases (IBD) [54, 55]. Metabolites produced by the microbiome contribute to epithelial barrier function and integrity. Butyrate, a SCFA increases mucus secretion by the intestinal goblet cells. The tryptophan metabolite indole increases the expression of tight junction proteins, occludins, and claudins [51]. AMPs are secreted by both the gut microbiota and host epithelial cells and have broad-spectrum antimicrobial properties vs. Gram-positive and Gram-negative bacteria [56]. In addition, AMPs are crucial for maintaining mucosal barrier function and inhibit the contact between the microbiota and the epithelium. Whereas in the small intestine AMPs are secreted by the Paneth cells, in the colon the mucosal barrier function is kept by the inner mucus layer of mucin 2, oligomeric mucus gel-forming (MUC2). The inner mucus layer also secretes AMPs. The gut microbiota protects the host barrier function by immune system regulation. In mice, segmented filamentous bacteria (SFB) are commensal bacteria that attach to the ileal epithelium and contribute to the differentiation of T helper 17 (Th17), and production of immunoglobulin A (IgA) and interleukin (IL)-22 by the type III innate lymphoid cells (ILC3). In turn, IL-17 and IL-22 upregulate RegIII production by IECs to control both commensal and pathogenic bacteria overgrowth [51].
Metabolic sequelae can follow due to hyperactivation of TLRs predisposes to production of sustained pro-inflammatory cytokines and chemokines, and risk of inflammatory diseases and autoimmune disorders. An example occurs with high-fat diet and weight gain, a condition predisposing to a type of metabolic endotoxemia, increased gut permeability along with elevation in plasma LPS [57]. This low-grade metabolically-associated inflammation becomes a feature in several chronic non-communicable diseases, namely obesity, T2D, liver steatosis, chronic kidney disease and atherosclerosis [58,59,60,61]. Gut microbiota composition and dysbiosis will change the LPS characteristics and effects on gut barrier, intestinal glucose absorption and glycemia, adipocyte inflammation, insulin and incretins [62]. Deranged PRRs expression can favour inflammatory changes, as in TLR5-deficient mice. The effect of microbial flagellin will not be controlled and animals develop dysbiosis, colitis or metabolic syndrome [63, 64]. The activation TLR downstream signalling pathways requires the activation of myeloid differentiation factor 88 protein (MyD88), except for TLR3 [65]. Notably, if intestinal MyD88 is deleted, during diet-induced obesity there will be protection against diet-induced obesity, diabetes and inflammation, with increased anti-inflammatory endocannabinoids (eCBs), restored antimicrobial peptide production and increased intestinal regulatory T cells [66].
3.2 Endocannabinoid system
The eCB signalling system involves metabolic functions such as energy, gluco-lipid metabolism, microbiota-host interactions, immunity, and inflammation [67, 68]. Beside the endogenous cannabinoid receptor type 1 (CB1) and CB2, sensing respectively anandamide (N-arachidonoylethanolamide (AEA)) and 2-arachidonoylglycerol (2-AG), the family of eCBs interact with other membrane receptors such as GPR55, PPARα and PPARγ, or transient receptor potential vanilloid type-1 (TRPV1). Other eCB-like compounds include bioactive lipids from the N-acylethanolamine (NAE) family or members of the acylglycerol family. eCB-like compounds have pharmacological activity, e.g., N-oleoylethanolamine (OEA) or N-palmitoylethanolamine (PEA) which activate PPARα and TRPV1, and OEA, N-linoleylethanolamine (LEA) and 2-oleoylglycerol (2-OG) which activate GPR119. The 1-palmitoylglycerol (1-PG) and 2-palmitoylglycerol (2-PG) act as agonists of PPARα. Current research indicates that there is a link between bioactive lipids of eCB system, gut microbiota, adipose tissue, and intestinal function.
In the metabolic background, the intestinal eCB system plays a role in obesity and diabetes. Increased AEA increases gut permeability, a mechanism requiring CB1-dependent pathways, and associated with changes in the gut microbiota [68].
Some potent eCB agonists increased adipogenesis and disrupted the gut barrier [69]. The eCB function and gut microbiota is deranged in genetically ob/ob obese mice and db/db diabetic mice (and) [70, 71]. The adipocyte-specific deletion of NAPE-PLD was associated with decreased thermogenic programme in adipose browning/beiging tissue with a shift in gut microbiota composition. The microbiota transplant from adipose tissue NAPE-PLD deleted mice to germ-free recipient mice was associated with a similar phenotype [72]. NAPE-PLD deleted in intestinal epithelial cells produced hyperphagic mice on a high-fat diet and developed obesity and hepatic steatosis, changes in both intestinal and plasma eCBs indicating a type of gut-to-brain axis defect plus gut microbiota changes [73] Liver deletion of NAPE-PLD caused a mice phenotype similar to that of a high-fat diet-like despite a normal diet with a gain in fat mass, liver steatosis and inflammation. There was a change in BA profile, likely related to a shift of gut microbiota [74]. Thus, the eCB system likely develops a crosstalk with the gut microbes and this mechanism involves NAPE-PLD and the production of bioactive lipids. The gut microbiota itself produces specific eCBs [75], a finding suggesting partly unexplored interactions involving the host, gut microbes and several regulatory patterns, including the endocannabinoidome (eCBome), a collection of several receptors and enzymes [76].
3.3 Aryl hydrocarbon receptor
The aryl hydrocarbon receptor (AhR) is a cellular transcription factor activated by ligands which include environmental triggers, nutrition-derived signals, phytochemicals, and bacterial metabolites such as tryptophan. The AhR pathway is at the crossroad between microbiota-epithelial barrier-metabolism and immune functions. Upon binding, AhR translocates into the nucleus where it is bound to its dimerization molecule AhR nuclear translocator. This step mediates the transcription of several genes responsible for immunity and inflammatory processes. The microbiome interacts with AhR [77, 78] with effects on energy metabolism and metabolic syndrome. The capacity of metabolising tryptophan into AhR binding derivates is decreased in metabolic syndrome [79]. By contrast, Lactobacillus strain increases AhR ligands, improves metabolic functions, the status of the gut barrier, and liver steatosis. Indigo binds AhR, has anti-inflammatory activities, protects against high-fat diet-induced obesity and metabolic disturbances. Lactobacillus spp are upregulated along with increased barrier cytokines interleukin (IL)-10 and IL-22 [80]. Microbial tryptophan metabolites such as indole-3-ethanol, indole-3-pyruvate and indole-3-aldehyde protect the gut epithelial barrier by preserving the integrity of the apical junctional complex which also includes myosin IIA and ezrin [81].
4 Products of the microbiome
In health and disease, the microbiome can produce a myriad of molecules and metabolites that continuously interact with the host (Fig. 3). An example is the production of pathogen-associated toxins and polysaccharides. Microbial molecules can have metabolic activity at a local and systemic level i.e., brain, heart, kidney, liver [82], white- and brown-adipose tissues [83]. Among the others, microbe-derived molecules include short-chain fatty acids (SCFAs), bile acids (BA), indole-3 acetic acid (IAA), p-cresol, p-cresyl-glucuronide (pCG), indoxyl sulphate (IS), hydrogen sulphide (H2S), trimethylamine N-oxide (TMAO), succinate. The ultimate translational value of such microbial products requires further studies in terms of specificity, stability, effectiveness, signalling, and pathways involved. Of special relevance are SCFAs and BA which will be discussed in the following paragraphs.

Crosstalk between gut microbiome, metabolites, and host within the gut, the microbiome inhabits the most superficial mucin layer with abundance estimated to reach 1014. At this level, there is an intense crosstalk between microbial populations, diet, metabolic processes with production of several molecules, enterohepatic circulation of primary and secondary bile acids, and membrane-associated and nuclear receptors especially in colonocytes and endocrine L cells. Among the others, Microbial/Pathogen-associated molecular patterns (PAMPs), lipopolysaccharides (LPS) from the microbiota are detected by pattern recognition receptors, including toll-like receptors (TLRs). Akkermansia muciniphila provides a protective effect on intestinal barrier and reduces the inflammation by producing Amuc_1100 on the outer membrane which interacts with TLR2. Other microbes secrete endocannabinoids from digestion of dietary components such as SCFAs or from metabolization of endogenous. Several receptors on the luminal plasmamembrane or nuclear receptors can sense the effects of such molecules. Abbreviations: AGEs, advanced glycation end products; AhR, aryl hydrocarbon receptor; CB1, CB2, cannabinoid receptor type 1 and type 2; BSCFAs, branched short chain fatty acids; CA, cholic acid; CDCA, chenodeoxycholic acid; DCA, deoxycholic acid; FXR, farnesoid X receptor; GPBAR1, G-protein-coupled bile acid receptor-1; GPR119, GPR43, GPR41, G-protein coupled receptor 119, 43 and 41; LCA, lithocholic acid; MYD88, myeloid differentiation primary response 88; PPARα/γ, peroxisome proliferator-activated receptors alpha and gamma; SCFAs, short chain fatty acid; TRPV1, transient receptor potential cation channel subfamily V member 1; UDCA, ursodeoxycholic acid
4.1 SCFAs
4.1.1 Production
SCFAs are carboxylic acids with an aliphatic chain of 1–5 carbons produced during degradation (fermentation) of nondigestible nutritional fibres and in small amounts also from gut peptides and proteins [21]. The list of SCFAs includes formic acid (C1), acetic acid (C2), propionic acid (C3), butyric acid (C4), and valeric acid (C5), with acetate, propionate, and butyrate as the most abundant in a proportion of 60:20:20. Several microbial species are involved in the production of SCFAs, namely Lactobacillus spp., Bifidobacterium spp., Akkermansia muciniphila, Bacteroides spp., Prevotella spp., Ruminococcus spp., Streptococcus spp. for acetate; Phascolarctobacterium succinatutens, Akkermansia muciniphila, Bacteroides spp., Dialister spp.,Megasphaera elsdenii, Veillonella spp., Coprococcus catus, Roseburia inulinivorans, Ruminococcus obeum, Salmonella spp. for propionate; Faecalibacterium prausnitzii, Clostridium leptum, Eubacterium rectale, Roseburia spp. for butyrate; Bifidobacterium spp., Prevotella spp., Parabacteroides spp., Bacteroides spp., Alistipes spp., Eubacterium spp., Erysipelatoclostridium spp., Blautia (Clostridium cluster XIVa) spp., Coprococcus, Dorea, Roseburia (Clostridium cluster XIVa) spp., Lactobacillus spp., Faecalibacterium (Clostridium cluster IV) spp., Ruminococcus (Clostridium cluster IV) spp., Streptococcus spp., Veillonella spp., Escherichia spp. for formate; Clostridium (Clostridium cluster I) spp. for valerate [34]. The main phyla involved in SCFAs are Firmicutes, Actinobacteria, Bacterioidetes, and Verrucomicrobia. Pathways involved derive from pyruvate via acetyl-CoA glycolytic pathway or the Wood–Ljungdahl pathway. Acetate originates from pyruvate-Acetyl-CoA or Wood–Ljungdahl pathway. Butyrate originates from pyruvate-acetyl-CoA or acetate. Carbohydrate metabolism in glycolysis, and amino acid metabolism (i.e., lysine or propionate from plant compounds such as phytate [84, 85]) contribute to butyrate and propionate synthesis, whereas acetate is produced from acetyl-CoA [21]. The SCFAs concentration depends on dietary intake, can vary among individuals, and decreases from the cecum and proximal colon to the distal colon, ileum, and jejunum [86]. For example, inulin is propionogenic, whereas resistant starches are more butyrogenic. Fermentation of the intestinal mucus that covers the IECs is another source of SCFAs from the host itself [87]. The branched SCFAs (BSCFAs) isobutyric and isovaleric acid, are produced when undigestible protein reach the colon and their branched amino acids, valine, leucine, and isoleucine undergo microbial fermentation. Certain dietary fibers such as polydextrose also contribute to generate BSCFAs such as isobutyric acid [88].
4.1.2 Absorption
The IECs absorb the luminal SCFAs as undissociated non-ionic molecules (passive diffusion) across the plasma membrane, and mainly as dissociated anionic molecules (by carrier-mediated transport involving the Monocarboxylic acid transporters -MCT1–4, the sodium-coupled monocarboxylate transporter—SMCT/SLC5A, and the SCFA/HCO3− exchangers. In humans MCT1, MCT3, and MCT4 are expressed in enterocytes, ileum, and colon at the apical/basolateral membrane (MCT1) or basolateral membrane (MCT3 and 4). Within the SMCT transporters, SMCT1(SLC5A8) is a Na-dependent SCFA transporter at the apical membrane in both small and large intestine. SMCT2 (SLC5A12) is a low-affinity SCFAs transporter localized in the apical membrane of the small intestine. SCFA/HCO3 exchangers are expressed on both the apical and basolateral membranes [12].
4.1.3 Receptors
The three main SCFAs activate extracellular mechanisms via the G-protein-coupled receptors (GPCRs) GPR41 (i.e., free fatty acid receptor FFAR-3), GPR43 (FFAR2), GPR109A (hydroxycarboxylic acid receptor HCAR-2), GPR119, GPR164. GPRs are expressed in the enteroendocrine L-cells of the intestine. i.e., mainly ileum and colon, in immune cells, i.e., mast cells, and leukocytes, and adipocytes. This close interaction between SCFs and GPRs is responsible for several local and systemic effects. The anti-inflammatory effects of SCFAs includes the interaction with regulatory T cells (Tregs), neutrophils, and macrophages by modulating migration, cytokine production, cytolytic activity, microbicidal activity, and production of reactive oxygen species (ROS) [89, 90]. GPR41 and GPR43 activate the downstream mitogen-activated protein kinase (MAPK) pathway which control inflammatory changes [91, 92]. SCFAs especially butyrate acts on the G-protein-coupled receptors (especially GPR109A) with downstream activation of signals leading to suppression of MAPK—NF-κB pathways and inhibition of INF-γ, IL-1, 2, 6, 8, TNF-α, activation of forkhead box protein P3 (Foxp3) and decreased inflammation, production of glutathione (GSH) and decreased ROS. Butyrate upregulates IL-10 expression and downregulate IL-6 expression. This effect develops by binding to GPCR109A on dendritic cells (DC) and macrophages. SCFAs also activate epigenetic mechanisms which influence gene expression [93] and inhibition of histone deacetylase (HDACs), activation of histone acetylase (HATs) and stabilization of hypoxia-inducible factors (HIFs). SCFAs contribute to the immune cell development, and maintenance of epithelial barrier function [91, 92, 94, 95]. This latter effect, especially from the main SFAs (C2, C3, C4) involves MUC2 production by goblet cells [96], enhanced tight junction protein expression like zona occludens (ZO)-1, ZO-2, occludin, junctional adhesion molecule A (JAMA), and claudin-7 [21].
SCFAs contribute to stool acidification and are energy source for enterocytes, with butyrate providing about 60–70% of energy requirements for proliferation and differentiation of colonocytes [97]. SCFAs (butyrate especially) contributes to control of the anaerobic condition in the colon via mitochondrial fatty acid metabolism (butyrate via beta-oxidation), oxidative phosphorylation and adenosine triphosphate (ATP) production [98]. This function keeps anaerobic bacteria close to the epithelium, and prevents the pro-inflammatory effect of facultative anaerobes such as Enterobacteriaceae i.e., phylum Proteobacteria [99, 100]. Butyrate also binds to peroxisome proliferator-activated receptor gamma (PPARγ) thereby repressing inducible nitric oxide synthase (iNOS), decreasing nitric oxide production (NO) and eventually nitrate production. This is a step which decreases NO production and ultimately luminal nitrate levels, acting as specific energy substrates used by putative pathogenic facultative anaerobes (Enterobacteriaceae) [12].
4.1.4 Systemic effects
At a systemic level, the remaining SCFAs entering the portal vein and the systemic circulation provide energy (about 10% of daily caloric intake) and substrates for key metabolic processes [101] (Fig. 4).

Pleiotropic effects of short chain fatty acids (SCFAs). Short chain fatty acids (SCFAs) have potent systemic effects which involve several organs and complex pathways referred to metabolic homeostasis. Main SCFAs are products of microbiome-dependent digestion from dietary fibers. Thus, qualitative, and quantitative qualitative shift in microbial diversity can derange several pathways at different levels, i.e., adipose tissue, immune cells, liver, pancreas, intestine, and brain involving gluco-lipid-inflammatory changes and food intake. Abbreviations: GLP-1, glucagon-like peptide-1; GLUT-4, activated glucose transporter protein-4; GPR, G-protein-coupled receptors; IL, interleukin; PYY; peptide YY; TNFa, tumor necrosis factor alpha; (+), increase; (-), decrease. After [12, 21, 102]
Especially acetate becomes a substrate for hepatic synthesis of cholesterol and modulates appetite and body weight via reduction of food intake. This action requires direct stimulation of the anorectic pathways in the hypothalamus and brainstem and increased expression of proopiomelanocortin (POMC) and decreased expression of agouti-related peptide (AgRP) with a change in appetite [103]. Most of the propionate (~ 80%) travels from the gut to the liver, and is used as a substrate for gluconeogenesis, lipogenesis, and protein synthesis, with important metabolic effects [104, 105]. In addition both propionate and butyrate contribute to regulates body weight, as reported in mice studies using high-fat diet [96, 106,107,108]. Butyrate and propionate via GPR41 receptor increase PYY secretion by the L-cells in the ileum and colon, a step slowing gastric emptying, increasing digestion and glucose absorption along with satiety and decreased food intake [109, 110]. The three SCFAs via GPCR43 receptors regulate the secretion of GLP-1, insulin, glucagon with effect on glycemia [111], and contribute to leptin secretion [112]. Lipid metabolism and energy expenditure are partly influenced by SCFAs, since acute oral administration of acetic acid [113] or the three SCFAs along with high fat diet increases energy expenditure and lipid oxidation in C57BL/6 J mice [114, 115]. Butyrate supplementation suppresses peroxisome proliferator-activated receptor gamma (PPARγ) expression and activity and increases lipid oxidation [114]. Clinical studies are in line with such results: acute oral sodium propionate supplementation in healthy volunteers enhanced resting energy expenditure and lipid oxidation compared with controls [115], and rectal infusion of 200 mM SCFAs resembling the physiological colonic concentration, stimulated lipid oxidation, reduced carbohydrate oxidation, and increased resting energy expenditure [115].
