
Abstract
Both somatostatin (SST) and somatostatin receptors (SSTRs) are proteins with important functions in both physiological tissue and in tumors, particularly in neuroendocrine tumors (NETs). NETs are frequently characterized by high SSTRs expression levels. SST analogues (SSAs) that bind and activate SSTR have anti-proliferative and anti-secretory activity, thereby reducing both the growth as well as the hormonal symptoms of NETs. Moreover, the high expression levels of SSTR type-2 (SSTR2) in NETs is a powerful target for therapy with radiolabeled SSAs. Due to the important role of both SST and SSTRs, it is of great importance to elucidate the mechanisms involved in regulating their expression in NETs, as well as in other types of tumors. The field of epigenetics recently gained interest in NET research, highlighting the importance of this process in regulating the expression of gene and protein expression. In this review we will discuss the role of the epigenetic machinery in controlling the expression of both SSTRs and the neuropeptide SST. Particular attention will be given to the epigenetic regulation of these proteins in NETs, whereas the involvement of the epigenetic machinery in other types of cancer will be discussed as well. In addition, we will discuss the possibility to target enzymes involved in the epigenetic machinery to modify the expression of the SST-system, thereby possibly improving therapeutic options.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Somatostatin receptors (SSTRs) are a family of G protein coupled receptors, of which different subtypes exist, i.e. SSTR1, SSTR2, SSTR3, SSTR4 and SSTR5. Alternative splicing of SSTR2 RNA generates two splice variants: SSTR2a and SSTR2b which differ in length. SSTRs can be activated by the neuropeptide somatostatin (SST), of which two isoforms are known, i.e. somatostatin-14 (SST-14) and somatostatin-28 (SST-28), both having high affinity for SSTRs [1, 2]. SST is produced by different organs in both the central nervous system, e.g. hypothalamus, and in other organs including pancreas, stomach and intestine. It is synthesized in response to multiple biological signals, for instance neurotransmitters, hormones and neuropeptides [3]. SSTR-expressing cells are found abundantly in human tissues, such as the brain, pituitary and the gastrointestinal tract [4]. The SST-system is therefore involved in regulating multiple physiological processes. This is mediated via several pathways that are activated upon binding of SST to SSTRs, which results in either inhibition of hormone secretion and cell proliferation, or induction of apoptosis [5, 6].
SSTR-mediated anti-secretory effects are induced via two main pathways: (1) inhibition of adenylyl cyclase (AC) resulting in reduced levels of cyclic AMP (cAMP) and (2) activation of K+-channels and inhibition of voltage-dependent Ca2+-channels resulting in reduced intracellular Ca2+ levels (Fig. 1a). Upon binding of SST to SSTR, src-homology phosphatase (SHP) proteins are activated which are involved in regulating cell proliferation and apoptosis. SHP type-1 (SHP-1) is involved in inducing apoptosis by increasing pro-apoptotic proteins such as the p53-Bax-caspase-3 pathway and by increasing JNK expression resulting in inhibition of anti-apoptotic proteins (Fig. 1b). SHP type-2 (SHP-2) activation results in Src activity, which phosphorylates PTPη. As a result, MAPK/ERK and PI3K/AKT proteins will be inactivated, causing upregulation of proteins involved in inhibiting proliferation (Fig. 1b).

(a) Upon binding of SST or SSAs, (1,2) K+ channels are activated and Ca2+ channels are inhibited, resulting in decreased Ca2+ levels, and (3) adenylyl cyclase (AC) activity is inhibited thereby reducing intracellular cAMP levels. This results in inhibition of hormone secretion. (b) Activation of the SST-system also results in anti-tumoral activity: (1) SHP-1 is activated, thereby increasing pro-apoptotic and reducing anti-apoptotic proteins, and (2) SHP-2 is activated which results in activation of PTPη by Src-mediated phosphorylation. PTPη causes inhibition of pathways physiologically involved in cell proliferation. In both figure a and b, effects induced by SSTR activation are indicated by green (enhanced) or red (reduced) arrows
In addition to its pivotal role in physiological processes, the SST-system also plays an important role in neuroendocrine tumors (NETs). SSTR2 is highly expressed on NETs, thereby being an important target for therapy [7, 8]. Moreover, aberrant SSTR expression has also been reported for other cancers, including breast cancer [9], colorectal cancer [10], prostate cancer [11, 12] and larynx cancer [13]. For prostate cancer, it was demonstrated that SSTR2 and SSTR5 are mostly expressed, i.e. in 34.8% and 56.5%, respectively [12]. In contrast, SSTR1 was expressed abundantly in 90% of primary breast cancer tissues, whereas SSTR2 and SSTR5 were expressed in a lower number of cases, i.e. in 34.4% and 44.4% of the examined cases, respectively [14]. In this review, we aim to highlight the role of epigenetic mechanisms involved in the regulation of the SST-system, consisting of both SSTRs and the neuropeptide SST. We will focus on the regulation of both proteins in different types of tumors, with particular emphasis on NETs. Moreover, we will discuss the possibility to target the epigenetic machinery in order to modulate either SSTR or SST expression. An improved understanding of the epigenetic regulation involved in the SST-system may result in new approaches to either improve the efficacy of current treatments or expand therapeutic options for patients with tumors expression low SSTR levels.
2 Somatostatin and somatostatin receptors in NETs
NETs arise from neuroendocrine cells found throughout the entire body. Consequently, NETs are a heterogeneous disease that can develop at different locations, such as the gastrointestinal tract, pancreas and lung, accounting for 54%, 22% and 12% of the NETs, respectively [15]. NETs are divided in functional and non-functional tumors. Functional NETs are frequently characterized by an overproduction of hormones, such as serotonin, gastrin or insulin. As the tumor is frequently already metastasized upon diagnosis [15], treatment options are limited. Currently, resection is still the only curative therapy option [16], and is only possible for the minority of NET patients due to the presence of metastases.
In the past decades, several treatment options were developed that improved patient outcome [17], of which some target the SSTR2. In literature, a high expression of SSTR2 has been described in NETs. For example, in a study performed by Mizutani et al. [18], SSTR mRNA levels were measured for 13 NET samples derived from several locations. It was shown that SSTR2 is expressed in all cases. In another study, examining 112 small intestine NETs (siNETs), 19 pancreatic NETs (pNETs) and 42 NETs derived from other locations, it was demonstrated that 65%, 76%, 90%, 86% and 93% of all cases were recognized by the expression of SSTR1, SSTR2, SSTR3, SSTR4 and SSTR5, respectively [7]. Upon discriminating low and high expression levels, SSTR2 was expressed most frequently, i.e. 51% of the examined NETs were recognized by high SSTR2 levels. Especially, pNETs and siNETs were most frequently characterized by the expression of SSTR2. These high SSTR2 expression levels paved the way for SSTR2-targeted treatments in NETs, including unlabeled somatostatin analogues (SSAs) [19] and peptide receptor radionuclide therapy (PRRT) [20, 21]. SSAs have potent anti-secretory effects and thereby reduce symptoms related to the overproduction of bioactive substances by tumors in a significant proportion of NET patients [22, 23]. In addition, various studies have demonstrated that SSAs have tumor growth inhibitory actions in NET patients. Octreotide long-acting release (LAR) and lanreotide autogel are both SSAs with high affinity for SSTR2 [24]. Patients with well-differentiated metastatic midgut NETs benefited from octreotide LAR treatment, as demonstrated in a placebo-controlled study with 85 patients enrolled. This study showed a significantly increased time to progression from 6 to 14.3 months between the control and octreotide LAR treatment groups, respectively [25]. Similar, a phase III study with lanreotide autogel in metastatic enteropancreatic NET patients reported a significantly increased progression-free survival compared to placebo [26].
The generation of radiolabeled SSAs further improved treatment options for patients with SSTR-positive tumors. PRRT with radiolabeled SSAs induced significant anti-tumor effects in NET patients with metastatic SSTR-expressing bronchial and gastroenteropancreatic NETs, in terms of response rates, progression-free survival, overall survival and safety profile [27, 28]. In the study by Brabander et al. [27], the objective tumor response rate was 39%, achieving 2% complete response and 37% partial response. Median overall survival rates depend on the primary location of the tumor, i.e. 52, 60 and 71 months for bronchial, midgut and pancreatic NETs, respectively. In the NETTER-I phase III clinical trial, it was demonstrated that 4 cycles of PRRT with [177Lu]Lu-[DOTA,Tyr3]-octreotide ([177Lu]Lu-DOTA-TATE) in combination with octreotide LAR resulted in longer progression-free survival and higher response rates in patients with advanced midgut NETs, compared to high-dose octreotide LAR treatment alone [29]. Together, these results led to the FDA and EMA approval of [177Lu]Lu-DOTA-TATE for SSTR-positive gastroenteropancreatic NETs. Additionally, studies demonstrated a possible role of PRRT as neo-adjuvant treatment option for non-functioning pNETs [30], emphasizing the broad scope of PRRT. Although treatment with SSAs and PRRT have both clearly proven their value for the treatment of NETs, complete responses are still rare [25, 31], leaving room for therapy improvement. Moreover, not all NET patients are eligible for SSTR-targeted treatments due to variable SSTR2 expression levels among patients [32, 33]. NETs display a low frequency of mutations and chromosomal aberrations [34], and no mutations in SSTR2 have been identified thus far. This suggests other mechanism(s) underlying these heterogeneous SSTR expression levels, e.g. epigenetic regulation.
2.1 Epigenetic regulation
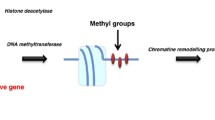
Cell-specific gene transcription is regulated by epigenetic modifications which are heritable during cell divisions. However, as the DNA sequence itself is not changed, these modifications are also characterized by their reversibility [35, 36]. Over the last years, epigenetic modifications have become an important field of interest, demonstrating their pivotal role in physiological processes, such as their role in cell differentiation and development [37]. Moreover, thorough investigations have demonstrated that the epigenetic machinery is also involved in the development of diseases, e.g. neurodevelopmental disorders and autoimmune diseases, as well as the development of cancer [38,39,40,41]. For its role in the development of cancer, the epigenetic machinery is involved in the activation of proto-oncogenes and/or inactivation of tumor suppressor genes, such as RB, P16 and BRCA1 [41]. Epigenetic modifications can regulate gene expression at different levels. In this review, we will specifically focus on modifications targeting the DNA and the histones (Fig. 2a).