4.2 Bile acids
BA are amphipathic molecules synthesized as primary BA, i.e., cholic acid (CA), chenodeoxycholic acid (CDCA) in humans and muricholic acid (MCA) in rodents from cholesterol in the pericentral hepatocytes [116]. Primary BA are then conjugated with taurine (2-aminoethanesulfonic acid) and glycine, actively secreted from into bile which is mainly stored and concentrated in the fasting state. Following postprandial gallbladder contraction, BA are secreted in the proximal intestine and actively absorbed in the terminal ileum to enter the portal vein. A small amount of BA enters the colon to undergo the microbiota-dependent biotransformation into secondary (deoxycholic acid, DCA; lithocholic acid, LCA) and tertiary BA (ursodeoxycholic acid, UDCA), which undergo passive absorption into the portal vein. This process of enterohepatic circulation of BA occurs 4–12 cycles/day [117,118,119]. The fecal loss is minimal at every cycle (5%) and must be compensated by the de novo synthesis of primary BA in the liver, i.e., ~ 200–600 mg/daily (Fig. 5a–b).

Metabolism of bile acids (BA). a Synthesis, conjugation, secretion, biotransformation, re-absorption and excretion of primary, secondary, and tertiary BA in humans. Primary BA are synthetized in the liver starting from cholesterol are the trihydroxy cholic acid (CA) and the dihydroxy chenodeoxycholic acid (CDCA). The two biosynthetic pathways are the classical pathway involving the 7a-hydroxylase which stimulates the 7a-hydroxylation of cholesterol. This major enzyme contributes to more than 75% of total production of primary BA. The alternative pathway is initiated by the sterol-27-hydroxylase which produces mainly CA. Once in the intestine, about 95% of primary BA are re-absorbed at the ileal level. The remaining part of primary BA enter the colon where the resident microbiota initiate BA deconjugation from taurine and glycine, dehydrogenation, dehydroxylation, and epimerization to produce «secondary» BA: the dihydroxy deoxycholic acid (DCA) and the monohydroxy lithocholic acid (LCA). The 7α-dehydrogenation of CDCA form the dihydroxy 7α-oxo-LCA which is metabolized to the “tertiary’ 7β-epimer, the dihydroxy hydrophylic ursodeoxycholic acid (UDCA). In the liver a small amount of LCA is quickly transformed in the sulphonated form (S-LCA). b Enrichment of hepatic and gallbladder bile in health as main solutes (left) and individual BA (right). Abbreviations: CA, cholic acid; CDCA, chenodeoxycholic acid; DCA, deoxycholic acid; LCA: lithocholic acid; UDCA, ursodeoxycholic acid
The synthesis of BA is tightly modulated by negative feedback mechanism controlled by the BA-farnesoid X nuclear receptor (FXR) at the enterocyte level and release of the fibroblast growth factor-19 (FGF19 in humans; FGF15 in rodents). FGF19 is the agonist of the hepatic FGF receptor 4 (FGFR4)/β-Klotho and activates the c-Jun N-terminal kinase/extracellular signal-regulated kinase (JNK/ERK) to inhibit CYP7A1 and CYP8B1-dependent BA synthesis in concert with the FXR- small heterodimer partner (SHP) inhibitory pathway [120, 121]. This pathway is strictly dependent on the enterohepatic circulation of BA [122, 123] and with the composition and abundance of gut microbiota [124]. BA are important signalling molecules [119, 125] on several receptors such as vitamin D receptor [126], pregnane X receptor [127] constitutive androstane, receptor [128], FXR [129], and G-protein-coupled bile acid receptor-1 (GPBAR1) [117]. For this BA modulate glucose and energy metabolism, epithelial cell proliferation, inflammation, gene expression, lipid homeostasis [117, 129, 130].
BA and microbiome share a bidirectional crosstalk in health and in disease [20, 131, 132]. Primary BA in the colon undergo bacterial deconjugation, i.e., removal of amino acid residues via bile salt hydrolase (BSH) activity, dehydroxylation via removal of hydroxyl groups, oxidation—dehydrogenation or epimerisation and transformation to secondary/tertiary BA. Notably, the BA bioavailability and bioactivities become highly dependent on such transformation [133] and BA become microbiota-derived signalling metabolites. This continuous process between BA, microbiome, and BA again is greatly dependent on spatial–temporal, and kinetic timing of the enterohepatic circulation of BA which encounter different microbial communities along the intestinal tract [18, 118]. For example, deconjugation is a widely available microbial feature [134] while other BA biotransformations require different and specialized bacterial species in the distal gut. The implication is that topographical, qualitative, and/or quantitative shift of gut microbiome profiles can greatly influence the human health and that the modulation of the gut microbiota composition is likely to oppose human diseases [135].
Gut dysbiosis can change the BA pool composition, with consequences at different levels, including signalling pathways [136]. Distinct BA-FXR binding can modulate the production of antimicrobial peptides (AMPs) such as angiogenin 1 and RNase family member 4 which inhibit gut microbial overgrowth and dysfunctional intestinal barrier [137]. Increased DCA has cytotoxic and detergent effects on the bacterial cell membranes with antimicrobial properties [138]. DCA can influence the enterocyte integrity and re-shape the microbial composition [138]. BA can therefore shift the gut microbiome and modify BA-metabolizing bacteria or inhibiting other BA-sensitive bacteria [139]. Microbes like Clostridium perfringens, Eubacterium lentum, Ruminococcus gnavus have antimicrobial effect via the use the iso-BA pathway and transformation of DCA and LCA to the 3b-OH epimers, i.e., iso-DCA and iso-LCA.
While vegetarian diets favor glycine conjugation, diets enriched in animal protein favor taurine conjugation. Thus, TCA favor gut microbes which convert taurine and CA to hydrogen sulphide and DCA, two genotoxin and tumor-promoters, respectively [140].
Dysbiosis and BA pool derangement have been involved in obesity, as well as NAFLD, and T2D [18, 141,142,143].
4.3 Other microbial molecules
Symbiotic microbes can produce immunomodulatory polysaccharides and sphingolipids (by Bacteroides spp) [144, 145] and muropeptides (by Enterococcus spp. [146]). The probiotics provide another source of molecules as in the case of L. acidophilus NCFM which dismiss a glycosylated surface layer protein signalling to the DC-SIGN receptor, [147] L. rhamnosus which produces a 90 kDa pilus protein SpaC of GG, a partly glycosylated mucus-binding protein with signalling capacity to DC-SIGN receptor on dendritic cells [147, 148]. Bifidobacterium sp provides a pilus-located Tad protein that promotes colonic persistence and epithelial proliferation [149, 150]. E. coli produces the caseinolytic protease B (ClpB), an antigen-mimetic of alpha-melanocyte-stimulating hormone which increases satiety via increased production of plasma GLP-1 and PYY [151]. Lactobacilli, Bifidobacteria also secrete ClpB which, in the case of Hafnia alvei inhibited satiety in a human trial [152]. A.muciniphila, effective in human trial, [153,154,155] secretes P9, an 84 kDa protein encoded by the Amuc_1831 gene. P9 given orally increases serum GLP-1 in mice, likely interacting with intercellular adhesion molecule 2 receptor [156, 157]. The 50 kDa Amuc_1434* protein inhibits the LS174T cell viability involving the apoptosis pathway via tumour-necrosis-factor-related apoptosis-inducing ligand (TRAIL) [158]. The 30 kDa Amuc_1100 protein is thermostable, can signal to TLR2 [159] and resembles a specific pilus-associated protein [160] which prevents the diet-induced obesity in a mouse model [159]. Novel molecules from the microbiome are being identified and investigated for their potential impact on health. The term enterosynes defines gut hormones, bioactive peptides, lipids, nutrients, microbiota, and immune factors able to modulate duodenal contraction by targeting the enteric nervous system (ENS) [161]. For example, T2D patients exhibit duodenal hypermotility which creates aberrant afferent nervous messages to the hypothalamus [162,163,164,165]. The impaired motility leads to excessive glucose absorption and hyperglycaemia [161, 165]. By contrast, a restored duodenal contraction by targeting ENS neurons and the gut-brain axis improves insulin sensitivity [161]. Another example is the use of dietary fibres able to reshape the gut microbiota and improve diabetes. In mice, oligofructose decreased duodenal contraction frequency by controlling enteric neurons activity. Both hyperglycaemia and inflammatory markers decreased in the adipose tissue [166] while the intestinal bioactive lipid (12-hydroxyeicosatetraenoic acid (12-HETE)) increased in colonic cells. Indeed, 12-HETE improved glucose metabolism in diabetic mice and the improved duodenal contractility via the mu-opioid receptors (MOR) which sense enkephalin and PPARγ. Notably, 12-HETE levels, expression of the proenkephalin and MOR were decreased in duodenal specimens of T2D patients compared with healthy subjects [166].
The probiotic Nissle 1917 (EcN) has anti-inflammatory properties in patients with IBD, and the effect is likely mediated by 3-hydroxyoctadecaenoic acid (C18-3OH) acting on PPARγ [167]. Notably, administration of oligofructose was associated with an increase in colonic C18-3OH concentration.
In parallel with the observed beneficial effects on health, other gut metabolites have harmful effects.
TMAO is a metabolite of quaternary amines, such as betaine, choline, and L-carnitine found in seafood, meat, vegetables, and fruits [168, 169]. Gut microbes use TMA lyase and its activating enzyme (CutCD) to convert quaternary ammonium ions to acetaldehyde and trimethylamine (TMA). The latter reaches the liver to be converted by flavin monooxygenase into TMAO which serum levels are strongly associated with atherosclerosis and cardiovascular risks [170]. In the mouse model TMAO promotes the growth of atherosclerotic plaques [171] and acts as uraemic toxin [172]. Microbial species can oppose the effects of TMAO such as Eubacterium limosum and E. maltosivorans which are able to deaminate betaine and other quaternary amines and produce beneficial SCFAs, namely acetate and butyrate [173,174,175]. The advanced glycation end products (AGEs) are molecules originating from thermal processing of foods when free amino groups of proteins and amino acids react with reducing carbohydrates. Binding to AGEs receptors [176, 177] mediate pro-inflammatory responses, promote the leaky gut and leakage of bacterial toxins into the systemic circulation [178]. The AGE fructoselysine originates from lysine and glucose. E. coli uses fructoselysine [179] while the genus Intestinimonas spp converts fructoselysine into butyrate [85]. Others have shown that another AGE, N-ε-carboxymethyllysine, was totally used by Cloacibacillus and Oscillibacter spp [180]. Thus, depending on distinct bacterial pathways fructoselysine and other AGEs can play a role in T2D and other diseases. The metabolism of histidine is partly dependent on intestinal bacteria to produce imidazole propionate (IMP), which increases in the serum of T2D patients [181]. In this scenario, insulin signalling and glucose tolerance become impaired via the mammalian target of rapamycin complex 1-dependent pathway [182]. Both Streptococcus mutans and Eggerthella lenta, although phylogenetically unrelated, produce IMP producers.
5 The gut microbiota in obesity
The gut microbiome is a key player in energy homeostasis [183, 184], and evidence in animal models and humans points to the close relationship between gut dysbiosis and obesity, including weight loss [185,186,187,188]. Seminal studies have shown that germ-free mice develop less adipose mass and are less prone to obesity, as compared with mice harvesting a normal microbiota [189, 190]. Genetically obese ob/ob mice develop a shift in microbiota composition, i.e., increased Firmicutes and reduced Bacteroidetes [191]. A similar shift in gut microbiota composition has been reported in humans, despite a huge interindividual variability and sometime poor methodological quality of the studies [188]. The “obese microbiota” has an increased capacity to harvest energy from the diet [183] and in animal models is a transmissible trait [183]. A large cohort study identified 34 bacterial taxa associated with BMI and concluded that microbiota explains ~ 4.5% of the variance in BMI, independent of age, sex, and genetic predisposition [192]. Early colonization with Bacteroidetes and Firmicutes with the maintenance of a stable ratio can prevent childhood obesity [193,194,195]. Compared to normal-weight subjects, obese individuals show reduced bacterial diversity, higher levels of Lactobacillus reuteri, Fusobacterium, Bacteroides fragilis, and Staphylococcus aureus, and lower levels of Lactobacillus plantarum, Methanobrevibacter, Akkermansia muciniphila, and Bifidobacterium animalis [188].
A recent systematic review indicated Proteobacteria as the most consistently obesity-associated phylum, and Faecalibacterium, Akkermansia, and Alistipes as lean-associated genera [196].
However, in humans, the association between the Bacteroidetes / Firmicutes ratio (i.e., increased Firmicutes vs. reduced Bacteroidetes) and the development of obesity is questionable [197,198,199,200]. In a group of children, increased Firmicutes (Clostridium and Lactobacillus) and decreased Bacteroidetes (Prevotella and Bacteroides) were only detected in obese, but not in overweight, as compared with lean subjects [197].
In adults, overweight and underweight subjects had a reduced gut microbial alpha-diversity, as compared with normal weight subjects, in the absence of significant differences in the overall gut microbiota composition [199]. In another cohort of adults, no difference was noticed between obese and non-obese subjects in the proportion of fecal Bacteroidetes, even after weight loss [200].
Further differences in gut microbiota diversity and composition emerge when overweight or obese subjects are divided according to the concurrent presence of metabolic disorders. Table 3 shows an example of recent studies trying to characterize gut microbiota according to specific phenotypes (i.e., metabolically healthy, or unhealthy subjects with normal weight or obesity). In this case the metabolically healthy obese subgroup shows heterogeneous results, with different microbiota diversity and composition as compared with both normal weight and metabolically unhealthy obese.
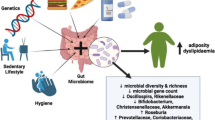
The apparent discrepant results in terms of both Bacteroidetes/Firmicutes ratio or links between specific gut microbiota features and the obesity phenotype can derive from methodological bias due to differences in sample processing and DNA sequence analysis or by the inadequate characterization of the recruited subjects, without precise assessment of confounding factors [196, 198] including gender, age, lifestyle, ethnicity, living area, dietary habits and socio-economic status [209, 210] (Fig. 6).

Potential factors conditioning the effects of gut microbiota on fat storage. Gender, age, lifestyle, dietary habits (including food type and possible chemical disruptors) and socio-economic status can change microbiota variety and composition over time, strongly affecting metabolic homeostasis and the risk of obesity onset and development. Abundance of the Bacteroides genus is lower in obese men than in women, and the abundance of Bilophila is lower in men compared to women, regardless of BMI [211]. Gut microbiota composition can vary with age, with an an age-dependent association with Bifidobacterium abundance [212]. The level and intensity of daily physical activity positively modulates the diversity of gut microbiota and the abundance of some species linked with beneficial effects, as Bifidobacteriaceae family, Bacteroides and Akkermansia genera [213]. High intake of fat and saturated fatty acids can decrease both microbiota richness and diversity, predisposing to symbiosis and obesity. High adherence to Mediterranean diet increases levels of fiber-degrading bacteria and leads to beneficial metabolic effects. Marked variations of gut microbiota are also possible according to ethnicity, living area and socio-economic status [209, 210]
Furthermore, whether changes in microbiota variety and composition precede or follow the onset and development of obesity is not totally understood, so far.
The relationships between changes of microbiota composition and obesity might depend on dietary habits (in particular high-fat diet) and subsequent increased energy-harvesting efficiency from the diet[183, 214, 215], with alterations in gut barrier and permeability leading to altered metabolic homeostasis [19], and with altered host gene expression generating chronic, low grade inflammation, insulin resistance, increased fat storage [216, 217]. In addition, microbiota variations driven by diet is not stable over time. In mice, high-fat diet increases Firmicutes, but changes in microbiota composition are dissociated from markers of energy harvest, which changed over time suggesting a possible microbial adaptation to diet and time [218]. Finally, although gut microbiota can influence fat accumulation, the adipose tissue can modulate the variety of gut microbes through adipokines [219]. In rats, leptin supplementation induced lower relative abundance of Sutterella and a higher proportion of Clostridium genus, whereas supplementation with adiponectin resulted in lower abundance of the Roseburiagenus and a higher proportion of the Enterococcus genus [219].
Thus, available evidence points to a complex and bidirectional scenario where gut microbiota is the critical living interface between external factors (mainly diet, lifestyle, toxic chemicals) and internal pathways. This interaction is able to create the pathogenic background for the onset and development of obesity and deranged metabolic homeostasis involving bacterial products, signalling molecules, epigenetic mechanisms modulating gene expression [220].
5.1 The role of the endocannabinoidome (eCBome)
The endocannabinoidome (eCBome) consists of complex interplays between endocannabinoid-related mediators, their receptors and metabolic enzymes, with potential effects on the metabolic homeostasis [221, 222]. The main players are bacterial metabolites structurally similar to the host endocannabinoid-like mediators (namely N-acylethanolamines [NAEs], N-acyl-amino acids [NAAs]), and interfering with the same receptors [75, 223]. Germ-free mice show age-dependent variations in intestinal eCBome gene expression and lipid mediator levels. These alterations can be reversed by fecal microbiota transplant from conventionally raised mice [76]. Manipulation of the microbiota-eCBome crosstalk is possible. High-fat diet-fed wild type mice developed increased levels of intestinal dehydroepiandrosterone (DHEA) and GLP-1 concentrations following treatment with A. muciniphila [76]. In obese humans, supplementation with the eCBome mediator Oleoylethanolamide (OEA) decreased the energy and carbohydrate intakes and increased the relative abundance of A. muciniphila [224]. In a cohort of overweight and obese subjects, the conversion from a habitual Western diet to isocaloric Mediterranean diet positively modulated the endocannabinoid system and increased the abundance of A. muciniphila, independently of body weight changes [225]. Furthermore, as recently reported, the production of endocannabinoid metabolites by gut microbiota is linked with enhanced motivation to engage in physical activity. These metabolites enhance activity of transient receptor potential cation channel subfamily V member 1 (TRPV1)-expressing sensory neurons, thereby elevating dopamine levels in the ventral striatum during exercise and improving running performance [226].
5.2 The BA – microbiota axis
External factors including nutrients (i.e., dietary fiber, proteins, fat, carbohydrates, [21]) and toxic agents (i.e., smoking [227], alcohol [228], environmental pollutants [229,230,231,232]) can affect both diversity and relative abundance of the gut microbiota [18]. Such changes will influence the enterohepatic circulation of BA, the dynamic system where primary hepatic BA will shift to secondary BA in the colon [18, 118]. Both qualitative/quantitative changes of gut microbiota and BA pool can influence metabolic homeostasis, as observed in NAFLD, obesity, and T2D [18, 129, 141,142,143] (Fig. 7).