(a) Epigenetic modifications can modify both DNA and histones. DNA methylation and inactivating histone methylation stimulate heterochromatin, resulting in inaccessible DNA and therefore no gene transcription. Histone acetylation and activating histone methylation stimulate euchromatin, thereby stimulating gene transcription. Histone methylation can therefore both be inactivating and activating, depending on which lysine residue is modified on which histone. Examples of inactivating histone methylation marks (indicated in red) are H3K9me2/3, H3K27me2/3 and H4K20me3, and activating histone methylation marks (indicated in green) are H3K4me2/3, H2K36me3 and H3K79me3. All epigenetic modifications are catalyzed by enzymes: (1) DNA methylation by DNMTs, (2) histone methylation marks by HMTs and HDMs, and (3) histone acetylation marks by HATs and HDACs. (b) DNMTs are involved in DNA methylation in which cytosine residues are converted into 5-methylcytosine residues. (c,d) Epigenetic drugs have been developed inhibiting certain groups of enzymes involved in epigenetic modifications, i.e. DNMTis and HDACis targeting DNMTs and HDACs, respectively, both stimulating transcriptionally active euchromatin. (e) HDACis often target multiple HDACs within HDAC class I, IIa and/or IIb. AB3, entinostat (ENT), tacedinaline (TAC), thailandepsin-A (TDP-A) and valproic acid (VPA) target HDAC1, 2 and 3; romidepsin (FK228) targets all HDAC protein within class I; LMK235 targets HDAC4 and 5 within class IIa; vorinostat (SAHA) and trichostatin A (TSA) target HDAC proteins within class I, IIa and IIb. [46, 104,105,106]
One of the major epigenetic mechanisms are histone modifications. Specific histone-modifying enzymes can stimulate the formation of either condensed, inactive heterochromatin or decondensed, active euchromatin (Fig. 2a). Acetylation and methylation of amino acids are the most frequently observed modifications at the N-terminal tails of the histones. Acetylation on lysine amino acids leads to a reduction of positive charges on the surface of histones, resulting in loss of interactions between DNA and histones. This in turn results in euchromatin formation which stimulates gene transcription. Histone acetyltransferases (HATs) and histone deacetylases (HDACs) are responsible for the addition and removal of acetyl groups on lysine residues, respectively. Whereas histone acetylation is linked to transcriptional activation, histone methylation can either be repressive or activating. This depends on which lysine residue (K) on which histone (H3, H4) is modified, and the extent of methylation, i.e. di- or tri-methylation (me2, me3). H3K9me2/3, H3K27me2/3 and H4K20me3 are inhibiting histone methylation marks, and H3K4me2/3, H2K36me3 and H3K79me3 are known as important activating histone marks. The process of histone methylation is mediated by histone lysine methyltransferases (HMTs) and histone demethylases (HDMs) [42, 43].
Another relevant epigenetic modification targets cytosine residues in the DNA. Methyl groups are transferred to the fifth carbon of the cytosine nucleobases by DNA methyltransferases (DNMTs) (Fig. 2a, b). This process is mainly catalyzed by three subtypes, i.e. DNMT1, DNMT3a and DNMT3b. DNMT1, interacting with ubiquitin-like with PHD and RING finger domain (UHRF) proteins, is involved in maintaining methylation profiles during replication. DNMT3a and DNMT3b are both involved in de novo transfer of methyl groups. The catalytic activity of DNMT3a and DNMT3b is increased upon association with DNMT3L, which does not have catalytic activity on its own. DNA methylation often occurs on cytosine residues followed by guanine residues or in CpG islands which are frequently present within the promotor region. In response to DNA methylation, transcription factors are no longer able to bind. Moreover, specific inhibitory proteins bind to the DNA upon methylation resulting in repression of transcription, such as methyl-CpG-binding domain (MBD) proteins and zinc-finger proteins. [44, 45].
Of note, there is a strong interplay between histone modifications and DNA methylation. For example, activating histone modifications prevent binding of DNMTs, thereby enhancing gene transcription. Moreover, DNMTs can interact with HMTs and HDAC, together stimulating the silenced heterochromatin state. Additionally, repressing MBD proteins interacting with methylated DNA are involved in regulating histone modifications, leading to transcriptional repression [44].
Based on the growing knowledge about the epigenetic machinery and its key role in gene transcription, drugs have been developed that target the enzymes involved in the above-mentioned processes. DNA methyltransferase inhibitors (DNMTis, Fig. 2c) and histone deacetylase inhibitors (HDACis, Fig. 2d) both stimulate gene transcription, as these epigenetic drugs inhibit DNMTs and HDACs, respectively. There are several HDAC subtypes, leading to the development of subtype-specific HDACis. In short, mainly based on both the homology with yeast HDACs and their function, human HDACs are divided in four classes; class I, II, III and IV, of which HDACs class II is subdivided in class IIa and IIb. The HDACis discussed in this review are targeting one or multiple classes, constituted of several HDAC proteins: epigenetic drugs targeting class I (HDAC1, 2, 3 and 8), class IIa (HDAC4, 5, 7 and 9) and/or class IIb (HDAC6 and 10) (Fig. 2e) [46, 47].
Summarizing, DNMTis and HDACis can both be used to specifically target and change the epigenetic machinery and profile. These epigenetic drugs can therefore create many possibilities to modify gene and thus protein expression levels, e.g. SSTR expression, in order to expand current therapy options for cancer (Fig. 3).

Activating histone marks (indicated in green) stimulate euchromatin, resulting in more gene transcription. Epigenetic drugs may be used to stimulate euchromatin, in order to increase the expression of certain proteins. Thereby, it may be possible to increase the expression of targets for therapy, e.g. SSTR2 in NET patients with insufficient expression levels for treatment
2.2 Epigenetic regulation in NETs
The important role of epigenetics in NET tissue has already been demonstrated in several clinical studies. In a cohort of pNET patients, significant upregulation of multiple HDAC subtypes was reported, including HDAC3, nuclear HDAC4, nuclear and cytoplasmic HDAC5, cytoplasmic HDAC8, HDAC9, nuclear HDAC10 and HDAC11. More specifically, HDAC1, HDAC2, nuclear HDAC5 and HDAC11 were significantly elevated in high grade (G3) pNETs, compared to low-grade pNETs (G1 and G2). Further analysis showed that especially upregulation of nuclear HDAC5 was significantly associated with a reduced disease-free survival and overall survival [48]. Additionally, the HDACi entinostat (ENT) reduced the activity of proteins involved in the progression from primary to metastatic disease [49]. This suggests that histone acetylation marks could be a target for therapy. In line with changes in histone acetylation patterns, aberrant DNA methylation patterns have been described in NET tissue, emphasizing the important role of epigenetics in regulating gene expression involved in numerous processes such as cell cycle, cell death, cell growth and DNA repair [34, 40]. In an extensive review by Mafficini et al. [50], the genetic and epigenetic alterations in pNETs and siNETs have been described. In this overview, promotor hypermethylation of multiple genes was described, for instance hypermethylation of RASSF1A and CDKN2A, both being tumor suppressor genes involved in regulating cell cycle arrest, apoptosis and/or senescence. Additionally, inactivating mutations in HMTs were described, further suggesting deregulation of the epigenetic machinery.
Altogether, there is strong evidence that the epigenetic machinery is highly involved in the pathophysiology of NETs. It might even be speculated that this system is also involved in the regulation of SST and SSTR expression and signaling. Elucidating the exact regulation of SSTR in NETs is of high importance, as this protein has an important function as target for therapy.
3 Epigenetic regulation of SSTR
3.1 Epigenetic regulation of SSTR in NETs
As mentioned above, SSAs are a cornerstone medical treatment modality for NETs, targeting SSTR2 which is often expressed at a high level in NETs. The genomic DNA of human SSTR2 contains multiple transcription start sites (TSSs). Two TSSs are located 82–93 nucleotides upstream [51, 52] from the translation start codon with an initiation element inr in close proximity. This inr is involved in regulating gene transcription in the absence of a TATA-box as transcription factors are able to bind to the E-box present within this inr [53]. Another TSS is located further upstream and contains a CpG island [54]. As CpGs are often the target for epigenetic modifications, it is likely that epigenetic regulation is involved in controlling SSTR2 gene expression via this TSS. This suggests that deregulation of the epigenetic machinery may also influence tumoral SSTR2 expression. To elucidate the role of epigenetic regulation in NET patients, different NET cell lines have been used. These include cell lines derived from pNETs (i.e. BON-1 and QGP-1), pulmonary NETs (i.e. NCI-H727), siNETs (i.e. GOT-1) and medullary thyroid cancer (i.e. TT and MZ-CRC-1), which are all characterized by their own basal SSTR2 expression levels.
In pNET cells lines BON-1 and QGP-1, both DNA methylation and histone modifications regulate SSTR2 expression. In comparison with other NET cell lines, BON-1 and QGP-1 cells are both characterized by relatively low SSTR2 expression levels. However, SSTR2 mRNA levels are still relatively high compared to cell lines derived from other types of cancer [54]. QGP-1 cells demonstrated low SSTR2 promotor methylation rates at only 2% in the 8 CpG islands examined [55]. Similar observations were made for the pancreatic BON-1 cells, characterized by slightly higher SSTR2 expression levels compared to QGP-1 cells. Low (~3%), or even unmeasurable levels of DNA methylation were found in the genomic region surrounding the TSS in BON-1 cells [54, 55]. The low levels of DNA methylation and relatively low SSTR2 expression levels could be related to the involvement of DNA methylation in other regions, as the above described studies only focus on specific areas in the promotor region. Torrisani et al. [54] showed an inverse association between the level of CpG island methylation and SSTR2 mRNA levels within several cell lines, including the pNET cell line BON-1. Additionally, transfection of a methylated SSTR2 promotor in BON-1 cells induced silencing of the SSTR2 promotor. This effect was caused by the absence of binding of transcription factor specificity protein-1, a protein involved in regulating the basal SSTR2 promotor activity [54]. Together, these observations support the potential of SSTR2 promotor methylation to suppress SSTR2 expression. Moreover, acetylation on histone 3 was present in both BON-1 and QGP-1 cells [56]. The involvement of histone acetylation was further confirmed by Veenstra et al. [55]. In conclusion, both DNA methylation and histone acetylation are likely involved in regulating SSTR2 expression, i.e. triggering heterochromatin and euchromatin, respectively.
The above-mentioned associations between epigenetic markers and SSTR2 expression levels suggest that epigenetic drugs could potentially stimulate SSTR expression in NET cells. The use of epigenetic drugs, such as DNMTis and HDACis, may stimulate euchromatin, thereby promoting SSTR2 gene transcription. This approach can especially be important for NET patients not eligible for SSTR2-mediated therapies due to insufficient or undetectable SSTR expression levels.