Crosstalk between bile acids (BA), microbiome, intestinal receptors, and systemic effects. In concert with several potential modulatory factors, the BA undergo the continuous recycling 4–12 times per day featuring the enterohepatic circulation. The microbial biotransformation of BA in the colon, shapes the final composition of the BA pool at equilibrium between primary, secondary, tertiary BA, synthesis, reabsorption, and fecal loss. During their travel through the ileum, BA act as potent signalling molecules at the level of the enterocyte nuclear Farnesoid X receptor (FXR) and the membrane-associated G protein-coupled bile acid receptor 1 (GPBAR-1). BA also control gut bacteria growth. This dynamic process, puts BA, microbiota, intestinal receptors in close contact with several systemic effects on metabolic. Immunological, and inflammatory homeostasis
In a cohort of Chinese adults with BMI ≥ 25 kg/m2, metabolically unhealthy subjects had reduced proportions of secondary BA, as compared with healthy, high BMI subjects [233]. As shown in a randomized, controlled trial in obese, metabolically healthy patients, fecal microbiota transplantation (FMT) from lean, healthy donor increased the relative abundance of Clostridium hylemonae, a bacterial species known to convert primary to secondary BA [234].
The altered metabolic homeostasis linked with BA unbalance is also due to dysregulated signalling pathways [136] involving the activation of the nuclear receptors Farnesoid X receptor (FXR) [129] and of the membrane-associated G protein-coupled BA receptor 1 (GPBAR-1) in the distal intestine, liver and extra hepatic tissue [117] (Figs. 8 and 9).

Systemic effects of bile acid (BA) as signalling molecules on the membrane-associated G protein-coupled bile acid receptor 1 (GPBAR-1) in targets tissues. Binding of BA to GPBAR-1 activates the stimulatory G α protein which, in turn, triggers adenylate cyclase (AC) activation and cyclic AMP (cAMP) production. For the GPBAR-1, the rank order of potency of BA is TLCA > TDCA > TCDCA > TCA. Systemic effect following GPBAR-1 activation occur in the intestine via secretion of the incretin GLP-1, GLP-2, PYY. Additional effects are documented in the pancreas, brown adipose tissue, skeletal muscle, macrophages, and Kupffer cells (in the liver). The combination of GPBAR-1 stimulation likely prevent/modulate the expression of several metabolic abnormalities, including obesity. Of note, multiple pathways operate in this complex scenario which involve microbiome eubiosis/dysbiosis, BA type and pool profile, and distinct effects on GPBAR-1. Abbreviations: cAMP, cyclic adenosine monophosphate; ATP, adenosine triphosphate; DIO2, type II iodothyronine deiodinase; GLP-1, glucagon-like peptide 1; GLP-2, glucagon-like peptide 2; IBD, inflammatory bowel disease; LPS, lipopolysaccharide; PYY, peptide YY; T3, active thyroid hormone; T4, inactive thyroxine; TCA, taurocholic acid; TCDCA, taurochenodeoxycholic acid; TDCA, taurodeoxycholic acid; TLCA, taurolithocholic acid. (+) activation; (-) inhibition. After [117]

Crosstalk between bile acids (BA), microbiota, intestinal farnesoid X receptor (FXR) and the metabolic effects of the fibroblast growth factor 19 (FGF19) in the liver. During the enterohepatic circulation BA undergo microbiota biotransformation and activate the intestinal nuclear receptor FXR. This step, in turn, promotes the secretion of FGF19 which signals the hepatocyte fibroblast growth factor receptor 4 (FGFR4) receptor with the co-receptor β-Klotho. Cytosolic adaptors are recruited, i.e., the fibroblast growth factor receptor substrate 2α (FRS2α) and growth factor receptor-bound protein 2 (GRB2). FGF19 can therefore produce potent metabolic effects via activation of major regulatory elements. The role of insulin is shown for comparison. Abbreviations; ACC2, acetyl-CoA carboxylase 2; CYP7A1, cholesterol-7α-hydroxylase; CREB cAMP-response element-binding protein; FXR, farnesoid X receptor; FXRE, FXR responsive element; GSK3, glycogen synthase kinase 3; Mnk1, protein kinase; p90 ribosomal S6 kinase; PXR, pregnane X receptor; PXRE, PXR response element; SCD1; stearoyl-CoA desaturase 1; SREBP1C, sterol regulatory element-binding protein 1C. (+) activation; (-) inhibition; ≠, unchanged. Adapted from [129]
The inactivation of FXR secondary to the altered BA homeostasis can promote hepatic de novo lipogenesis [129] and, as shown in animal models, the pathways involving FXR signalling are able to promote diet-induced obesity [137, 235, 236].
In mice, the genetic deletion of intestinal FXR has a protective role against diet-induced diabetes and obesity [235, 236]. Evidence obtained by the administration of the FXR agonist GW4064 suggests that this effect might also involve the intestinal incretin Glucagon-like peptide 1 (GLP-1). In particular, the lack of intestinal FXR promotes the intestinal release of GLP-1 which, in turn, improves glycemic control and induces weight loss [237]. A similar mechanism occurs in mice following the activation of GPBAR-1 by the specific agonist INT-777, which stimulates GLP-release and, therefore, exerts a protective role against diabetes and obesity [238]. In mice, the gut microbiota-BA crosstalk has a role in weight regain following calorie restriction.
In obese humans with insulin resistance, a 2-week very-low-calorie diet, increased peak postprandial deoxycholate levels and decreased resting and postprandial energy expenditure by 7% and 11%, respectively [239]. As shown by animal models, calorie restriction induces a remodeling of gut microbiota (mainly reducing Parabacteroides distasonis) and of the profile of serum BA (decreased proportion of non-12alpha-hydroxylated BA, ursodeoxycholic acid and lithocholic acid). In weight-rebound mice, treatment with Parabacteroides distasonis or non-12alpha-hydroxylated BA decreased weight regain via increased thermogenesis [240].
5.3 Other microbial products
The role of the gut-brain axis consists of a bilateral communication network between the gastrointestinal tract and the central nervous system. In this context the capability of the microbiota to metabolize nutrients and produce active molecules must be considered. The interplay between gut microbiota, dietary fiber and production of SCFAs can contribute to evolution of obesity, since SCFAs are able to decrease hepatic production of lipid and glucose [21]. SCFAs also stimulate anorexigenic gut peptides, such as GLP-1, and PYY [96]. Decreased abundance of bacteria producing SCFAs [96] occurs with intestinal inflammation and metabolic disorders [241].
In overweight/obese humans, a Mediterranean diet enriched in fiber increased alpha-diversities of fecal microbiota, with increased abundance of different species such as Akkermansia muciniphila and Intestinimonas butyriciproducens, and increased postprandial serum concentrations of plasma butyrate [242].
Gut microbiota also modulates tryptophan metabolism, with effects on appetite and on the gut-brain axis with effects on body weight and metabolic homeostasis [243]. These actions are mainly due to bacterial catabolites of tryptophan as serotonin (5-hydroxytryptamine), kynurenine, and indole derivatives [243, 244].
Serotonin has anorectic effect in brain [245]. At peripheral level, however, this signalling molecule contributes with the development of obesity in mice and rats [246,247,248]. In humans, obese have an increased capacity to produce and release serotonin, which is associated with higher BMI [249]. The gram-positive bacterium Clostridium ramosum has been linked, in animal models, with the development of obesity following high-fat diet [250]. In germ-free mice, Clostridium ramosum stimulates serotonin secretion, facilitating intestinal lipid absorption and the development of obesity [251].
Decreased serum levels of kynurenic acid, one of the main catabolites of tryptophane, have been reported in obese, with a negative correlation between serum concentrations of this catabolite and BMI [252]. However, the link between kynurenic acid and obesity is controversial, since a previous study documented, in another cohort of obese, a positive association between kynurenine levels, BMI and insulin resistance [253]. Increased serum levels of kynurenic acid occur in obese women with type 2 diabetes, as compared with normoglycemic individuals [254]. Increased kynurenic pathway metabolites occurred in a cohort of overweight and obese Asian children, with significant positive associations with body fat percentage [255]. Of note, kynurenine metabolites produce differential modulatory effects on microbiome. At lower concentrations kynurenic acid shows no effect on bacterial viability or stimulates growth of Lactobacillus reuteri and Lactobacillus rhamnosus. Conversely, at high concentrations this tryptophan catabolite can negatively impact the growth of most products and strains [256].
Indole, another product deriving from the microbial catabolism of tryptophan, modulates the colonic secretion of GLP-1, potentially influencing the host metabolism [257]. Furthermore, the microbial catabolites indole-3-acetic acid and 3-indolepropionic acid binds the aryl hydrocarbon receptor (AhR) [79], expanding highly adaptive lactobacilli [258] and modulating gut barrier function, GLP-1 secretion and metabolic homeostasis, with protective effects against obesity [79, 258, 259].
A decreased production of AhR agonists by gut microbiota has been reported in humans with metabolic syndrome. In these subjects, restoring AhR signalling through a supplementation with AhR agonist or a Lactobacillus strain with high AhR ligand-production capacity generated beneficial metabolic effects, with improved gut barrier function and GLP-1 secretion [79].
Lastly, the specific bacterial components LPS are able to trigger low-grade inflammation and insulin resistance following high-fat diet [57]. However, beneficial metabolic effects can derive from LPS of specific strains (i.e., R. sphaeroides), mainly through antagonistic actions on Toll-like receptors [62].
6 The modulation of gut microbiota-fat axis by external factors
The abundance and variety of gut microbiota depends on a complex interplay between several external factors as dietary habits and diet components, lifestyle, and ingestion of toxic chemicals negatively affecting gut microbes. All these components have a potential role in influencing the relationships between gut microbiota and excessive fat production and storage.
6.1 Dietary habits
Dietary habits can shape the composition of gut microbiota in obesity. Eating a western-like diet is a predisposing factor for gut dysbiosis, gut barrier dysfunction, and low-grade systemic inflammation [260]. In animals, the dietary composition in fat and sugar consisting of high-sugar and high-fat diet, can modulate gut microbiota, promoting obesity [261]. In humans, high intake of fat and saturated fatty acids can decrease both microbiota richness and diversity [262]. By contrast, the supplementation with omega-3 fatty acids plays a protective role against insulin resistance and inflammation in white adipose tissue. Such beneficial effects relate to microbiota composition, i.e., increased relative abundance of Bifidobacterium spp., Lactobacillus spp. and Akkermansia muciniphila [263]). The beneficial effects of Akkermansia muciniphila on obesity have been reported by a number of animal and human studies supporting a causal role between the relative abundance of this bacterium and obesity [264].
As shown by a randomized controlled trial in obese and overweight subjects, adherence to Mediterranean diet had beneficial metabolic effects (i.e., decreased plasma cholesterol and circulating levels of saturated fatty acids, increased levels of polyunsaturated fatty acids, decreased insulin resistance) and changed microbiome. Treated subjects showed increased levels of fiber degrading Faecalibacterium prausnitzii and Roseburia, and decreased levels of the potentially proinflammatory Ruminococcus gnavus and Ruminococcus torques. Increased levels of genes for microbial carbohydrate degradation linked to butyrate metabolism were also noticed [265]. Of note, the beneficial effects of Mediterranean dietary habits seem independent by caloric intake [266]. Beneficial effects on gut microbiota have been also reported following intermittent fasting, a dietary regimen linked with weight loss and beneficial metabolic effects (i.e., improved glycemic control) [267], increased bacterial alpha diversity, increased Lactobacillus and Bifidobacterium and decreased pathogenic bacteria [268]. During Ramadan (a popular example of intermittent fasting), increased proportion of A. muciniphila, F. prausnitzii and Roseburia have been reported [269, 270].
6.2 Other lifestyle factors
Low level of physical activity [213, 271] and smoking [272, 273] can induce changes of gut microbiota and can therefore put the individuals at increased risk of obesity.
In overweight or obese adults, the level and intensity of daily physical activity were positively associated with the relative abundance of Faecalibacterium and negatively with the abundances of Alistipes, Parabacteroides, and Gemmiger, as well as the concentrations of SCFAs [271]. A recent systematic review revealed that exercise consisting of mainly aerobic exercises at moderate or high intensity for at least 6 weeks, positively modulates the diversity of gut microbiota and the abundance of some species of bacteria linked with beneficial metabolic effects, as Bifidobacteriaceae family, Bacteroides and Akkermansia genera [213].
Available evidence suggests that smoking is able to change microbiota composition [273] by decreasing diversity, increasing Proteobacteria and Bacteroidetes phyla and the genera Clostridium, Bacteroides and Prevotella, and decreasing Actinobacteria and Firmicutes phyla, Bifidobacteria and Lactococcus genera [272]. Changes are similar to those previously described in overweight and obese subjects. On the other hand, in mice, microbiome depletion induced by antibiotics prevents smoking-cessation-induced weight gain, whereas fecal microbiome transplantation from mice previously exposed to cigarette smoke into germ-free mice naive to smoke exposure induces excessive weight gain. In humans, smokers had a distinct composition of fecal microbiota, with elevated levels of plasma choline, betaine and dimethylglycine [273]. These findings are comparable to those obtained in the described mouse model. During active smoking, excessive production of dimethylglycine and related suppression of aceturic acid should contribute to a non-microbiome dependent anorexia. Following smoking cessation, the obesogenic “smoking microbiome” should lead to weight gain [273].
6.3 Toxic chemicals contaminating food
Gut microbiota can come in contact with ingested foods contaminated with toxic chemicals, which include persistent organic pollutants (polychlorinated biphenyls, polybrominated diphenyl ethers) [274] or pesticides [275, 276]. These molecules can induce gut dysbiosis which, in turn, can predispose to altered metabolic homeostasis and obesity.
Exposure to organochlorine pesticides such as cis-nonachlor, oxychlordane, and trans-nonachlor has been linked with the amount of methanobacteriales which, in turn, is related with higher body weight, waist circumference [277], and obesity [278].
Long term exposure to chlorpyrifos, a widely diffused pesticide ingested with contaminated food, can increase Proteobacteria and decrease Bacteroidetes. Changes are likely linked with obesity and insulin resistance, mainly through altered gut barrier and LPS-mediated low-grade inflammation [275, 276]. In mice, exposure to chlorpyrifos for 12 weeks increased body fat and insulin resistance, generating a low-grade inflammation and a metabolic syndrome. These changes were paralleled by decreased bacteria diversity and richness, with increased Firmicutes and Proteobacteria and decreased Bacteroidetes abundance. Dietary manipulations can play a role, since a western diet enhances the effects of chlorpyrifos, while a Mediterranean and vegetarian diets are protective with pathways involving the gut microbiota, SCFAs production and improved intestinal barrier integrity [276]. In animal studies a type of gut dysbiosis predisposing to obesity follows the ingestion of metals (in particular cadmium [279], lead [280]) or endocrine disrupting chemicals as Bisphenol A (BPA) [281], phthalates [282], triclosan [283].
In early life male mice exposed to low-dose cadmium developed gut microbiota changes testified by reduced diversity, increased Bacteroidetes, decreased Firmicutes, decreased levels of Bifidobacterium and Prevotella, and fat accumulation in adult age [279].
In mice on high fat diet, chronic Pb administration increased body weight, visceral obesity, insulin resistance, and liver fat accumulation increasing pathogenic genera (Desulfovibrio, Alistipes and Helicobacter), and decreasing beneficial microbes as Akkermansia and Barnesiella [280]. Mice exposed to BPA showed decreased Bifidobacteria, Firmicutes phylum, specifically Clostridium butyricum, Clostridium Cluster XIV, Clostridium butyricum, and Clostridium Cluster XIVa [281].
Mice chronically exposed to di-(2-ethylhexyl)-phthalate showed weight gain, increased hepatic lipogenesis and decreased Firmicutes to Bacteroidetes ratio [282].
In high fat diet-fed mice, exposure to triclosan, a widely employed antiseptic and disinfectant with endocrine disrupting activity, caused a reduction in fecal species richness and diversity, further shifting gut microbiota toward lower Bacteriodetes and higher Firmicutes [283].
7 Concluding remarks
The worldwide obesity epidemic results from the complex interplay of genetic and environmental factors. In eubiosis, the gut microbiota works at the interface of several interactions connecting food, nutrients, calories with metabolic homeostasis and fat accumulation. Lean and metabolically health phenotype host Akkermansia muciniphila, and species from genera Faecalibacterium, Alistipes, Roseburia. Conditions of dysbiosis contribute to abnormal energy expenditure and deranged metabolic homeostasis and occur with overweight, obesity, and type 2 diabetes (Fig. 10).

Potential consequences of impaired body function homeostasis on gut microbiome, bile acid (BA) pool microbial products and metabolic consequences. The onset of dysbiosis can profoundly affect the composition of the BA pool and ratio between primary vs. secondary BA. The onset of a chronic condition of dysbiosis can also impact the production/metabolization of several active molecules and metabolites. This qualitative a/o quantitative shift of composition with changes of key metabolic pathways and activation of pro-inflammatory responses which pave the way to obesity and other related conditions. Progression and complications of such conditions, in turn, have the capability of perpetuating the changes of microbiome and related aspects
Furthermore, several bacterial products are involved in the pathogenesis of chronic, low-grade inflammation and insulin resistance, the two major pathologic conditions associated with obesity. On the other hand, dietary habits, lifestyle, food contaminants, and excessive fat storage can affect microbiota composition. The potential association between gut microbiota and obesity stems from early animal studies. Germ-free mice have less weight than mice with a normal gut microbiome [183]. Studies mainly on rodents suggest that gut microbial transplant influences adiposity. An association exists between the proportion of Bacteroidetes and Firmicutes bacteria in the gut and the development of obesity. The gut microbiota composition might affect both adiposity and the response to dietary intervention in humans as well, but the picture becomes more complex. It is unclear if a causal relationship exists between microbiome and obesity [284]. Indeed, individuals with a higher versus lower gut Prevotella to Bacteroides ratio lost significantly more weight on a calorie-restricted, high-fiber diet [285]. Nevertheless, obese individuals after losing weight host a microbiome like that of lean individuals, with a higher Firmicutes to Bacteroidetes ratio. This profile, however, is reversed with weight regain [286]. Despite the gut microbiome likely affects the risk of adiposity-related comorbidities such as type 2 diabetes and inflammation, no evidence suggests that manipulation of the gut microbiome will be an effective obesity treatment [287]
Whilst searching the ultimate role of gut microbiota in different obesity phenotypes in humans, we must be aware that the study heterogeneity, lack of robust longitudinal studies, the changing gut microbiota over time, and several confounders limit the translational value of animal studies connecting the “obesogenic” microbiota with obesity phenotypes. Further longitudinal studies based on metagenomics, transcriptomics, proteomics and metabolomics, and precise characterization of confounders are urgently needed to link gut microbiota function and metabolic phenotypes. More evidence must confirm that microbial-derived metabolites such as SCFAs and BA in the long-term can represent precision interventions against impaired metabolic homeostasis, overweight and obesity.