3.1.1 Modulation of SSTR expression in vitro in NET cell lines
Successful stimulation of SSTR2 through attenuation of methylation has been demonstrated in BON-1 cells by treatment with DNMTis 5-aza-2′-deoxycytidine (5-AZA-dC) or 5-azacitidine (5-AZA), as shown by significantly enhanced uptake of [68Ga]Ga-DOTA-TOC [57]. Further analysis of 5-AZA-dC pretreatment demonstrated increased SSTR2 mRNA and SSTR2 protein expression levels, which increased over time. Moreover, the uptake of [68Ga]Ga-DOTA-TOC was clearly enhanced at human 5-AZA-dC therapeutic serum concentrations, whereas effects were barely observed at lower concentrations. Based on these data, a time- and dose-dependency was suggested. The efficacy of 5-AZA-dC in modulating SSTR2 expression is investigated in several other studies. A seven-day treatment schedule resulted in enhanced SSTR2 mRNA expression levels in both BON-1 and QGP-1 cells using 100 nM and 50 nM, respectively. Receptor functionality was subsequently demonstrated with internalization studies using [125I]I-[Tyr3]octreotide, reporting a significantly increased 1.85-fold uptake in BON-1 cells [55]. In line with this, significantly increased SSTR2 protein levels in BON-1 cells after a 3 day exposure to 2.5 μM 5-AZA-dC were also observed in the study by Jin et al. [58]. However, in another study, it was demonstrated that a 3 day exposure to a lower dose of 5-AZA-dC (2 μM) had no significant effects on SSTR2 mRNA expression levels in BON-1 cells [54]. As the experimental set-up is similar in terms of cell line and exposure time, the results support data the above mentioned of Taelman et al. [57] of a dose-dependent response. As both time- and dose-dependency are clearly suggested, a precise treatment regimen may be important parameter for study outcome.
In addition to DNA methylation, histone acetylation is also likely involved in regulating SSTR2. HDACis therefore also gained great interest as a novel therapeutic strategy to stimulate SSTR2 expression. Several HDACis have been tested in QGP-1 cells, which has led to contradictory results. Whereas Veenstra et al. [55] demonstrated increased internalization of radiolabeled SSAs after valproic acid (VPA) treatment, SSTR2 mRNA levels were significantly decreased by 1 mM VPA. This indicated other modes of action, e.g. fast redirection of the receptor to the membrane after internalization, via yet unknown epigenetic mechanisms. Contrary to these findings, significantly increased SSTR2 mRNA levels were reported after VPA treatment by Guenter et al. [59] when using a higher VPA dosage (4 mM). Other HDACis, such as romidepsin (FK228), vorinostat (SAHA) and AB3, provided similar results as significant upregulation was demonstrated on SSTR2 mRNA expression level. Unfortunately, western blot analysis could not confirm SSTR2 upregulation in QGP-1 cells upon HDACi treatment [59]. In contrast to this, the use of the HDACi LMK235 provided more convincing results, as this treatment resulted in increased SSTR2 protein expression levels [56]. An epigenetic mechanism of action was confirmed by an augmented acetylation of histone 3 upon HDACi-treatment. Of note, LMK235 has high affinity for HDAC4 and HDAC5, both belonging to HDAC class IIa, whereas all the other tested HDACis either target multiple HDAC-classes or specifically target HDAC class I. This may suggest that HDAC4 and HDAC5 are highly involved in inducing euchromatin, thereby enabling SSTR2 transcription in QGP-1 cells. The effects of HDACi-treatment in BON-1 cells were more consistent than the results in the QGP-1 cell line. A screen of several HDACis (i.e. scriptaid, dacinostat, panobinostat, trichostatin A (TSA), SAHA, phenylbutyrate, FK228 and tacedinaline (TAC)) demonstrated enhanced uptake of radiolabeled SSAs by BON-1 cells, reaching statistical significance for most HDACis [57]. Further analysis of cells treated with TAC demonstrated significantly increased SSTR2 mRNA and SSTR2 protein expression levels. In line with these results, significantly increased SSTR2 mRNA levels were described after TSA treatment by Torrisani et al. [54]. Furthermore, protein expression levels were significantly increased upon TAC treatment [58], specifically inhibiting HDAC1–3, further supporting the enhanced uptake described by Taelman et al. [57]. FK228, SAHA and AB3 were also able to enhance SSTR2 mRNA significantly within 24 h, and even demonstrated increased protein expression levels after 48 h treatment [59]. Moreover, it was shown that upon LMK235 treatment the level of acetylation on histone 3 was increased, providing a dose-dependent increase of SSTR2 protein after a one day treatment [56].
Furthermore, the effects of VPA in BON-1 cells were evaluated in various studies. VPA treatment resulted in an increased level of acetylation on histone 4, thereby confirming changes in the epigenetic machinery. In line with this, SSTR2b protein expression was increased [60]. This VPA-augmented SSTR2 expression level was confirmed at mRNA level upon short- (24–28 h) and long-term (7 days) VPA treatment [55, 59, 61]. Furthermore, a 7.2-fold stimulated SSTR2 protein expression level was observed [59], while the functionality of increased SSTR2 expression was further confirmed by a significantly increased uptake of radiolabeled SSAs [55, 57]. Receptor functionality was also confirmed by an increased efficacy of camptothecin-somatostatin conjugates after VPA treatment, as demonstrated by reduced BON-1 cell proliferation [60]. These results suggest that SSTR2 expression is more easily modified in BON-1 cells than in QGP-1 cells, and the effects seem to be less dependent on the targeted HDAC classes.
In addition to pNETs, the effect of HDACis was examined in both small intestinal (i.e. GOT-1 and KRJ-I) and pulmonary (i.e. NCI-H727) NETs, characterized by variable SSTR2 expression levels. Expression levels in GOT-1 cells exceed that of NCI-H727 cells, which are both characterized by higher expression levels compared to BON-1 and KRJ-I cells [58, 62]. Although there has been some debate about the origin of KRJ-I cells [63, 64], VPA treatment increased SSTR2 mRNA levels in these cells [61]. Studies with the small intestinal cell line GOT-1 are still limited. The effect of HDACi treatment was solely examined upon monotherapy with either VPA [61] or TAC [58], resulting in a statistically significant ~2-fold increased SSTR2 protein expression level after TAC treatment, while no significant changes in mRNA expression were observed after VPA treatment. For comparison, a similar treatment schedule did change SSTR2 significantly in BON-1 and KRJ-I cells.
In the pulmonary NET cell line NCI-H727, SSTR2 protein expression level was not changed significantly upon TAC treatment [58]. However, in another study, HDACis thailandepsin-A (TDP-A), FK228, SAHA, VPA and AB3 were able to increase SSTR2 mRNA levels significantly when high dosages were used [65]. Western blot analysis confirmed over 2.5-fold upregulation in all conditions. Further examination of TDP-A-treated NCI-H727 cells demonstrated both receptor functionality and increased receptor-mediated uptake of [68Ga]Ga-DOTA-TATE. Equal concentrations of TDP-A, FK228, SAHA, VPA, and AB3 showed SSTR2 protein upregulation in the TT medullary thyroid cancer cell line up to 3-fold, whereas these effects were not observed in the MZ-CRC-1 medullary thyroid cancer cell line. Of note, MZ-CRC-1 cells are characterized by high basal SSTR2 expression level compared to TT cells.
Studies have also focused on combining epigenetic treatments, e.g. the combination of DNMTis and HDACis. In BON-1 cells, the combination treatment of 5-AZA-dC and VPA had additive or synergetic effects as demonstrated by higher SSTR2 mRNA levels and higher uptake of [125I]I-[Tyr3]octreotide compared to either monotherapy [55]. Moreover, this combination of epigenetic drugs also significantly increased [125I]I-[Tyr3]octreotide uptake in QGP-1 cells, while effects of both monotherapies didn’t result in significant changes. In addition, combination of 5-AZA-dC and TAC gave synergistic effects as well, in terms of [68Ga]Ga-DOTA-TOC uptake and cell survival [57]. A similar combination treatment of 5-AZA-dC and TAC was examined in the BON-1, NCI-H727 and GOT-1 cell line by another research group, demonstrating significantly increased SSTR2 protein expression levels of 8.31-, 1.56- and 2.06-fold, respectively [58]. Additive effects were demonstrated for BON-1 cells, whereas this was not evidently observed for H727 and GOT-1. Thus, the effects in H727 cells and GOT-1 cells were less pronounced compared to results obtained with BON-1 cells.
Altogether these studies clearly suggest the involvement of the epigenetic machinery in the regulation of SSTR2 expression in NET cells. Although convincing results in QGP-1 were only induced upon LMK235 treatment, results obtained with other cell lines suggest that especially NET cell lines with low (i.e. BON-1) or intermediate (i.e. NCI-H727 and TT cells) SSTR2 expression levels are susceptible to epigenetic drug treatment, whereas upregulation in NET cell lines with high SSTR2 expression levels is more limited (i.e. GOT-1 and MZ-CRC-1 cells). This supports the concept of epigenetic therapy for NET patients with insufficient SSTR2 expression, thereby potentially making more patients eligible for treatment with (radiolabeled) SSAs. The studies described above are summarized in Table 1. Of note, DNMTis (i.e. 5-AZA-dC) and some of the HDACis (i.e. VPA, TAC, SAHA, FK228) have been tested in clinical trials. Based on published pharmacokinetic parameters, it can be concluded that the drug concentrations used in the studies described above are within the same order of magnitude or even within the achievable human therapeutic range.
3.1.2 Modulation of SSTR expression in vivo in NET xenograft-models
Based on the in vitro results discussed above, the effects of epigenetic drugs were also tested in vivo using NET-bearing mice. Direct anti-proliferative effects can be induced by HDACi treatment. Reduced xenograft growth was observed after AB3, VPA, TDP-A, FK229 and ENT treatment in BON-1, GOT-1, TT and/or H-STS NET tumor-bearing mice, although statistical significance was not reached in all studies [49, 60, 61, 66,67,68]. The combination treatment of VPA and camptothecin-somatostatin conjugate significantly reduced BON-1 tumor growth by 66%, compared to 17% and 42% for both monotherapies, respectively [60]. Tumors were not resected in this study and HDACi-upregulated SSTR2 expression was therefore not confirmed. Encapsulation of the HDACi TDP-A in micelles functionalized with either KE108 [66] or octreotide [68] reduced tumor volume with 92% and 74%, respectively. In both these studies, significant differences were found between the effects observed after treatment with TDP-A-loaded targeted micelles compared to TDP-A-loaded non-targeted micelles. Similar results were described for AB3-encapsulted KE108-functionalized micelles tested in medullary thyroid cancer TT xenografts [67]. According to the authors, the enhanced effects for TDP-A-loaded targeted micelles can be explained by the combination of both passive and active tumor targeting ability, i.e. enhanced permeation retention effect and efficient targeting of SSTRs, respectively. Unfortunately, tumors were not further analyzed to confirm changes in SSTR2 expression levels upon HDACi treatment. Although these data are not available, it may also be hypothesized that the enhanced effects upon treatment with TDP-A-loaded or AB3-loaded targeted micelles are caused by the fact that the HDACis are targeted to the SSTR2-expressing tumor cells, resulting in enhanced receptor expression due to HDACi-mediated changes in the epigenetic machinery. This SSTR2 upregulation may lead to increased therapeutic efficacy as more functionalized micelles will be targeted to the tumor cells. However, this hypothesis requires further investigations.