Abbreviations
- AGEs:
-
Advanced glycation end products
- AMPs:
-
Antimicrobial peptides
- BPA:
-
Bisphenol A
- BA:
-
Bile acids
- BMI:
-
Body Mass Index
- CA:
-
Cholic acid
- CDCA:
-
Chenodeoxycholic acid
- DAMPs:
-
Damage-associated molecular patterns
- DCA:
-
Deoxycholic acid
- DHEA:
-
Dehydroepiandrosterone
- eCBome:
-
Endocannabinoidome
- ENS:
-
Enteric nervous system
- FXR:
-
Farnesoid X receptor
- GLP-1:
-
Glucagon-like peptide-1
- GPBAR-1:
-
G protein-coupled bile acid receptor 1
- IECs:
-
Intestinal epithelial cells
- LCA:
-
Lithocholic acid
- LPS:
-
Lipopolysaccharides
- MAMPs:
-
Metabolism-associated molecular patterns
- NAFLD:
-
Nonalcoholic fatty liver disease
- NODs:
-
Nucleotide-binding oligomerization domain-containing proteins
- OEA:
-
Oleoylethanolamide
- PAMPs:
-
Pathogen- associated molecular patterns
- PRRs:
-
Pathogen recognition receptors
- PYY:
-
Peptide YY
- SCFAs:
-
Short chain fatty acids
- T2D:
-
Type 2 diabetes
- TLRs:
-
Toll like receptors
- TMA:
-
Trimethylamine
- TMAO:
-
Trimethylamine N-oxide
- TRPV1:
-
Transient receptor potential cation channel subfamily V member 1
- UDCA:
-
Ursodeoxycholic acid
- VIP:
-
Vasoactive intestinal peptide
References
World Health Organization. ICD-11 for Mortality and Morbidity Statistics. 2023.
World Health Organization. Obesity and Overweight. 2023. https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight. https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight. Accessed 26 Apr 2023.
Collaboration NCDRF. Rising rural body-mass index is the main driver of the global obesity epidemic in adults. Nature. 2019;569(7755):260–4. https://doi.org/10.1038/s41586-019-1171-x.
Vecchie A, Dallegri F, Carbone F, Bonaventura A, Liberale L, Portincasa P, Fruhbeck G, Montecucco F. Obesity phenotypes and their paradoxical association with cardiovascular diseases. Eur J Intern Med. 2018;48:6–17. https://doi.org/10.1016/j.ejim.2017.10.020.
Bhaskaran K, Douglas I, Forbes H, dos-Santos-Silva I, Leon DA, Smeeth L. Body-mass index and risk of 22 specific cancers: a population-based cohort study of 5.24 million UK adults. Lancet. 2014;384(9945):755–65. https://doi.org/10.1016/S0140-6736(14)60892-8.
Dietz W, Santos-Burgoa C. Obesity and its Implications for COVID-19 mortality. Obesity. 2020;28(6):1005. https://doi.org/10.1002/oby.22818.
Swinburn B, Sacks G, Ravussin E. Increased food energy supply is more than sufficient to explain the US epidemic of obesity. Am J Clin Nutr. 2009;90(6):1453–6. https://doi.org/10.3945/ajcn.2009.28595.
Church TS, Thomas DM, Tudor-Locke C, Katzmarzyk PT, Earnest CP, Rodarte RQ, Martin CK, Blair SN, Bouchard C. Trends over 5 decades in U.S. occupation-related physical activity and their associations with obesity. PloS One. 2011;6(5):e19657. https://doi.org/10.1371/journal.pone.0019657.
Faienza MF, Wang DQH, Frühbeck G, Garruti G, Portincasa P. The dangerous link between childhood and adulthood predictors of obesity and metabolic syndrome. Int Emerg Med. 2016;11(2):175–82.
Swinburn BA, Sacks G, Hall KD, McPherson K, Finegood DT, Moodie ML, Gortmaker SL. The global obesity pandemic: shaped by global drivers and local environments. Lancet. 2011;378(9793):804–14. https://doi.org/10.1016/S0140-6736(11)60813-1.
Di Ciaula A, Portincasa P. Diet and contaminants: Driving the rise to obesity epidemics? Curr Med Chem. 2019;26(19):3471–82. https://doi.org/10.2174/0929867324666170518095736.
de Vos WM, Tilg H, Van Hul M, Cani PD. Gut microbiome and health: mechanistic insights. Gut. 2022;71(5):1020–32. https://doi.org/10.1136/gutjnl-2021-326789.
Cani PD, Van Hul M. Mediterranean diet, gut microbiota and health: when age and calories do not add up! Gut. 2020;69(7):1167–8. https://doi.org/10.1136/gutjnl-2020-320781.
Cani PD, Moens de Hase E, Van Hul M. Gut microbiota and host metabolism: From proof of concept to therapeutic intervention. Microorganisms. 2021;9(6):1302. https://doi.org/10.3390/microorganisms9061302.
Cani PD, Van Hul M. Do diet and microbes really “PREDICT” cardiometabolic risks? Nature reviews Endocrinology. 2021;17(5):259–60. https://doi.org/10.1038/s41574-021-00480-7.
Torres-Fuentes C, Schellekens H, Dinan TG, Cryan JF. The microbiota-gut-brain axis in obesity. Lancet Gastroenterol Hepatol. 2017;2(10):747–56. https://doi.org/10.1016/S2468-1253(17)30147-4.
Dommett R, Zilbauer M, George JT, Bajaj-Elliott M. Innate immune defence in the human gastrointestinal tract. Mol Immunol. 2005;42(8):903–12. https://doi.org/10.1016/j.molimm.2004.12.004.
Di Ciaula A, Bonfrate L, Portincasa P. The role of microbiota in nonalcoholic fatty liver disease. Eur J Clin Investig. 2022;52(7):e13768. https://doi.org/10.1111/eci.13768.
Portincasa P, Bonfrate L, Khalil M, Angelis M, Calabrese FM, D’Amato M, Wang DQ, Di Ciaula A. Intestinal barrier and permeability in health, obesity and NAFLD. Biomedicines. 2021;10(1):83. https://doi.org/10.3390/biomedicines10010083.
Di Ciaula A, Baj J, Garruti G, Celano G, De Angelis M, Wang HH, Di Palo DM, Bonfrate L, Wang DQ, Portincasa P. Liver steatosis, gut-liver axis, microbiome and environmental factors. A never-ending bidirectional cross-talk. J Clin Med. 2020;9(8):2648. https://doi.org/10.3390/jcm9082648.
Portincasa P, Bonfrate L, Vacca M, De Angelis M, Farella I, Lanza E, Khalil M, Wang DQ, Sperandio M, Di Ciaula A. Gut microbiota and short chain fatty acids: Implications in glucose homeostasis. Int J Mol Sci. 2022;23(3):1105. https://doi.org/10.3390/ijms23031105.
Savage DC. Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol. 1977;31:107–33. https://doi.org/10.1146/annurev.mi.31.100177.000543.
Chelakkot C, Ghim J, Ryu SH. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp Mol Med. 2018;50(8):1–9. https://doi.org/10.1038/s12276-018-0126-x.
Konig J, Wells J, Cani PD, Garcia-Rodenas CL, MacDonald T, Mercenier A, Whyte J, Troost F, Brummer RJ. Human intestinal barrier function in health and disease. Clin Transl Gastroenterol. 2016;7(10):e196. https://doi.org/10.1038/ctg.2016.54.
McGhee JR, Fujihashi K. Inside the mucosal immune system. PLoS Biol. 2012;10(9):e1001397. https://doi.org/10.1371/journal.pbio.1001397.
Cresci GA, Bawden E. Gut microbiome: What we do and don’t know. Nutr Clin Pract Official Publ Am Soc Parent Enteral Nutr. 2015;30(6):734–46. https://doi.org/10.1177/0884533615609899.
den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud DJ, Bakker BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res. 2013;54(9):2325–40. https://doi.org/10.1194/jlr.R036012.
Iacob S, Iacob DG, Luminos LM. Intestinal microbiota as a host defense mechanism to infectious threats. Front Microbiol. 2018;9:3328. https://doi.org/10.3389/fmicb.2018.03328.
Maslowski KM, Mackay CR. Diet, gut microbiota and immune responses. Nat Immunol. 2011;12(1):5–9. https://doi.org/10.1038/ni0111-5.
Hooper LV, Macpherson AJ. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat Rev Immunol. 2010;10(3):159–69. https://doi.org/10.1038/nri2710.
Xu J, Gordon JI. Honor thy symbionts. Proc Natl Acad Sci USA. 2003;100(18):10452–9. https://doi.org/10.1073/pnas.1734063100.
Pickard JM, Zeng MY, Caruso R, Nunez G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol Rev. 2017;279(1):70–89. https://doi.org/10.1111/imr.12567.
Sender R, Fuchs S, Milo R. Are we really vastly outnumbered? Revisiting the ratio of bacterial to host cells in humans. Cell. 2016;164(3):337–40. https://doi.org/10.1016/j.cell.2016.01.013.
Islam MR, Arthur S, Haynes J, Butts MR, Nepal N, Sundaram U. The role of gut microbiota and metabolites in obesity-associated chronic gastrointestinal disorders. Nutrients. 2022;14(3). https://doi.org/10.3390/nu14030624.
Hill MJ. Intestinal flora and endogenous vitamin synthesis. Eur J Cancer Prevent Official J Eur Cancer Prevent Organ. 1997;6 Suppl 1(2):S43-45. https://doi.org/10.1097/00008469-199703001-00009.
De Angelis M, Garruti G, Minervini F, Bonfrate L, Portincasa P, Gobbetti M. The food-gut human axis: the effects of diet on gut microbiota and metabolome. Curr Med Chem. 2019;26(19):3567–83. https://doi.org/10.2174/0929867324666170428103848.
Portincasa P, Celano G, Serale N, Vitellio P, Calabrese FM, Chira A, David L, Dumitrascu DL, De Angelis M. Clinical and metabolomic effects of lactiplantibacillus and plantarum and pediococcus acidilactici in fructose intolerant patients. Nutrients. 2022;14(12). https://doi.org/10.3390/nu14122488.
Vacca M, Celano G, Calabrese FM, Portincasa P, Gobbetti M, De Angelis M. The controversial role of human gut lachnospiraceae. Microorganisms. 2020;8(4). https://doi.org/10.3390/microorganisms8040573.
Zoetendal EG, Raes J, van den Bogert B, Arumugam M, Booijink CC, Troost FJ, Bork P, Wels M, de Vos WM, Kleerebezem M. The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates. ISME J. 2012;6(7):1415–26. https://doi.org/10.1038/ismej.2011.212.
Wilson KH, Perini F. Role of competition for nutrients in suppression of Clostridium difficile by the colonic microflora. Infect Immun. 1988;56(10):2610–4. https://doi.org/10.1128/iai.56.10.2610-2614.1988.
Gantois I, Ducatelle R, Pasmans F, Haesebrouck F, Hautefort I, Thompson A, Hinton JC, Van Immerseel F. Butyrate specifically down-regulates salmonella pathogenicity island 1 gene expression. Appl Environ Microbiol. 2006;72(1):946–9. https://doi.org/10.1128/AEM.72.1.946-949.2006.
Lawley TD, Walker AW. Intestinal colonization resistance Immunology. 2013;138(1):1–11. https://doi.org/10.1111/j.1365-2567.2012.03616.x.
Ford SA, Kao D, Williams D, King KC. Microbe-mediated host defence drives the evolution of reduced pathogen virulence. Nat Commun. 2016;7(1):13430. https://doi.org/10.1038/ncomms13430.
Fridman CM, Keppel K, Gerlic M, Bosis E, Salomon D. A comparative genomics methodology reveals a widespread family of membrane-disrupting T6SS effectors. Nat Commun. 2020;11(1):1085. https://doi.org/10.1038/s41467-020-14951-4.
Zhang LJ, Gallo RL. Antimicrobial peptides. Curr Biol CB. 2016;26(1):R14-19. https://doi.org/10.1016/j.cub.2015.11.017.
Walsh D, McCarthy J, O’Driscoll C, Melgar S. Pattern recognition receptors–molecular orchestrators of inflammation in inflammatory bowel disease. Cytokine Growth Factor Rev. 2013;24(2):91–104. https://doi.org/10.1016/j.cytogfr.2012.09.003.
Wang X, Wang Y, Antony V, Sun H, Liang G. Metabolism-associated molecular patterns (MAMPs). Trends Endocrinol Metabol TEM. 2020;31(10):712–24. https://doi.org/10.1016/j.tem.2020.07.001.
Bertani B, Ruiz N. Function and biogenesis of lipopolysaccharides. EcoSal Plus. 2018;8(1). https://doi.org/10.1128/ecosalplus.ESP-0001-2018.
Belkaid Y, Harrison OJ. Homeostatic immunity and the microbiota. Immunity. 2017;46(4):562–76. https://doi.org/10.1016/j.immuni.2017.04.008.
Fitzgerald KA, Kagan JC. Toll-like receptors and the control of immunity. Cell. 2020;180(6):1044–66. https://doi.org/10.1016/j.cell.2020.02.041.
Okumura R, Takeda K. Maintenance of intestinal homeostasis by mucosal barriers. Inflamm Regen. 2018;38(1):5. https://doi.org/10.1186/s41232-018-0063-z.
Quesniaux VJ, Nicolle DM, Torres D, Kremer L, Guerardel Y, Nigou J, Puzo G, Erard F, Ryffel B. Toll-like receptor 2 (TLR2)-dependent-positive and TLR2-independent-negative regulation of proinflammatory cytokines by mycobacterial lipomannans. J Immunol. 2004;172(7):4425–34. https://doi.org/10.4049/jimmunol.172.7.4425.
Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–8. https://doi.org/10.1126/science.282.5396.2085.
Bhinder G, Stahl M, Sham HP, Crowley SM, Morampudi V, Dalwadi U, Ma C, Jacobson K, Vallance BA. Intestinal epithelium-specific MyD88 signaling impacts host susceptibility to infectious colitis by promoting protective goblet cell and antimicrobial responses. Infect Immun. 2014;82(9):3753–63. https://doi.org/10.1128/IAI.02045-14.
Frantz AL, Rogier EW, Weber CR, Shen L, Cohen DA, Fenton LA, Bruno ME, Kaetzel CS. Targeted deletion of MyD88 in intestinal epithelial cells results in compromised antibacterial immunity associated with downregulation of polymeric immunoglobulin receptor, mucin-2, and antibacterial peptides. Mucosal Immunol. 2012;5(5):501–12. https://doi.org/10.1038/mi.2012.23.
Lei J, Sun L, Huang S, Zhu C, Li P, He J, Mackey V, Coy DH, He Q. The antimicrobial peptides and their potential clinical applications. Am J Transl Res. 2019;11(7):3919.
Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmee E, Cousin B, Sulpice T, Chamontin B, Ferrieres J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761–72. https://doi.org/10.2337/db06-1491.
Tilg H, Burcelin R, Tremaroli V. Liver tissue microbiome in NAFLD: next step in understanding the gut-liver axis? Gut. 2020;69(8):1373–4. https://doi.org/10.1136/gutjnl-2019-320490.
Regnier M, Van Hul M, Knauf C, Cani PD. Gut microbiome, endocrine control of gut barrier function and metabolic diseases. J Endocrinol. 2021;248(2):R67–82. https://doi.org/10.1530/JOE-20-0473.
Tilg H, Cani PD, Mayer EA. Gut microbiome and liver diseases. Gut. 2016;65(12):2035–44. https://doi.org/10.1136/gutjnl-2016-312729.
Tilg H, Adolph TE, Gerner RR, Moschen AR. The Intestinal Microbiota in Colorectal Cancer. Cancer Cell. 2018;33(6):954–64. https://doi.org/10.1016/j.ccell.2018.03.004.
Anhe FF, Barra NG, Cavallari JF, Henriksbo BD, Schertzer JD. Metabolic endotoxemia is dictated by the type of lipopolysaccharide. Cell Rep. 2021;36(11):109691. https://doi.org/10.1016/j.celrep.2021.109691.
Vijay-Kumar M, Sanders CJ, Taylor RT, Kumar A, Aitken JD, Sitaraman SV, Neish AS, Uematsu S, Akira S, Williams IR, Gewirtz AT. Deletion of TLR5 results in spontaneous colitis in mice. J Clin Investig. 2007;117(12):3909–21. https://doi.org/10.1172/JCI33084.
Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328(5975):228–31. https://doi.org/10.1126/science.1179721.
Takeda K, Akira S. Microbial recognition by Toll-like receptors. J Dermatol Sci. 2004;34(2):73–82. https://doi.org/10.1016/j.jdermsci.2003.10.002.
Everard A, Geurts L, Caesar R, Van Hul M, Matamoros S, Duparc T, Denis RG, Cochez P, Pierard F, Castel J, Bindels LB, Plovier H, Robine S, Muccioli GG, Renauld JC, Dumoutier L, Delzenne NM, Luquet S, Backhed F, Cani PD. Intestinal epithelial MyD88 is a sensor switching host metabolism towards obesity according to nutritional status. Nat Commun. 2014;5(1):5648. https://doi.org/10.1038/ncomms6648.
Cani PD, Plovier H, Van Hul M, Geurts L, Delzenne NM, Druart C, Everard A. Endocannabinoids–at the crossroads between the gut microbiota and host metabolism. Nature reviews Endocrinology. 2016;12(3):133–43. https://doi.org/10.1038/nrendo.2015.211.
Muccioli GG, Naslain D, Backhed F, Reigstad CS, Lambert DM, Delzenne NM, Cani PD. The endocannabinoid system links gut microbiota to adipogenesis. Mol Syst Biol. 2010;6(1):392. https://doi.org/10.1038/msb.2010.46.
Geurts L, Muccioli GG, Delzenne NM, Cani PD. Chronic endocannabinoid system stimulation induces muscle macrophage and lipid accumulation in type 2 diabetic mice independently of metabolic endotoxaemia. PloS One. 2013;8(2):e55963. https://doi.org/10.1371/journal.pone.0055963.
Suriano F, Manca C, Flamand N, Depommier C, Van Hul M, Delzenne NM, Silvestri C, Cani PD, Di Marzo V. Exploring the endocannabinoidome in genetically obese (ob/ob) and diabetic (db/db) mice: Links with inflammation and gut microbiota. Biochim Biophys Acta Mol Cell Biol Lipids. 2022;1867(1):159056. https://doi.org/10.1016/j.bbalip.2021.159056.