In the study published by Taelman et al. [57], it was demonstrated that 5-AZA-dC significantly increased the uptake of [68Ga]Ga-DOTA-TOC in BON-1 tumor-bearing mice in a dose-dependent manner, resulting in increased tumor-to-background and tumor-to-kidney ratios. Moreover, a blocking study demonstrated SSTR-specific uptake after HDACi treatment, indicating SSTR-upregulation. As a result, tumors could be visualized using PET/CT-imaging modality. In addition to this study using a DNMTi, two studies have been published in which SSTR2 expression levels were examined by PET/CT-scans upon inhibition of HDAC class I proteins. For BON-1 tumor-bearing mice, significantly increased standard uptake values (SUVs) were observed on a PET/CT-scan after [68Ga]Ga-DOTA-TATE injection when mice were pre-treated with FK228 [59]. A similar effect was observed in mice with NCI-H727 xenografts that were treated with TDP-A. This study showed a trend towards SSTR upregulation following HDACi-treatment, although statistical significance was not reached due to differences in individual tumor size and uptake of [68Ga]Ga-DOTA-TATE [65].
3.2 Epigenetic regulation of SSTR in other cancer types
Studies focusing on other types of cancer showed that deregulation of SSTR is also often established by epigenetic mechanisms. For colorectal cancer (CRC), the SSTR2 promotor was characterized by enhanced methylation levels, which was associated with reduced SSTR2 expression [69]. Similar results were obtained in head and neck squamous cell carcinomas (HNSCC). Here, higher methylation levels of SSTR1 were detected compared to adjacent normal mucosal tissue, which correlated with several clinicopathologic features [70]. This was confirmed in squamous cell carcinoma cell lines, exhibiting low SSTR1 mRNA levels and high levels of promotor methylation in comparison to normal cell lines. In line with these results, increased methylation levels on CpG sites present within the SSTR2 promotor region were also described for laryngeal squamous cell carcinomas [71]. Moreover, the SSTR1 promotor was frequently methylated in Epstein-Barr virus (EBV) positive primary gastric cancer samples (67%), whereas this was not the case in EBV-negative primary gastric cancer samples [72]. In line with this result, SSTR2, SSTR3 and SSTR5 mRNA expression levels were also reported to be reduced in gastric cancer samples compared to paired normal gastric tissue [73]. For SSTR2, this was confirmed by Kim et al. [74] by a negative correlation between SSTR2 methylation and gene expression in human gastric tumor tissue. In general, SSTR expression is reduced in cancer because of methylation of the promotor region. This suggests the involvement of the epigenetic machinery in deregulated SSTR expression in cancer. Epigenetic drugs can therefore potentially modulate SSTR expression and thus therapeutic opportunities.
3.2.1 Modulation of SSTR expression in vitro and in vivo in other types of cancer
The involvement of the epigenetic machinery in other cancer types is further supported by studies aiming to increase SSTR expression using epigenetic drugs. The effect of VPA was evaluated in several human cell lines, i.e. hepatocellular carcinoma cells [75], small cell lung cancer cells [76] and cervical cancer cells [77, 78]. The epigenetic mechanism of action of VPA was confirmed by western blot analysis in hepatocellular carcinoma and lung cancer cells, as demonstrated by a decreased expression of HDAC4 protein and increased acetylation on histone 4. Likely as a result of this altered acetylation pattern, SSTR2 was upregulated, i.e. a 20.6 and 7.4-fold increase, respectively. Therapeutic efficacy was increased in small cell lung cancer cells when VPA treatment was combined with camptothecin- or colchicine-somatostatin conjugates as shown by decreased cell growth in vitro. For cervical cancer cells, expression of SSTR subtypes were also changed upon VPA treatment. Here, VPA increased expression of SSTR2, SSTR3 and SSTR5 in a dose-dependent manner, while SSTR1 expression levels were downregulated. The VPA-induced SSTR2 upregulation, resulted in enhanced effects of cytotoxic-somatostatin conjugates, both in vitro and in vivo [77, 78].
The epigenetic machinery was also shown to be involved in the regulation of SSTR2 expression in pancreatic cancer cell lines. Upon epigenetic drug treatment with DNMTi 5-AZA-dC or HDACi TSA, SSTR2 mRNA levels were increased, with even stronger upregulation observed for the combination treatment. These results suggest the involvement of both DNA methylation and histone acetylation [54]. The possibility to modulate SSTR2 transcription by DNA methylation was confirmed by Gailhouste et al. [79], as SSTR2 mRNA levels were upregulated after 5-AZA treatment, resulting in reduced cell growth upon treatment with SSAs. Upregulation of SSTR4 and SSTR5 was also reported in response to DNMTi treatment, thereby emphasizing the important role of DNA methylation in controlling the expression of several receptors within the SST-pathway.