Geurts L, Lazarevic V, Derrien M, Everard A, Van Roye M, Knauf C, Valet P, Girard M, Muccioli GG, Francois P, de Vos WM, Schrenzel J, Delzenne NM, Cani PD. Altered gut microbiota and endocannabinoid system tone in obese and diabetic leptin-resistant mice: impact on apelin regulation in adipose tissue. Front Microbiol. 2011;2:149. https://doi.org/10.3389/fmicb.2011.00149.
Geurts L, Everard A, Van Hul M, Essaghir A, Duparc T, Matamoros S, Plovier H, Castel J, Denis RG, Bergiers M, Druart C, Alhouayek M, Delzenne NM, Muccioli GG, Demoulin JB, Luquet S, Cani PD. Adipose tissue NAPE-PLD controls fat mass development by altering the browning process and gut microbiota. Nat Commun. 2015;6(1):6495. https://doi.org/10.1038/ncomms7495.
Everard A, Plovier H, Rastelli M, Van Hul M, de Wouters d’Oplinter A, Geurts L, Druart C, Robine S, Delzenne NM, Muccioli GG, de Vos WM, Luquet S, Flamand N, Di Marzo V, Cani PD. Intestinal epithelial N-acylphosphatidylethanolamine phospholipase D links dietary fat to metabolic adaptations in obesity and steatosis. Nat Commun. 2019;10(1):457. https://doi.org/10.1038/s41467-018-08051-7.
Lefort C, Roumain M, Van Hul M, Rastelli M, Manco R, Leclercq I, Delzenne NM, Marzo VD, Flamand N, Luquet S, Silvestri C, Muccioli GG, Cani PD. Hepatic NAPE-PLD Is a key regulator of liver lipid metabolism. Cells. 2020;9(5):1247. https://doi.org/10.3390/cells9051247.
Cohen LJ, Esterhazy D, Kim SH, Lemetre C, Aguilar RR, Gordon EA, Pickard AJ, Cross JR, Emiliano AB, Han SM, Chu J, Vila-Farres X, Kaplitt J, Rogoz A, Calle PY, Hunter C, Bitok JK, Brady SF. Commensal bacteria make GPCR ligands that mimic human signalling molecules. Nature. 2017;549(7670):48–53. https://doi.org/10.1038/nature23874.
Manca C, Boubertakh B, Leblanc N, Deschenes T, Lacroix S, Martin C, Houde A, Veilleux A, Flamand N, Muccioli GG, Raymond F, Cani PD, Di Marzo V, Silvestri C. Germ-free mice exhibit profound gut microbiota-dependent alterations of intestinal endocannabinoidome signaling. J Lipid Res. 2020;61(1):70–85. https://doi.org/10.1194/jlr.RA119000424.
Frost F, Kacprowski T, Ruhlemann M, Pietzner M, Bang C, Franke A, Nauck M, Volker U, Volzke H, Dorr M, Baumbach J, Sendler M, Schulz C, Mayerle J, Weiss FU, Homuth G, Lerch MM. Long-term instability of the intestinal microbiome is associated with metabolic liver disease, low microbiota diversity, diabetes mellitus and impaired exocrine pancreatic function. Gut. 2021;70(3):522–30. https://doi.org/10.1136/gutjnl-2020-322753.
Dong F, Perdew GH. The aryl hydrocarbon receptor as a mediator of host-microbiota interplay. Gut Microbes. 2020;12(1):1859812. https://doi.org/10.1080/19490976.2020.1859812.
Natividad JM, Agus A, Planchais J, Lamas B, Jarry AC, Martin R, Michel ML, Chong-Nguyen C, Roussel R, Straube M, Jegou S, McQuitty C, Le Gall M, da Costa G, Lecornet E, Michaudel C, Modoux M, Glodt J, Bridonneau C, Sovran B, Dupraz L, Bado A, Richard ML, Langella P, Hansel B, Launay JM, Xavier RJ, Duboc H, Sokol H. Impaired aryl hydrocarbon receptor ligand production by the gut microbiota is a key factor in metabolic syndrome. Cell metabolism. 2018;28(5):737–49 e734. https://doi.org/10.1016/j.cmet.2018.07.001.
Lin YH, Luck H, Khan S, Schneeberger PHH, Tsai S, Clemente-Casares X, Lei H, Leu YL, Chan YT, Chen HY, Yang SH, Coburn B, Winer S, Winer DA. Aryl hydrocarbon receptor agonist indigo protects against obesity-related insulin resistance through modulation of intestinal and metabolic tissue immunity. Int J Obes. 2019;43(12):2407–21. https://doi.org/10.1038/s41366-019-0340-1.
Scott SA, Fu J, Chang PV. Microbial tryptophan metabolites regulate gut barrier function via the aryl hydrocarbon receptor. Proc Natl Acad Sci USA. 2020;117(32):19376–87. https://doi.org/10.1073/pnas.2000047117.
Guarner F, Malagelada JR. Gut flora in health and disease. Lancet. 2003;361(9356):512–9. https://doi.org/10.1016/S0140-6736(03)12489-0.
Reynes B, Palou M, Rodriguez AM, Palou A. Regulation of adaptive thermogenesis and browning by prebiotics and postbiotics. Front Physiol. 2018;9:1908. https://doi.org/10.3389/fphys.2018.01908.
Bui TPN, Manneras-Holm L, Puschmann R, Wu H, Troise AD, Nijsse B, Boeren S, Backhed F, Fiedler D, deVos WM. Conversion of dietary inositol into propionate and acetate by commensal Anaerostipes associates with host health. Nat Commun. 2021;12(1):4798. https://doi.org/10.1038/s41467-021-25081-w.
Bui TP, Ritari J, Boeren S, de Waard P, Plugge CM, de Vos WM. Production of butyrate from lysine and the Amadori product fructoselysine by a human gut commensal. Nat Commun. 2015;6(1):10062. https://doi.org/10.1038/ncomms10062.
Topping DL, Clifton PM. Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol Rev. 2001;81(3):1031–64. https://doi.org/10.1152/physrev.2001.81.3.1031.
Rajilic-Stojanovic M, Shanahan F, Guarner F, de Vos WM. Phylogenetic analysis of dysbiosis in ulcerative colitis during remission. Inflamm Bowel Dis. 2013;19(3):481–8. https://doi.org/10.1097/MIB.0b013e31827fec6d.
Heimann E, Nyman M, Palbrink AK, Lindkvist-Petersson K, Degerman E. Branched short-chain fatty acids modulate glucose and lipid metabolism in primary adipocytes. Adipocyte. 2016;5(4):359–68. https://doi.org/10.1080/21623945.2016.1252011.
Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157(1):121–41. https://doi.org/10.1016/j.cell.2014.03.011.
Park J-H, Eberl G. Type 3 regulatory T cells at the interface of symbiosis. J Microbiol. 2018;56(3):163–71.
Stanhope KL, Schwarz JM, Havel PJ. Adverse metabolic effects of dietary fructose: results from the recent epidemiological, clinical, and mechanistic studies. Curr Opin Lipidol. 2013;24(3):198–206. https://doi.org/10.1097/MOL.0b013e3283613bca.
Le Poul E, Loison C, Struyf S, Springael JY, Lannoy V, Decobecq ME, Brezillon S, Dupriez V, Vassart G, Van Damme J, Parmentier M, Detheux M. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem. 2003;278(28):25481–9. https://doi.org/10.1074/jbc.M301403200.
Biesalski HK. Nutrition meets the microbiome: micronutrients and the microbiota. Ann New York Acad Sci. 2016;1372(1):53–64. https://doi.org/10.1111/nyas.13145.
Tyagi S, Venugopalakrishnan J, Ramaswamy K, Dudeja PK. Mechanism of n-butyrate uptake in the human proximal colonic basolateral membranes. Am J Physiol Gastrointest Liver Physiol. 2002;282(4):G676-682. https://doi.org/10.1152/ajpgi.00173.2000.
Reynolds DA, Rajendran VM, Binder HJ. Bicarbonate-stimulated [14C]butyrate uptake in basolateral membrane vesicles of rat distal colon. Gastroenterology. 1993;105(3):725–32. https://doi.org/10.1016/0016-5085(93)90889-k.
Blaak EE, Canfora EE, Theis S, Frost G, Groen AK, Mithieux G, Nauta A, Scott K, Stahl B, van Harsselaar J, van Tol R, Vaughan EE, Verbeke K. Short chain fatty acids in human gut and metabolic health. Benef Microbes. 2020;11(5):411–55. https://doi.org/10.3920/BM2020.0057.
Roediger WE. Utilization of nutrients by isolated epithelial cells of the rat colon. Gastroenterology. 1982;83(2):424–9. https://doi.org/10.1016/S0016-5085(82)80339-9.
Tazoe H, Otomo Y, Kaji I, Tanaka R, Karaki SI, Kuwahara A. Roles of short-chain fatty acids receptors, GPR41 and GPR43 on colonic functions. J Physiol Pharmacol. 2008;59(Suppl 2):251–62.
Litvak Y, Mon KKZ, Nguyen H, Chanthavixay G, Liou M, Velazquez EM, Kutter L, Alcantara MA, Byndloss MX, Tiffany CR, Walker GT, Faber F, Zhu Y, Bronner DN, Byndloss AJ, Tsolis RM, Zhou H, Baumler AJ. Commensal Enterobacteriaceae Protect against Salmonella Colonization through Oxygen Competition. Cell Host Microbe. 2019;25(1):128–39 e125. https://doi.org/10.1016/j.chom.2018.12.003.
Litvak Y, Byndloss MX, Tsolis RM, Baumler AJ. Dysbiotic Proteobacteria expansion: a microbial signature of epithelial dysfunction. Curr Opin Microbiol. 2017;39:1–6. https://doi.org/10.1016/j.mib.2017.07.003.
Bergman E. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol Rev. 1990;70(2):567–90.
Salamone D, Rivellese AA, Vetrani C. The relationship between gut microbiota, short-chain fatty acids and type 2 diabetes mellitus: the possible role of dietary fibre. Acta Diabetol. 2021;58(9):1131–8. https://doi.org/10.1007/s00592-021-01727-5.
Frost G, Sleeth ML, Sahuri-Arisoylu M, Lizarbe B, Cerdan S, Brody L, Anastasovska J, Ghourab S, Hankir M, Zhang S, Carling D, Swann JR, Gibson G, Viardot A, Morrison D, Louise Thomas E, Bell JD. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat Commun. 2014;5(1):3611. https://doi.org/10.1038/ncomms4611.
Koh A, De Vadder F, Kovatcheva-Datchary P, Backhed F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell. 2016;165(6):1332–45. https://doi.org/10.1016/j.cell.2016.05.041.
van der Beek CM, Dejong CHC, Troost FJ, Masclee AAM, Lenaerts K. Role of short-chain fatty acids in colonic inflammation, carcinogenesis, and mucosal protection and healing. Nutr Rev. 2017;75(4):286–305. https://doi.org/10.1093/nutrit/nuw067.
Li Z, Yi CX, Katiraei S, Kooijman S, Zhou E, Chung CK, Gao Y, van den Heuvel JK, Meijer OC, Berbee JFP, Heijink M, Giera M, Willems van Dijk K, Groen AK, Rensen PCN, Wang Y. Butyrate reduces appetite and activates brown adipose tissue via the gut-brain neural circuit. Gut. 2018;67(7):1269–79. https://doi.org/10.1136/gutjnl-2017-314050.
Lu Y, Fan C, Li P, Lu Y, Chang X, Qi K. Short chain fatty acids prevent high-fat-diet-induced obesity in mice by regulating G protein-coupled receptors and gut microbiota. Sci Rep. 2016;6(1):37589; https://doi.org/10.1038/srep37589.
Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, Cefalu WT, Ye J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes. 2009;58(7):1509–17. https://doi.org/10.2337/db08-1637.
van den Hoek AM, Heijboer AC, Corssmit EP, Voshol PJ, Romijn JA, Havekes LM, Pijl H. PYY3–36 reinforces insulin action on glucose disposal in mice fed a high-fat diet. Diabetes. 2004;53(8):1949–52. https://doi.org/10.2337/diabetes.53.8.1949.
Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL, Wren AM, Brynes AE, Low MJ, Ghatei MA, Cone RD, Bloom SR. Gut hormone PYY(3–36) physiologically inhibits food intake. Nature. 2002;418(6898):650–4. https://doi.org/10.1038/nature00887.
Barrera JG, Sandoval DA, D’Alessio DA, Seeley RJ. GLP-1 and energy balance: an integrated model of short-term and long-term control. Nat Rev Endocrinol. 2011;7(9):507–16. https://doi.org/10.1038/nrendo.2011.77.
Sakakibara S, Yamauchi T, Oshima Y, Tsukamoto Y, Kadowaki T. Acetic acid activates hepatic AMPK and reduces hyperglycemia in diabetic KK-A(y) mice. Biochem Biophys Res Commun. 2006;344(2):597–604. https://doi.org/10.1016/j.bbrc.2006.03.176.
Hattori M, Kondo T, Kishi M, Yamagami K. A single oral administration of acetic acid increased energy expenditure in C57BL/6J mice. Biosci Biotechnol Biochem. 2010;74(10):2158–9. https://doi.org/10.1271/bbb.100486.
den Besten G, Bleeker A, Gerding A, van Eunen K, Havinga R, van Dijk TH, Oosterveer MH, Jonker JW, Groen AK, Reijngoud DJ, Bakker BM. Short-chain fatty acids protect against high-fat diet-induced obesity via a PPARgamma-dependent switch from lipogenesis to fat oxidation. Diabetes. 2015;64(7):2398–408. https://doi.org/10.2337/db14-1213.
Sukkar AH, Lett AM, Frost G, Chambers ES. Regulation of energy expenditure and substrate oxidation by short-chain fatty acids. J Endocrinol. 2019;242(2):R1–8. https://doi.org/10.1530/JOE-19-0098.
Lefort C, Cani PD. The liver under the spotlight: Bile acids and oxysterols as pivotal actors controlling metabolism. Cells. 2021;10(2):400.
Portincasa P, Di Ciaula A, Garruti G, Vacca M, De Angelis M, Wang DQ. Bile acids and GPBAR-1: Dynamic interaction involving genes, environment and gut microbiome. Nutrients. 2020;12(12):3709. https://doi.org/10.3390/nu12123709.
Garruti G, Di Ciaula A, Wang HH, Wang DQ, Portincasa P. Cross-talk between bile acids and gastro-intestinal and thermogenic hormones: Clues from bariatric surgery. Ann Hepatol. 2017;16(Suppl. 1: s3–105.):s68–82. https://doi.org/10.5604/01.3001.0010.5499.
Di Ciaula A, Garruti G, Lunardi Baccetto R, Molina-Molina E, Bonfrate L, Wang DQ, Portincasa P. Bile acid physiology. Ann Hepatol. 2017;16(Suppl. 1: s3–105.):s4–14. https://doi.org/10.5604/01.3001.0010.5493.
Gonzalez FJ. Nuclear receptor control of enterohepatic circulation. Compr Physiol. 2012;2(4):2811–28. https://doi.org/10.1002/cphy.c120007.
Matsubara T, Li F, Gonzalez FJ. FXR signaling in the enterohepatic system. Mol Cell Endocrinol. 2013;368(1–2):17–29. https://doi.org/10.1016/j.mce.2012.05.004.
Sun L, Cai J, Gonzalez FJ. The role of farnesoid X receptor in metabolic diseases, and gastrointestinal and liver cancer. Nat Rev Gastroenterol Hepatol. 2021;18(5):335–47. https://doi.org/10.1038/s41575-020-00404-2.
Nguyen JT, Shaw RPH, Anakk S. Bile acids—a peek into their history and signaling. Endocrinology. 2022;163(11):bqac155.
Poland JC, Flynn CR. Bile acids, their receptors, and the gut microbiota. Physiology. 2021;36(4):235–45.
Chiang JYL, Ferrell JM. Bile acids as metabolic regulators and nutrient sensors. Ann Rev Nutr. 2019;39(1):175–200. https://doi.org/10.1146/annurev-nutr-082018-124344.
Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, Haussler MR, Mangelsdorf DJ. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002;296(5571):1313–6.
Ihunnah CA, Jiang M, Xie W. Nuclear receptor PXR, transcriptional circuits and metabolic relevance. Biochim Biophys Acta. 2011;1812(8):956–63. https://doi.org/10.1016/j.bbadis.2011.01.014.
Wagner M, Halilbasic E, Marschall HU, Zollner G, Fickert P, Langner C, Zatloukal K, Denk H, Trauner M. CAR and PXR agonists stimulate hepatic bile acid and bilirubin detoxification and elimination pathways in mice. Hepatology. 2005;42(2):420–30. https://doi.org/10.1002/hep.20784.
Di Ciaula A, Bonfrate L, Baj J, Khalil M, Garruti G, Stellaard F, Wang HH, Wang DQ, Portincasa P. Recent advances in the digestive, metabolic and therapeutic effects of farnesoid X receptor and fibroblast growth factor 19: from cholesterol to bile acid signaling. Nutrients. 2022;14(23). https://doi.org/10.3390/nu14234950.
Ahmad TR, Haeusler RA. Bile acids in glucose metabolism and insulin signalling - mechanisms and research needs. Nat Rev Endocrinol. 2019;15(12):701–12. https://doi.org/10.1038/s41574-019-0266-7.
Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall HU, Bamberg K, Angelin B, Hyotylainen T, Oresic M, Backhed F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metabol. 2013;17(2):225–35. https://doi.org/10.1016/j.cmet.2013.01.003.
Long SL, Gahan CGM, Joyce SA. Interactions between gut bacteria and bile in health and disease. Mol Aspects Med. 2017;56:54–65. https://doi.org/10.1016/j.mam.2017.06.002.
de Aguiar Vallim TQ, Tarling EJ, Edwards PA. Pleiotropic roles of bile acids in metabolism. Cell Metabol. 2013;17(5):657–69. https://doi.org/10.1016/j.cmet.2013.03.013.
Jones BV, Begley M, Hill C, Gahan CG, Marchesi JR. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Pro Natl Acad Sci USA. 2008;105(36):13580–5. https://doi.org/10.1073/pnas.0804437105.
Sato Y, Atarashi K, Plichta DR, Arai Y, Sasajima S, Kearney SM, Suda W, Takeshita K, Sasaki T, Okamoto S, Skelly AN, Okamura Y, Vlamakis H, Li Y, Tanoue T, Takei H, Nittono H, Narushima S, Irie J, Itoh H, Moriya K, Sugiura Y, Suematsu M, Moritoki N, Shibata S, Littman DR, Fischbach MA, Uwamino Y, Inoue T, Honda A, Hattori M, Murai T, Xavier RJ, Hirose N, Honda K. Novel bile acid biosynthetic pathways are enriched in the microbiome of centenarians. Nature. 2021;599(7885):458–64. https://doi.org/10.1038/s41586-021-03832-5.
Zhou H, Hylemon PB. Bile acids are nutrient signaling hormones. Steroids. 2014;86:62–8. https://doi.org/10.1016/j.steroids.2014.04.016.
Parseus A, Sommer N, Sommer F, Caesar R, Molinaro A, Stahlman M, Greiner TU, Perkins R, Backhed F. Microbiota-induced obesity requires farnesoid X receptor. Gut. 2017;66(3):429–37. https://doi.org/10.1136/gutjnl-2015-310283.
Yokota A, Fukiya S, Islam KB, Ooka T, Ogura Y, Hayashi T, Hagio M, Ishizuka S. Is bile acid a determinant of the gut microbiota on a high-fat diet? Gut Microbes. 2012;3(5):455–9. https://doi.org/10.4161/gmic.21216.
Jiao N, Baker SS, Chapa-Rodriguez A, Liu W, Nugent CA, Tsompana M, Mastrandrea L, Buck MJ, Baker RD, Genco RJ, Zhu R, Zhu L. Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut. 2018;67(10):1881–91. https://doi.org/10.1136/gutjnl-2017-314307.
Ridlon JM, Wolf PG, Gaskins HR. Taurocholic acid metabolism by gut microbes and colon cancer. Gut Microbes. 2016;7(3):201–15. https://doi.org/10.1080/19490976.2016.1150414.
Boursier J, Diehl AM. Implication of gut microbiota in nonalcoholic fatty liver disease. PLoS Pathogens. 2015;11(1):e1004559. https://doi.org/10.1371/journal.ppat.1004559.
Lake AD, Novak P, Shipkova P, Aranibar N, Robertson D, Reily MD, Lu Z, Lehman-McKeeman LD, Cherrington NJ. Decreased hepatotoxic bile acid composition and altered synthesis in progressive human nonalcoholic fatty liver disease. Toxicol Appl Pharmacol. 2013;268(2):132–40. https://doi.org/10.1016/j.taap.2013.01.022.
Chavez-Talavera O, Tailleux A, Lefebvre P, Staels B. Bile acid control of metabolism and inflammation in obesity, type 2 diabetes, dyslipidemia, and nonalcoholic fatty liver disease. Gastroenterology. 2017;152(7):1679–94 e1673. https://doi.org/10.1053/j.gastro.2017.01.055.
Oh SF, Praveena T, Song H, Yoo JS, Jung DJ, Erturk-Hasdemir D, Hwang YS, Lee CC, Le Nours J, Kim H, Lee J, Blumberg RS, Rossjohn J, Park SB, Kasper DL. Host immunomodulatory lipids created by symbionts from dietary amino acids. Nature. 2021;600(7888):302–7. https://doi.org/10.1038/s41586-021-04083-0.
Erturk-Hasdemir D, Oh SF, Okan NA, Stefanetti G, Gazzaniga FS, Seeberger PH, Plevy SE, Kasper DL. Symbionts exploit complex signaling to educate the immune system. Proc Natl Acad Sci USA. 2019;116(52):26157–66. https://doi.org/10.1073/pnas.1915978116.
Griffin ME, Espinosa J, Becker JL, Luo JD, Carroll TS, Jha JK, Fanger GR, Hang HC. Enterococcus peptidoglycan remodeling promotes checkpoint inhibitor cancer immunotherapy. Science. 2021;373(6558):1040–6. https://doi.org/10.1126/science.abc9113.
Konstantinov SR, Smidt H, de Vos WM, Bruijns SC, Singh SK, Valence F, Molle D, Lortal S, Altermann E, Klaenhammer TR, van Kooyk Y. S layer protein A of Lactobacillus acidophilus NCFM regulates immature dendritic cell and T cell functions. Proc Natl Acad Sci USA. 2008;105(49):19474–9. https://doi.org/10.1073/pnas.0810305105.
Tytgat HL, van Teijlingen NH, Sullan RM, Douillard FP, Rasinkangas P, Messing M, Reunanen J, Satokari R, Vanderleyden J, Dufrene YF, Geijtenbeek TB, de Vos WM, Lebeer S. Probiotic gut microbiota isolate interacts with dendritic cells via glycosylated heterotrimeric pili. PloS one. 2016;11(3):e0151824. https://doi.org/10.1371/journal.pone.0151824.
O’Connell Motherway M, Houston A, O’Callaghan G, Reunanen J, O’Brien F, O’Driscoll T, Casey PG, de Vos WM, van Sinderen D, Shanahan F. A Bifidobacterial pilus-associated protein promotes colonic epithelial proliferation. Mol Microbiol. 2019;111(1):287–301. https://doi.org/10.1111/mmi.14155.
O’Connell Motherway M, Zomer A, Leahy SC, Reunanen J, Bottacini F, Claesson MJ, O’Brien F, Flynn K, Casey PG, Munoz JA, Kearney B, Houston AM, O’Mahony C, Higgins DG, Shanahan F, Palva A, de Vos WM, Fitzgerald GF, Ventura M, O’Toole PW, van Sinderen D. Functional genome analysis of Bifidobacterium breve UCC2003 reveals type IVb tight adherence (Tad) pili as an essential and conserved host-colonization factor. Proc Natl Acad Sci USA. 2011;108(27):11217–22. https://doi.org/10.1073/pnas.1105380108.
Breton J, Tennoune N, Lucas N, Francois M, Legrand R, Jacquemot J, Goichon A, Guerin C, Peltier J, Pestel-Caron M, Chan P, Vaudry D, do Rego JC, Lienard F, Penicaud L, Fioramonti X, Ebenezer IS, Hokfelt T, Dechelotte P, and Fetissov SO,. Gut commensal E. coli proteins activate host satiety pathways following nutrient-induced bacterial growth. Cell Metabol. 2016;23(2):324–34. https://doi.org/10.1016/j.cmet.2015.10.017.
Legrand R, Lucas N, Dominique M, Azhar S, Deroissart C, Le Solliec MA, Rondeaux J, Nobis S, Guerin C, Leon F, do do Rego JC, Pons N, Le Chatelier E, Ehrlich SD, Lambert G, Dechelotte P, Fetissov SO. Commensal Hafnia alvei strain reduces food intake and fat mass in obese mice-a new potential probiotic for appetite and body weight management. Int J Obes. 2020;44(5):1041–51. https://doi.org/10.1038/s41366-019-0515-9.
Depommier C, Everard A, Druart C, Plovier H, Van Hul M, Vieira-Silva S, Falony G, Raes J, Maiter D, Delzenne NM, de Barsy M, Loumaye A, Hermans MP, Thissen JP, de Vos WM, Cani PD. Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: a proof-of-concept exploratory study. Nat Med. 2019;25(7):1096–103. https://doi.org/10.1038/s41591-019-0495-2.
Depommier C, Vitale RM, Iannotti FA, Silvestri C, Flamand N, Druart C, Everard A, Pelicaen R, Maiter D, Thissen JP, Loumaye A, Hermans MP, Delzenne NM, de Vos WM, Di Marzo V, Cani PD. Beneficial effects of Akkermansia muciniphila are not associated with major changes in the circulating Endocannabinoidome but linked to higher Mono-Palmitoyl-Glycerol Levels as New PPARalpha Agonists. Cells. 2021;10(1):185. https://doi.org/10.3390/cells10010185.
Depommier C, Everard A, Druart C, Maiter D, Thissen JP, Loumaye A, Hermans MP, Delzenne NM, de Vos WM, Cani PD. Serum metabolite profiling yields insights into health promoting effect of A. muciniphila in human volunteers with a metabolic syndrome. Gut Microbes. 2021;13(1):1994270. https://doi.org/10.1080/19490976.2021.1994270.
Yoon HS, Cho CH, Yun MS, Jang SJ, You HJ, Kim JH, Han D, Cha KH, Moon SH, Lee K, Kim YJ, Lee SJ, Nam TW, Ko G. Akkermansia muciniphila secretes a glucagon-like peptide-1-inducing protein that improves glucose homeostasis and ameliorates metabolic disease in mice. Nat Microbiol. 2021;6(5):563–73. https://doi.org/10.1038/s41564-021-00880-5.
Cani PD, Knauf C. A newly identified protein from Akkermansia muciniphila stimulates GLP-1 secretion. Cell Metabol. 2021;33(6):1073–5. https://doi.org/10.1016/j.cmet.2021.05.004.
Meng X, Zhang J, Wu H, Yu D, Fang X. Akkermansia muciniphila aspartic protease Amuc_1434* inhibits human colorectal cancer LS174T cell viability via TRAIL-mediated apoptosis pathway. Int J Mol Sci. 2020;21(9):3385. https://doi.org/10.3390/ijms21093385.
Plovier H, Everard A, Druart C, Depommier C, Van Hul M, Geurts L, Chilloux J, Ottman N, Duparc T, Lichtenstein L, Myridakis A, Delzenne NM, Klievink J, Bhattacharjee A, van der Ark KC, Aalvink S, Martinez LO, Dumas ME, Maiter D, Loumaye A, Hermans MP, Thissen JP, Belzer C, de Vos WM, Cani PD. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat Med. 2017;23(1):107–13. https://doi.org/10.1038/nm.4236.
Ottman N, Reunanen J, Meijerink M, Pietila TE, Kainulainen V, Klievink J, Huuskonen L, Aalvink S, Skurnik M, Boeren S, Satokari R, Mercenier A, Palva A, Smidt H, de Vos WM, Belzer C. Pili-like proteins of Akkermansia muciniphila modulate host immune responses and gut barrier function. PloS One. 2017;12(3):e0173004. https://doi.org/10.1371/journal.pone.0173004.
Knauf C, Abot A, Wemelle E, Cani PD. Targeting the enteric nervous system to treat metabolic disorders? “Enterosynes” as therapeutic gut factors. Neuroendocrinology. 2020;110(1–2):139–46. https://doi.org/10.1159/000500602.
Abot A, Cani PD, Knauf C. Impact of intestinal peptides on the enteric nervous system: Novel approaches to control glucose metabolism and food intake. Front Endocrinol (Lausanne). 2018;9:328. https://doi.org/10.3389/fendo.2018.00328.
Abot A, Lucas A, Bautzova T, Bessac A, Fournel A, Le-Gonidec S, Valet P, Moro C, Cani PD, Knauf C. Galanin enhances systemic glucose metabolism through enteric Nitric Oxide Synthase-expressed neurons. Mol Metab. 2018;10:100–8. https://doi.org/10.1016/j.molmet.2018.01.020.
Knauf C, Cani PD, Perrin C, Iglesias MA, Maury JF, Bernard E, Benhamed F, Gremeaux T, Drucker DJ, Kahn CR, Girard J, Tanti JF, Delzenne NM, Postic C, Burcelin R. Brain glucagon-like peptide-1 increases insulin secretion and muscle insulin resistance to favor hepatic glycogen storage. J Clin Investig. 2005;115(12):3554–3. https://doi.org/10.1172/JCI25764.
Fournel A, Drougard A, Duparc T, Marlin A, Brierley SM, Castro J, Le-Gonidec S, Masri B, Colom A, Lucas A, Rousset P, Cenac N, Vergnolle N, Valet P, Cani PD, Knauf C. Apelin targets gut contraction to control glucose metabolism via the brain. Gut. 2017;66(2):258–69. https://doi.org/10.1136/gutjnl-2015-310230.
Abot A, Wemelle E, Laurens C, Paquot A, Pomie N, Carper D, Bessac A, Mas Orea X, Fremez C, Fontanie M, Lucas A, Lesage J, Everard A, Meunier E, Dietrich G, Muccioli GG, Moro C, Cani PD, Knauf C. Identification of new enterosynes using prebiotics: roles of bioactive lipids and mu-opioid receptor signalling in humans and mice. Gut. 2021;70(6):1078–87. https://doi.org/10.1136/gutjnl-2019-320230.
Pujo J, Petitfils C, Le Faouder P, Eeckhaut V, Payros G, Maurel S, Perez-Berezo T, Van Hul M, Barreau F, Blanpied C, Chavanas S, Van Immerseel F, Bertrand-Michel J, Oswald E, Knauf C, Dietrich G, Cani PD, Cenac N. Bacteria-derived long chain fatty acid exhibits anti-inflammatory properties in colitis. Gut. 2021;70(6):1088–97. https://doi.org/10.1136/gutjnl-2020-321173.
Demarquoy J, Georges B, Rigault C, Royer M-C, Clairet A, Soty M, Lekounoungou S, Le Borgne F. Radioisotopic determination of l-carnitine content in foods commonly eaten in Western countries. Food Chem. 2004;86(1):137–42. https://doi.org/10.1016/j.foodchem.2003.09.023.
Zeisel SH, Mar MH, Howe JC, Holden JM. Concentrations of choline-containing compounds and betaine in common foods. J Nutr. 2003;133(5):1302–7. https://doi.org/10.1093/jn/133.5.1302.
Brown JM, Hazen SL. Microbial modulation of cardiovascular disease. Nat Rev Microbiol. 2018;16(3):171–81. https://doi.org/10.1038/nrmicro.2017.149.
Koay YC, Chen YC, Wali JA, Luk AWS, Li M, Doma H, Reimark R, Zaldivia MTK, Habtom HT, Franks AE, Fusco-Allison G, Yang J, Holmes A, Simpson SJ, Peter K, O’Sullivan JF. Plasma levels of trimethylamine-N-oxide can be increased with “healthy” and “unhealthy” diets and do not correlate with the extent of atherosclerosis but with plaque instability. Cardiovasc Res. 2021;117(2):435–49. https://doi.org/10.1093/cvr/cvaa094.
Velasquez MT, Ramezani A, Manal A, Raj DS. Trimethylamine N-oxide: The good, the bad and the unknown. Toxins. 2016;8(11):326. https://doi.org/10.3390/toxins8110326.
Feng Y, Bui TPN, Stams AJM, Boeren S, Sanchez-Andrea I, de Vos WM. Comparative genomics and proteomics of Eubacterium maltosivorans: functional identification of trimethylamine methyltransferases and bacterial microcompartments in a human intestinal bacterium with a versatile lifestyle. Environ Microbiol. 2022;24(1):517–34. https://doi.org/10.1111/1462-2920.15886.
Kountz DJ, Behrman EJ, Zhang L, Krzycki JA. MtcB, a member of the MttB superfamily from the human gut acetogen Eubacterium limosum, is a cobalamin-dependent carnitine demethylase. J Biol Chem. 2020;295(34):11971–81. https://doi.org/10.1074/jbc.RA120.012934.
Picking JW, Behrman EJ, Zhang L, Krzycki JA. MtpB, a member of the MttB superfamily from the human intestinal acetogen Eubacterium limosum, catalyzes proline betaine demethylation. J Biol Chem. 2019;294(37):13697–707. https://doi.org/10.1074/jbc.RA119.009886.
Peppa M, Mavroeidi I. Experimental animal studies support the role of dietary advanced glycation end products in health and disease. Nutrients. 2021;13(10):3467. https://doi.org/10.3390/nu13103467.
Sakellariou S, Fragkou P, Levidou G, Gargalionis AN, Piperi C, Dalagiorgou G, Adamopoulos C, Saetta A, Agrogiannis G, Theohari I, Sougioultzis S, Tsioli P, Karavokyros I, Tsavaris N, Kostakis ID, Zizi-Serbetzoglou A, Vandoros GP, Patsouris E, Korkolopoulou P. Clinical significance of AGE-RAGE axis in colorectal cancer: associations with glyoxalase-I, adiponectin receptor expression and prognosis. BMC Cancer. 2016;16(1):174. https://doi.org/10.1186/s12885-016-2213-5.
Qu W, Yuan X, Zhao J, Zhang Y, Hu J, Wang J, Li J. Dietary advanced glycation end products modify gut microbial composition and partially increase colon permeability in rats. Mol Nutr Food Res. 2017;61(10):1700118. https://doi.org/10.1002/mnfr.201700118.
Wiame E, Delpierre G, Collard F, Van Schaftingen E. Identification of a pathway for the utilization of the Amadori product fructoselysine in Escherichia coli. J Biol Chem. 2002;277(45):42523–9. https://doi.org/10.1074/jbc.M200863200.
Bui TPN, Troise AD, Fogliano V, de Vos WM. Anaerobic degradation of N-epsilon-carboxymethyllysine, a major glycation end-product, by human intestinal bacteria. J Agric Food Chem. 2019;67(23):6594–602. https://doi.org/10.1021/acs.jafc.9b02208.
Molinaro A, Bel Lassen P, Henricsson M, Wu H, Adriouch S, Belda E, Chakaroun R, Nielsen T, Bergh PO, Rouault C, Andre S, Marquet F, Andreelli F, Salem JE, Assmann K, Bastard JP, Forslund S, Le Chatelier E, Falony G, Pons N, Prifti E, Quinquis B, Roume H, Vieira-Silva S, Hansen TH, Pedersen HK, Lewinter C, Sonderskov NB, MetaCardis C, Kober L, Vestergaard H, Hansen T, Zucker JD, Galan P, Dumas ME, Raes J, Oppert JM, Letunic I, Nielsen J, Bork P, Ehrlich SD, Stumvoll M, Pedersen O, Aron-Wisnewsky J, Clement K, Backhed F. Imidazole propionate is increased in diabetes and associated with dietary patterns and altered microbial ecology. Nat Commun. 2020;11(1):5881. https://doi.org/10.1038/s41467-020-19589-w.
Koh A, Molinaro A, Stahlman M, Khan MT, Schmidt C, Manneras-Holm L, Wu H, Carreras A, Jeong H, Olofsson LE, Bergh PO, Gerdes V, Hartstra A, de Brauw M, Perkins R, Nieuwdorp M, Bergstrom G, Backhed F. Microbially produced imidazole propionate impairs insulin signaling through mTORC1. Cell. 2018;175(4):947–61 e917. https://doi.org/10.1016/j.cell.2018.09.055.
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–31. https://doi.org/10.1038/nature05414.
Pinart M, Dotsch A, Schlicht K, Laudes M, Bouwman J, Forslund SK, Pischon T, Nimptsch K. Gut microbiome composition in obese and non-obese persons: A systematic review and meta-analysis. Nutrients. 2021;14(1):12. https://doi.org/10.3390/nu14010012.
Furet JP, Kong LC, Tap J, Poitou C, Basdevant A, Bouillot JL, Mariat D, Corthier G, Dore J, Henegar C, Rizkalla S, Clement K. Differential adaptation of human gut microbiota to bariatric surgery-induced weight loss: links with metabolic and low-grade inflammation markers. Diabetes. 2010;59(12):3049–57. https://doi.org/10.2337/db10-0253.
Liou AP, Paziuk M, Luevano JM Jr, Machineni S, Turnbaugh PJ, Kaplan LM. Conserved shifts in the gut microbiota due to gastric bypass reduce host weight and adiposity. Sci Transl Med. 2013;5(178):178ra141. https://doi.org/10.1126/scitranslmed.3005687.
Ciobarca D, Catoi AF, Copaescu C, Miere D, Crisan G. Bariatric surgery in obesity: Effects on gut microbiota and micronutrient status. Nutrients. 2020;12(1):235. https://doi.org/10.3390/nu12010235.
Michels N, Zouiouich S, Vanderbauwhede B, Vanacker J, Indave Ruiz BI, Huybrechts I. Human microbiome and metabolic health: An overview of systematic reviews. Obes Rev Official J Int Assoc Stud Obes. 2022;23(4):e13409. https://doi.org/10.1111/obr.13409.
Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA. 2004;101(44):15718–23. https://doi.org/10.1073/pnas.0407076101.
Backhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci USA. 2007;104(3):979–84. https://doi.org/10.1073/pnas.0605374104.
Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA. 2005;102(31):11070–5. https://doi.org/10.1073/pnas.0504978102.
Fu J, Bonder MJ, Cenit MC, Tigchelaar EF, Maatman A, Dekens JA, Brandsma E, Marczynska J, Imhann F, Weersma RK, Franke L, Poon TW, Xavier RJ, Gevers D, Hofker MH, Wijmenga C, Zhernakova A. The gut microbiome contributes to a substantial proportion of the variation in blood lipids. Circ Res. 2015;117(9):817–24. https://doi.org/10.1161/CIRCRESAHA.115.306807.
Dreyer JL, Liebl AL. Early colonization of the gut microbiome and its relationship with obesity. Hum Microbiome J. 2018;10:1–5. https://doi.org/10.1016/j.humic.2018.08.002.
Sanchez M, Panahi S, Tremblay A. Childhood obesity: A role for gut microbiota? Int J Environ Res Public Health. 2015;12(1):162–75.
Mueller NT, Bakacs E, Combellick J, Grigoryan Z, Dominguez-Bello MG. The infant microbiome development: mom matters. Trends Mol Med. 2015;21(2):109–17. https://doi.org/10.1016/j.molmed.2014.12.002.
Xu Z, Jiang W, Huang W, Lin Y, Chan FKL, Ng SC. Gut microbiota in patients with obesity and metabolic disorders - a systematic review. Genes Nutr. 2022;17(1):2. https://doi.org/10.1186/s12263-021-00703-6.
Barczynska R, Litwin M, Slizewska K, Szalecki M, Berdowska A, Bandurska K, Libudzisz Z, Kapusniak J. Bacterial microbiota and fatty acids in the faeces of overweight and obese children. Pol J Microbiol. 2018;67(3):339–45. https://doi.org/10.21307/pjm-2018-041.
Magne F, Gotteland M, Gauthier L, Zazueta A, Pesoa S, Navarrete P, Balamurugan R. The firmicutes/bacteroidetes ratio: A relevant marker of gut dysbiosis in obese patients? Nutrients. 2020;12(5). https://doi.org/10.3390/nu12051474.
Wan Y, Yuan J, Li J, Li H, Yin K, Wang F, Li D. Overweight and underweight status are linked to specific gut microbiota and intestinal tricarboxylic acid cycle intermediates. Clin Nutr. 2020;39(10):3189–98. https://doi.org/10.1016/j.clnu.2020.02.014.
Duncan SH, Lobley GE, Holtrop G, Ince J, Johnstone AM, Louis P, Flint HJ. Human colonic microbiota associated with diet, obesity and weight loss. Int J Obes. 2008;32(11):1720–4. https://doi.org/10.1038/ijo.2008.155.
Olivares P, Pacheco ABF, Aranha LN, Oliveira BDS, Santos AA, Santos P, Neto JFN, Rosa G, Oliveira GMM. Gut microbiota of adults with different metabolic phenotypes. Nutrition. 2021;90:111293. https://doi.org/10.1016/j.nut.2021.111293.
Zhong X, Harrington JM, Millar SR, Perry IJ, O'Toole PW, Phillips CM. Gut microbiota associations with metabolic health and obesity status in older adults. Nutrients. 2020;12(8). https://doi.org/10.3390/nu12082364.
Yuan X, Chen R, McCormick KL, Zhang Y, Lin X, Yang X. The role of the gut microbiota on the metabolic status of obese children. Microb Cell Fact. 2021;20(1):53. https://doi.org/10.1186/s12934-021-01548-9.
Chen H, Tang N, Ye Q, Yu X, Yang R, Cheng H, Zhang G, Zhou X. Alternation of the gut microbiota in metabolically healthy obesity: An integrated multiomics analysis. Front Cell Infect Microbiol. 2022;12:1012028. https://doi.org/10.3389/fcimb.2022.1012028.
Zeng Q, Yang Z, Wang F, Li D, Liu Y, Wang D, Zhao X, Li Y, Wang Y, Feng X, Chen J, Li Y, Zheng Y, Kenney T, Gu H, Feng S, Li S, He Y, Xu X, Dai W. Association between metabolic status and gut microbiome in obese populations. Microb Genom. 2021;7(8). https://doi.org/10.1099/mgen.0.000639.
Kim MH, Yun KE, Kim J, Park E, Chang Y, Ryu S, Kim HL, Kim HN. Gut microbiota and metabolic health among overweight and obese individuals. Sci Rep. 2020;10(1):19417. https://doi.org/10.1038/s41598-020-76474-8.
Uema T, Millman JF, Okamoto S, Nakamura T, Yamashiro K, Uehara M, Honma KI, Miyazato M, Ashikari A, Saito S, Maeda S, Imamura M, Ishida H, Matsushita M, Nakamura K, Masuzaki H. Profile of gut microbiota and serum metabolites associated with metabolic syndrome in a remote island most afflicted by obesity in Japan. Sci Rep. 2022;12(1):17292. https://doi.org/10.1038/s41598-022-21708-0.
Alcazar M, Escribano J, Ferre N, Closa-Monasterolo R, Selma-Royo M, Feliu A, Castillejo G, Luque V, and Obemat2.0 Study G. Gut microbiota is associated with metabolic health in children with obesity. Clin Nutr. 2022;41(8):1680–8. https://doi.org/10.1016/j.clnu.2022.06.007.
He Y, Wu W, Zheng HM, Li P, McDonald D, Sheng HF, Chen MX, Chen ZH, Ji GY, Zheng ZD, Mujagond P, Chen XJ, Rong ZH, Chen P, Lyu LY, Wang X, Wu CB, Yu N, Xu YJ, Yin J, Raes J, Knight R, Ma WJ, Zhou HW. Regional variation limits applications of healthy gut microbiome reference ranges and disease models. Nat Med. 2018;24(10):1532–5. https://doi.org/10.1038/s41591-018-0164-x.
Stanislawski MA, Dabelea D, Lange LA, Wagner BD, Lozupone CA. Gut microbiota phenotypes of obesity. NPJ Biofilms Microbiomes. 2019;5(1):18. https://doi.org/10.1038/s41522-019-0091-8.
Haro C, Rangel-Zuniga OA, Alcala-Diaz JF, Gomez-Delgado F, Perez-Martinez P, Delgado-Lista J, Quintana-Navarro GM, Landa BB, Navas-Cortes JA, Tena-Sempere M, Clemente JC, Lopez-Miranda J, Perez-Jimenez F, Camargo A. Intestinal microbiota is influenced by gender and body mass index. PloS One. 2016;11(5):e0154090. https://doi.org/10.1371/journal.pone.0154090.
Kurilshikov A, Medina-Gomez C, Bacigalupe R, Radjabzadeh D, Wang J, Demirkan A, Le Roy CI, Raygoza Garay JA, Finnicum CT, Liu X, Zhernakova DV, Bonder MJ, Hansen TH, Frost F, Ruhlemann MC, Turpin W, Moon JY, Kim HN, Lull K, Barkan E, Shah SA, Fornage M, Szopinska-Tokov J, Wallen ZD, Borisevich D, Agreus L, Andreasson A, Bang C, Bedrani L, Bell JT, Bisgaard H, Boehnke M, Boomsma DI, Burk RD, Claringbould A, Croitoru K, Davies GE, van Duijn CM, Duijts L, Falony G, Fu J, van der Graaf A, Hansen T, Homuth G, Hughes DA, Ijzerman RG, Jackson MA, Jaddoe VWV, Joossens M, Jorgensen T, Keszthelyi D, Knight R, Laakso M, Laudes M, Launer LJ, Lieb W, Lusis AJ, Masclee AAM, Moll HA, Mujagic Z, Qibin Q, Rothschild D, Shin H, Sorensen SJ, Steves CJ, Thorsen J, Timpson NJ, Tito RY, Vieira-Silva S, Volker U, Volzke H, Vosa U, Wade KH, Walter S, Watanabe K, Weiss S, Weiss FU, Weissbrod O, Westra HJ, Willemsen G, Payami H, Jonkers D, Arias Vasquez A, de Geus EJC, Meyer KA, Stokholm J, Segal E, Org E, Wijmenga C, Kim HL, Kaplan RC, Spector TD, Uitterlinden AG, Rivadeneira F, Franke A, Lerch MM, Franke L, Sanna S, D’Amato M, Pedersen O, Paterson AD, Kraaij R, Raes J, Zhernakova A. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat Genet. 2021;53(2):156–65. https://doi.org/10.1038/s41588-020-00763-1.
Silva JSC, Seguro CS, Naves MMV. Gut microbiota and physical exercise in obesity and diabetes - A systematic review. Nutr Metabol Cardiovasc Dis NMCD. 2022;32(4):863–77. https://doi.org/10.1016/j.numecd.2022.01.023.
Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444(7122):1022–3. https://doi.org/10.1038/4441022a.
Turnbaugh PJ, Backhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 2008;3(4):213–23. https://doi.org/10.1016/j.chom.2008.02.015.
Snedeker SM, Hay AG. Do interactions between gut ecology and environmental chemicals contribute to obesity and diabetes? Environ Health Perspect. 2012;120(3):332–9. https://doi.org/10.1289/ehp.1104204.
Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, Camporez JP, Shulman GI, Gordon JI, Hoffman HM, Flavell RA. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482(7384):179–85. https://doi.org/10.1038/nature10809.
Murphy EF, Cotter PD, Healy S, Marques TM, O’Sullivan O, Fouhy F, Clarke SF, O’Toole PW, Quigley EM, Stanton C, Ross PR, O’Doherty RM, Shanahan F. Composition and energy harvesting capacity of the gut microbiota: relationship to diet, obesity and time in mouse models. Gut. 2010;59(12):1635–42. https://doi.org/10.1136/gut.2010.215665.
Grases-Pinto B, Abril-Gil M, Castell M, Rodriguez-Lagunas MJ, Burleigh S, Fak Hallenius F, Prykhodko O, Perez-Cano FJ, Franch A. Influence of leptin and adiponectin supplementation on intraepithelial lymphocyte and microbiota composition in suckling rats. Front Immunol. 2019;10:2369. https://doi.org/10.3389/fimmu.2019.02369.
Kasubuchi M, Hasegawa S, Hiramatsu T, Ichimura A, Kimura I. Dietary gut microbial metabolites, short-chain fatty acids, and host metabolic regulation. Nutrients. 2015;7(4):2839–49. https://doi.org/10.3390/nu7042839.
Sihag J, Di Marzo V. (Wh)olistic (E)ndocannabinoidome-microbiome-axis modulation through (N)utrition (WHEN) to curb obesity and related disorders. Lipids Health Dis. 2022;21(1):9. https://doi.org/10.1186/s12944-021-01609-3.
Iannotti FA, Di Marzo V. The gut microbiome, endocannabinoids and metabolic disorders. J Endocrinol. 2021;248(2):R83–97. https://doi.org/10.1530/JOE-20-0444.
Chang FY, Siuti P, Laurent S, Williams T, Glassey E, Sailer AW, Gordon DB, Hemmerle H, Voigt CA. Gut-inhabiting Clostridia build human GPCR ligands by conjugating neurotransmitters with diet- and human-derived fatty acids. Nat Microbiol. 2021;6(6):792–805. https://doi.org/10.1038/s41564-021-00887-y.
Payahoo L, Khajebishak Y, Alivand MR, Soleimanzade H, Alipour S, Barzegari A, Ostadrahimi A. Investigation the effect of oleoylethanolamide supplementation on the abundance of Akkermansia muciniphila bacterium and the dietary intakes in people with obesity: A randomized clinical trial. Appetite. 2019;141:104301. https://doi.org/10.1016/j.appet.2019.05.032.
Tagliamonte S, Laiola M, Ferracane R, Vitale M, Gallo MA, Meslier V, Pons N, Ercolini D, Vitaglione P. Mediterranean diet consumption affects the endocannabinoid system in overweight and obese subjects: possible links with gut microbiome, insulin resistance and inflammation. Eur J Nutr. 2021;60(7):3703–16. https://doi.org/10.1007/s00394-021-02538-8.
Dohnalova L, Lundgren P, Carty JRE, Goldstein N, Wenski SL, Nanudorn P, Thiengmag S, Huang KP, Litichevskiy L, Descamps HC, Chellappa K, Glassman A, Kessler S, Kim J, Cox TO, Dmitrieva-Posocco O, Wong AC, Allman EL, Ghosh S, Sharma N, Sengupta K, Cornes B, Dean N, Churchill GA, Khurana TS, Sellmyer MA, FitzGerald GA, Patterson AD, Baur JA, Alhadeff AL, Helfrich EJN, Levy M, Betley JN, Thaiss CA. A microbiome-dependent gut-brain pathway regulates motivation for exercise. Nature. 2022;612(7941):739–47. https://doi.org/10.1038/s41586-022-05525-z.
Biedermann L, Zeitz J, Mwinyi J, Sutter-Minder E, Rehman A, Ott SJ, Steurer-Stey C, Frei A, Frei P, Scharl M, Loessner MJ, Vavricka SR, Fried M, Schreiber S, Schuppler M, Rogler G. Smoking cessation induces profound changes in the composition of the intestinal microbiota in humans. PloS One. 2013;8(3):e59260. https://doi.org/10.1371/journal.pone.0059260.
Mutlu EA, Gillevet PM, Rangwala H, Sikaroodi M, Naqvi A, Engen PA, Kwasny M, Lau CK, Keshavarzian A. Colonic microbiome is altered in alcoholism. Am J Physiol Gastrointest Liver Physiol. 2012;302(9):G966-978. https://doi.org/10.1152/ajpgi.00380.2011.
Gao B, Chi L, Mahbub R, Bian X, Tu P, Ru H, Lu K. Multi-omics reveals that lead exposure disturbs gut microbiome development, key metabolites, and metabolic pathways. Chem Res Toxicol. 2017;30(4):996–1005. https://doi.org/10.1021/acs.chemrestox.6b00401.
Gao B, Bian X, Mahbub R, Lu K. Sex-specific effects of organophosphate diazinon on the gut microbiome and its metabolic functions. Environ Health Perspect. 2017;125(2):198–206. https://doi.org/10.1289/EHP202.
Joly C, Gay-Queheillard J, Leke A, Chardon K, Delanaud S, Bach V, Khorsi-Cauet H. Impact of chronic exposure to low doses of chlorpyrifos on the intestinal microbiota in the Simulator of the Human Intestinal Microbial Ecosystem (SHIME) and in the rat. Environ Sci Pollut Res Int. 2013;20(5):2726–34. https://doi.org/10.1007/s11356-012-1283-4.
Joly Condette C, Bach V, Mayeur C, Gay-Queheillard J, Khorsi-Cauet H. Chlorpyrifos exposure during perinatal period affects intestinal microbiota associated with delay of maturation of digestive tract in rats. J Pediatr Gastroenterol Nutr. 2015;61(1):30–40. https://doi.org/10.1097/MPG.0000000000000734.
Wei M, Huang F, Zhao L, Zhang Y, Yang W, Wang S, Li M, Han X, Ge K, Qu C, Rajani C, Xie G, Zheng X, Zhao A, Bian Z, Jia W. A dysregulated bile acid-gut microbiota axis contributes to obesity susceptibility. EBioMedicine. 2020;55:102766. https://doi.org/10.1016/j.ebiom.2020.102766.
Bustamante JM, Dawson T, Loeffler C, Marfori Z, Marchesi JR, Mullish BH, Thompson CC, Crandall KA, Rahnavard A, Allegretti JR, Cummings BP. Impact of fecal microbiota transplantation on gut bacterial bile acid metabolism in humans. Nutrients. 2022;14(24). https://doi.org/10.3390/nu14245200.
Jiang C, Xie C, Li F, Zhang L, Nichols RG, Krausz KW, Cai J, Qi Y, Fang ZZ, Takahashi S, Tanaka N, Desai D, Amin SG, Albert I, Patterson AD, Gonzalez FJ. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J Clin Investig. 2015;125(1):386–402. https://doi.org/10.1172/JCI76738.
Li F, Jiang C, Krausz KW, Li Y, Albert I, Hao H, Fabre KM, Mitchell JB, Patterson AD, Gonzalez FJ. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat Commun. 2013;4:2384. https://doi.org/10.1038/ncomms3384.
Trabelsi MS, Daoudi M, Prawitt J, Ducastel S, Touche V, Sayin SI, Perino A, Brighton CA, Sebti Y, Kluza J, Briand O, Dehondt H, Vallez E, Dorchies E, Baud G, Spinelli V, Hennuyer N, Caron S, Bantubungi K, Caiazzo R, Reimann F, Marchetti P, Lefebvre P, Backhed F, Gribble FM, Schoonjans K, Pattou F, Tailleux A, Staels B, Lestavel S. Farnesoid X receptor inhibits glucagon-like peptide-1 production by enteroendocrine L cells. Nat Commun. 2015;6(1):7629. https://doi.org/10.1038/ncomms8629.
Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, Macchiarulo A, Yamamoto H, Mataki C, Pruzanski M, Pellicciari R, Auwerx J, Schoonjans K. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metabol. 2009;10(3):167–77. https://doi.org/10.1016/j.cmet.2009.08.001.
van Nierop FS, Kulik W, Endert E, Schaap FG, Olde Damink SW, Romijn JA, Soeters MR. Effects of acute dietary weight loss on postprandial plasma bile acid responses in obese insulin resistant subjects. Clin Nutr. 2017;36(6):1615–20. https://doi.org/10.1016/j.clnu.2016.10.006.
Li M, Wang S, Li Y, Zhao M, Kuang J, Liang D, Wang J, Wei M, Rajani C, Ma X, Tang Y, Ren Z, Chen T, Zhao A, Hu C, Shen C, Jia W, Liu P, Zheng X, Jia W. Gut microbiota-bile acid crosstalk contributes to the rebound weight gain after calorie restriction in mice. Nat Commun. 2022;13(1):2060. https://doi.org/10.1038/s41467-022-29589-7.
Le Roy T, Moens de Hase E, Van Hul M, Paquot A, Pelicaen R, Regnier M, Depommier C, Druart C, Everard A, Maiter D, Delzenne NM, Bindels LB, de Barsy M, Loumaye A, Hermans MP, Thissen JP, Vieira-Silva S, Falony G, Raes J, Muccioli GG, Cani PD. Dysosmobacter welbionis is a newly isolated human commensal bacterium preventing diet-induced obesity and metabolic disorders in mice. Gut. 2022;71(3):534–43. https://doi.org/10.1136/gutjnl-2020-323778.
Vitale M, Giacco R, Laiola M, Della Pepa G, Luongo D, Mangione A, Salamone D, Vitaglione P, Ercolini D, Rivellese AA. Acute and chronic improvement in postprandial glucose metabolism by a diet resembling the traditional Mediterranean dietary pattern: Can SCFAs play a role? Clin Nutr. 2021;40(2):428–37.
Gao K, Mu CL, Farzi A, Zhu WY. Tryptophan metabolism: A link between the gut microbiota and brain. Adv Nutr. 2020;11(3):709–23. https://doi.org/10.1093/advances/nmz127.
Agus A, Planchais J, Sokol H. Gut microbiota regulation of tryptophan metabolism in health and disease. Cell Host Microbe. 2018;23(6):716–24. https://doi.org/10.1016/j.chom.2018.05.003.
Wurtman RJ, Wurtman JJ. Brain serotonin, carbohydrate-craving, obesity and depression. Obes Res. 1995;3(Suppl 4):477S-480S. https://doi.org/10.1002/j.1550-8528.1995.tb00215.x.
Bertrand RL, Senadheera S, Markus I, Liu L, Howitt L, Chen H, Murphy TV, Sandow SL, Bertrand PP. A Western diet increases serotonin availability in rat small intestine. Endocrinology. 2011;152(1):36–47. https://doi.org/10.1210/en.2010-0377.
Crane JD, Palanivel R, Mottillo EP, Bujak AL, Wang H, Ford RJ, Collins A, Blumer RM, Fullerton MD, Yabut JM, Kim JJ, Ghia JE, Hamza SM, Morrison KM, Schertzer JD, Dyck JR, Khan WI, Steinberg GR. Inhibiting peripheral serotonin synthesis reduces obesity and metabolic dysfunction by promoting brown adipose tissue thermogenesis. Nat Med. 2015;21(2):166–72. https://doi.org/10.1038/nm.3766.
Kim HJ, Kim JH, Noh S, Hur HJ, Sung MJ, Hwang JT, Park JH, Yang HJ, Kim MS, Kwon DY, Yoon SH. Metabolomic analysis of livers and serum from high-fat diet induced obese mice. J Proteome Res. 2011;10(2):722–31. https://doi.org/10.1021/pr100892r.
Young RL, Lumsden AL, Martin AM, Schober G, Pezos N, Thazhath SS, Isaacs NJ, Cvijanovic N, Sun EWL, Wu T, Rayner CK, Nguyen NQ, Fontgalland D, Rabbitt P, Hollington P, Sposato L, Due SL, Wattchow DA, Liou AP, Jackson VM, Keating DJ. Augmented capacity for peripheral serotonin release in human obesity. Int J Obes. 2018;42(11):1880–9. https://doi.org/10.1038/s41366-018-0047-8.
Woting A, Pfeiffer N, Loh G, Klaus S, Blaut M. Clostridium ramosum promotes high-fat diet-induced obesity in gnotobiotic mouse models. mBio. 2014;5(5):e01530–01514. https://doi.org/10.1128/mBio.01530-14.
Mandic AD, Woting A, Jaenicke T, Sander A, Sabrowski W, Rolle-Kampcyk U, von Bergen M, Blaut M. Clostridium ramosum regulates enterochromaffin cell development and serotonin release. Sci Rep. 2019;9(1):1177. https://doi.org/10.1038/s41598-018-38018-z.
Pyun DH, Kim TJ, Kim MJ, Hong SA, Abd El-Aty AM, Jeong JH, Jung TW. Endogenous metabolite, kynurenic acid, attenuates nonalcoholic fatty liver disease via AMPK/autophagy- and AMPK/ORP150-mediated signaling. J Cell Physiol. 2021;236(7):4902–12. https://doi.org/10.1002/jcp.30199.
Favennec M, Hennart B, Caiazzo R, Leloire A, Yengo L, Verbanck M, Arredouani A, Marre M, Pigeyre M, Bessede A, Guillemin GJ, Chinetti G, Staels B, Pattou F, Balkau B, Allorge D, Froguel P, Poulain-Godefroy O. The kynurenine pathway is activated in human obesity and shifted toward kynurenine monooxygenase activation. Obesity. 2015;23(10):2066–74. https://doi.org/10.1002/oby.21199.
Kubacka J, Staniszewska M, Sadok I, Sypniewska G, Stefanska A. The kynurenine pathway in obese middle-aged women with normoglycemia and type 2 diabetes. Metabolites. 2022;12(6). https://doi.org/10.3390/metabo12060492.
Tan KM, Tint MT, Kothandaraman N, Yap F, Godfrey KM, Lee YS, Tan KH, Gluckman PD, Chong YS, Chong MFF, Eriksson JG, Cameron-Smith D. Association of plasma kynurenine pathway metabolite concentrations with metabolic health risk in prepubertal Asian children. Int J Obes. 2022;46(6):1128–37. https://doi.org/10.1038/s41366-022-01085-4.
Dolecka J, Urbanik-Sypniewska T, Skrzydlo-Radomanska B, Parada-Turska J. Effect of kynurenic acid on the viability of probiotics in vitro. Pharmacol Rep. 2011;63(2):548–51. https://doi.org/10.1016/s1734-1140(11)70522-9.
Chimerel C, Emery E, Summers DK, Keyser U, Gribble FM, Reimann F. Bacterial metabolite indole modulates incretin secretion from intestinal enteroendocrine L cells. Cell Rep. 2014;9(4):1202–8. https://doi.org/10.1016/j.celrep.2014.10.032.
Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, Zecchi R, D’Angelo C, Massi-Benedetti C, Fallarino F, Carvalho A, Puccetti P, Romani L. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity. 2013;39(2):372–85. https://doi.org/10.1016/j.immuni.2013.08.003.
Postal BG, Ghezzal S, Aguanno D, Andre S, Garbin K, Genser L, Brot-Laroche E, Poitou C, Soula H, Leturque A, Clement K, Carriere V. AhR activation defends gut barrier integrity against damage occurring in obesity. Mol Metab. 2020;39:101007. https://doi.org/10.1016/j.molmet.2020.101007.
Malesza IJ, Malesza M, Walkowiak J, Mussin N, Walkowiak D, Aringazina R, Bartkowiak-Wieczorek J, Madry E. High-fat, western-style diet, systemic inflammation, and gut microbiota: A narrative review. Cells. 2021;10(11). https://doi.org/10.3390/cells10113164.
Suriano F, Vieira-Silva S, Falony G, de Wouters d’Oplinter A, Paone P, Delzenne NM, Everard A, Raes J, Van Hul M, Cani PD. Fat and not sugar as the determining factor for gut microbiota changes, obesity, and related metabolic disorders in mice. American journal of physiology Endocrinol Metabol. 2023;324(1):E85–96. https://doi.org/10.1152/ajpendo.00141.2022.
Wolters M, Ahrens J, Romani-Perez M, Watkins C, Sanz Y, Benitez-Paez A, Stanton C, Gunther K. Dietary fat, the gut microbiota, and metabolic health - A systematic review conducted within the MyNewGut project. Clin Nutr. 2019;38(6):2504–20. https://doi.org/10.1016/j.clnu.2018.12.024.
Caesar R, Tremaroli V, Kovatcheva-Datchary P, Cani PD, Backhed F. Crosstalk between gut microbiota and dietary lipids aggravates WAT inflammation through TLR signaling. Cell Metabol. 2015;22(4):658–68. https://doi.org/10.1016/j.cmet.2015.07.026.
Cani PD, Depommier C, Derrien M, Everard A, de Vos WM. Author Correction: Akkermansia muciniphila: paradigm for next-generation beneficial microorganisms. Nat Rev Gastroenterol Hepatol. 2022;19(10):682. https://doi.org/10.1038/s41575-022-00650-6.
Meslier V, Laiola M, Roager HM, De Filippis F, Roume H, Quinquis B, Giacco R, Mennella I, Ferracane R, Pons N, Pasolli E, Rivellese A, Dragsted LO, Vitaglione P, Ehrlich SD, Ercolini D. Mediterranean diet intervention in overweight and obese subjects lowers plasma cholesterol and causes changes in the gut microbiome and metabolome independently of energy intake. Gut. 2020;69(7):1258–68. https://doi.org/10.1136/gutjnl-2019-320438.
Van Hul M, Le Roy T, Prifti E, Dao MC, Paquot A, Zucker JD, Delzenne NM, Muccioli G, Clement K, Cani PD. From correlation to causality: the case of Subdoligranulum. Gut Microbes. 2020;12(1):1–13. https://doi.org/10.1080/19490976.2020.1849998.
Welton S, Minty R, O’Driscoll T, Willms H, Poirier D, Madden S, Kelly L. Intermittent fasting and weight loss: Systematic review. Can Fam Phys. 2020;66(2):117–25.
Khan MN, Khan SI, Rana MI, Ayyaz A, Khan MY, Imran M. Intermittent fasting positively modulates human gut microbial diversity and ameliorates blood lipid profile. Front Microbiol. 2022;13:922727. https://doi.org/10.3389/fmicb.2022.922727.
Ozkul C, Yalinay M, Karakan T. Structural changes in gut microbiome after Ramadan fasting: a pilot study. Benef Microbes. 2020;11(3):227–33. https://doi.org/10.3920/BM2019.0039.
Su J, Wang Y, Zhang X, Ma M, Xie Z, Pan Q, Ma Z, Peppelenbosch MP. Remodeling of the gut microbiome during Ramadan-associated intermittent fasting. Am J Clin Nutr. 2021;113(5):1332–42. https://doi.org/10.1093/ajcn/nqaa388.
Hughes RL, Pindus DM, Khan NA, Burd NA, Holscher HD. Associations between accelerometer-measured physical activity and fecal microbiota in adults with overweight and obesity. Med Sci Sports Exerc. 2022. https://doi.org/10.1249/MSS.0000000000003096.
Savin Z, Kivity S, Yonath H, Yehuda S. Smoking and the intestinal microbiome. Arch Microbiol. 2018;200(5):677–84. https://doi.org/10.1007/s00203-018-1506-2.
Fluhr L, Mor U, Kolodziejczyk AA, Dori-Bachash M, Leshem A, Itav S, Cohen Y, Suez J, Zmora N, Moresi C, Molina S, Ayalon N, Valdes-Mas R, Hornstein S, Karbi H, Kviatcovsky D, Livne A, Bukimer A, Eliyahu-Miller S, Metz A, Brandis A, Mehlman T, Kuperman Y, Tsoory M, Stettner N, Harmelin A, Shapiro H, Elinav E. Publisher Correction: Gut microbiota modulates weight gain in mice after discontinued smoke exposure. Nature. 2022;603(7903):E35. https://doi.org/10.1038/s41586-022-04611-6.
Popli S, Badgujar PC, Agarwal T, Bhushan B, Mishra V. Persistent organic pollutants in foods, their interplay with gut microbiota and resultant toxicity. Scie Total Environ. 2022;832:155084. https://doi.org/10.1016/j.scitotenv.2022.155084.
Liang Y, Zhan J, Liu D, Luo M, Han J, Liu X, Liu C, Cheng Z, Zhou Z, Wang P. Organophosphorus pesticide chlorpyrifos intake promotes obesity and insulin resistance through impacting gut and gut microbiota. Microbiome. 2019;7(1):19. https://doi.org/10.1186/s40168-019-0635-4.
Chen AS, Liu DH, Hou HN, Yao JN, Xiao SC, Ma XR, Li PZ, Cao Q, Liu XK, Zhou ZQ, Wang P. Dietary pattern interfered with the impacts of pesticide exposure by regulating the bioavailability and gut microbiota. Sci Total Environ. 2023;858(Pt 2):159936. https://doi.org/10.1016/j.scitotenv.2022.159936.
Lee HS, Lee JC, Lee IK, Moon HB, Chang YS, Jacobs DR Jr, Lee DH. Associations among organochlorine pesticides, methanobacteriales, and obesity in Korean women. PloS One. 2011;6(11):e27773. https://doi.org/10.1371/journal.pone.0027773.
Zhang H, DiBaise JK, Zuccolo A, Kudrna D, Braidotti M, Yu Y, Parameswaran P, Crowell MD, Wing R, Rittmann BE, Krajmalnik-Brown R. Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci USA. 2009;106(7):2365–70. https://doi.org/10.1073/pnas.0812600106.
Ba Q, Li M, Chen P, Huang C, Duan X, Lu L, Li J, Chu R, Xie D, Song H, Wu Y, Ying H, Jia X, Wang H. Sex-dependent effects of cadmium exposure in early life on gut microbiota and fat accumulation in mice. Environ Health Perspect. 2017;125(3):437–46. https://doi.org/10.1289/EHP360.
Wang N, Sheng Z, Zhou S, Jiang F, Zhang Z. Chronic lead exposure exacerbates hepatic glucolipid metabolism disorder and gut microbiota dysbiosis in high-fat-diet mice. Food Chem Toxicol Int J Publ Br Ind Biol Re Assoc. 2022;170:113451. https://doi.org/10.1016/j.fct.2022.113451.
Malaise Y, Menard S, Cartier C, Gaultier E, Lasserre F, Lencina C, Harkat C, Geoffre N, Lakhal L, Castan I, Olier M, Houdeau E, Guzylack-Piriou L. Gut dysbiosis and impairment of immune system homeostasis in perinatally-exposed mice to Bisphenol A precede obese phenotype development. Sci Rep. 2017;7(1):14472. https://doi.org/10.1038/s41598-017-15196-w.
Su H, Yuan P, Lei H, Zhang L, Deng D, Zhang L, Chen X. Long-term chronic exposure to di-(2-ethylhexyl)-phthalate induces obesity via disruption of host lipid metabolism and gut microbiota in mice. Chemosphere. 2022;287(Pt 4):132414. https://doi.org/10.1016/j.chemosphere.2021.132414.
Yueh MF, He F, Chen C, Vu C, Tripathi A, Knight R, Karin M, Chen S, Tukey RH. Triclosan leads to dysregulation of the metabolic regulator FGF21 exacerbating high fat diet-induced nonalcoholic fatty liver disease. Proc Natl Acad USA. 2020;117(49):31259–66. https://doi.org/10.1073/pnas.2017129117.
Maruvada P, Leone V, Kaplan LM, Chang EB. The human microbiome and obesity: Moving beyond associations. Cell Host Microbe. 2017;22(5):589–99. https://doi.org/10.1016/j.chom.2017.10.005.
Hjorth MF, Blaedel T, Bendtsen LQ, Lorenzen JK, Holm JB, Kiilerich P, Roager HM, Kristiansen K, Larsen LH, Astrup A. Prevotella-to-Bacteroides ratio predicts body weight and fat loss success on 24-week diets varying in macronutrient composition and dietary fiber: results from a post-hoc analysis. Int J Obes. 2019;43(1):149–57. https://doi.org/10.1038/s41366-018-0093-2.
Mathur R, Barlow GM. Obesity and the microbiome. Exp Rev Gastroenterol Hepatol. 2015;9(8):1087–99. https://doi.org/10.1586/17474124.2015.1051029.
Rosenbaum M, Knight R, Leibel RL. The gut microbiota in human energy homeostasis and obesity. Trends Endocrinol Metabol TEM. 2015;26(9):493–501. https://doi.org/10.1016/j.tem.2015.07.002.
Acknowledgements
This paper has been partly supported by funding to P.P. from the European Union’s Horizon 2020 Research and Innovation program under the Marie Skłodowska-Curie Grant Agreement No. 722619 (FOIE GRAS), Grant Agreement No. 734719 (mtFOIE GRAS), Grant Regione Puglia, CUP H99C20000340002 (Fever Apulia), and Grant EUROSEEDS Uniba—S56—By-products Sustainable Recovery 4 Health (BSR-4H): University of Bari Aldo Moro, 2022. The project SYS-TEMIC “an integrated approach to the challenge of sustainable food systems: adaptive and mitigatory strategies to address climate change and malnutrition", Knowledge hub on Nutrition and Food Security, has received funding from national research funding parties in Belgium (FWO), France (INRA), Germany (BLE), Italy (MIPAAF), Latvia (IZM), Norway (RCN), Portugal (FCT), and Spain (AEI) in a joint action of JPI HDHL, JPI-OCEANS and FACCE-JPI launched in 2019 under the ERA-NET ERA-HDHL (n° 696295). PP is grant recipient in Project funded under the National Recovery and Resilience Plan (NRRP), Mission 4 Component 2 Investment 1.3—Call for tender No. 341 of 15/03/2022 of Italian Ministry of University and Research funded by the European Union – NextGenerationEU Award Number: Project code PE0000003, Concession Decree No. 1550 of 11/10/2022 adopted by the Italian Ministry of University and Research, CUP D93C22000890001, Project title “Research and innovation network on food and nutrition Sustainability, Safety and Security – Working ON Foods” (ONFoods). The authors are indebted to the Consortium of Mediterranean Universities (CMU), Bari (www.cmungo.eu) for helpful discussion. The figures were partly generated by using materials from Servier Medical Art (Servier) under consideration of a Creative Commons Attribution 3.0 Unported License.
Author information
Authors and Affiliations
Contributions
ADC and PP conceptualized the article, LB, MK, and GG performed the literature search, ADC and LB provided the first draft, PP critically revised the work.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest in the authorship or publication of this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Di Ciaula, A., Bonfrate, L., Khalil, M. et al. Contribution of the microbiome for better phenotyping of people living with obesity. Rev Endocr Metab Disord 24, 839–870 (2023). https://doi.org/10.1007/s11154-023-09798-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11154-023-09798-1