SSTR5 expression in primary human laryngeal squamous cell carcinoma tissue demonstrated to be significantly lower compared to corresponding normal tissue. Low expression levels were confirmed in cell lines. Further analysis of these cancer cell lines demonstrated that methylation of exon 1 of the SSTR5 gene is likely involved in downregulation of the protein. The involvement of histone modifications was also confirmed, as treatment with 5-AZA and/or TSA upregulated SSTR5 mRNA expression levels. For the AMC-HN-8 cell line, the presence of active and inactive histone modifications in the SSTR5 promotor were examined. Activating histone mark H3K4me3 was enriched upon 5-AZA-dC or combination treatment, activating histone mark H3K9ac was enriched upon TSA or combination treatment, and repressive histone mark H3K9me2 was decreased upon 5-AZA-dC or combination treatment. In this extensive study by Wang et al. [80], the involvement of both DNA methylation and histone modifications was therefore clearly demonstrated in the regulation of SSTR5 in laryngeal squamous cell carcinomas cell lines.
Moreover, 5-AZA-dC and TSA treatment increased SSTR5 mRNA expression levels in a castration-resistant prostate cancer cell line [81]. Combining DNMTi and HDACi treatment had additive effects in this cell line in terms of SSTR5 expression, whereas such effects were not observed in androgen-sensitive cell lines, suggesting cell type-specific responses. This cell type-specific response was further confirmed as 5-AZA-dC treatment was associated with SSTR1 hypomethylation in androgen receptor-positive prostate cancer cell line, whereas this was not observed in an androgen-receptor negative prostate cancer cell line [82].
Data has also suggested the involvement of epigenetic mechanisms in the controlling SSTR2 expression in gastric cancer. In line with this, 5-AZA-dC and/or TSA treatment restored both SSTR2 and SSTR4 mRNA expression in 75% of the examined gastric cell lines, with the greatest effects observed upon combination therapy [74]. Upon comparing EBV-positive AGS gastric cancer cells and EBV-negative AGS gastric cancer cells, it was demonstrated that EBV-positive AGS cells are characterized by enhanced activity of DNMT3b and higher SSTR1 CpG island methylation levels. Further analysis showed that the viral latent membrane protein 2A (LMP2A), expressed upon EBV infection, is involved in DNMT3b upregulation. Treatment of EBV-positive AGS cells with 5-AZA-dC resulted in increased SSTR1 mRNA levels. Of note, this was not observed in the EBV-negative gastric cell line. The latter cell line is characterized by very low SSTR1 CpG island methylation levels compared to EBV-positive AGS cells [72, 83]. This suggests that DNMTis are only efficient in enhancing SSTR levels in cells characterized by high DNA methylation levels.
Summarizing, these data demonstrated that, in line with the epigenetic regulation involved in SSTR2 expression in NETs, the epigenetic machinery plays an important role in the regulation of multiple SSTRs in other cancer types as well. Moreover, it is possible to increase SSTR expression in a number of cancer types by epigenetic drug treatment.
4 Epigenetic regulation of SST in cancer
As discussed above, activation of SSTR by SST can induce several effects, e.g. inhibiting cell proliferation and hormone secretion, and promoting apoptosis. SST is therefore known has a protein with anti-proliferative and anti-secretory activity. This was further supported in a recently published paper, showing that knock out of SST in the BGC823 gastric cancer cell line resulted in an increased capacity for migration and invasion in vitro [84]. Due to its role, SST expression in cancer is evaluated extensively in order to find new biomarkers or to expand current therapeutic options. For NET patients, long-acting SSAs increase progression-free survival [25, 26], confirming the anti-proliferative activity of SST upon SSTR activation. In addition, preclinical studies showed that the SST-SSTR interaction has tumor suppressor activity in certain tumors [85]. This raises the question whether deregulation of SST expression in NETs has impact of NET function as well. However, to the best of our knowledge, no reports have been published yet about the epigenetic regulation of SST in NETs.
On the other hand, the regulation of SST expression in gastric cancer has been subject to research. SST knock-down experiments in the GES-1 cell line resulted in a lowering of cells in the G0/G1 phase, suggesting an important role of SST in cell proliferation [86]. Moreover, a high DNA methylation level of the SST promotor and its assocation with undetectable SST expression has been described in seven gastric cancer cell lines [87]. Li. et al. [88] confirmed SST promotor hypermethylation in gastric cancer tissue. However, the authors were unable to validate a reduction in SST mRNA expression in human tissue using 10 pairs of tumor and adjacent non-tumorous tissue (p = 0.074). Contradictory to this, reduced SST mRNA and SST protein expression levels have been described in gastric carcinoma samples throughout multiple studies using larger cohorts of patients. Reduced SST mRNA and SST protein expression levels thereby both correlated with increased SST DNA methylation levels [73, 87, 89].
For renal cell carcinomas, published results are equivocal. Ricketts et al. [90] demonstrated hypermethylation both in cell lines and in primary tissue samples. In these tissue samples, it was shown that tumor-specific hypermethylation of the SST promotor was associated with reduced SST mRNA expression levels. Contradictory, Morris et al. [91] reported promotor hypermethylation only in renal cancer cell lines, whereas this was not observed in any of the analyzed primary renal cell carcinoma tissue samples.
The involvement of the epigenetic machinery in the regulation of SST has also been demonstrated in several studies focusing on colon cancer and CRC. CpG sites within the SST gene were hypermethylated in different stages of tumorous samples, i.e. 94%, 100%, 94% and 57% for adenomas with low-grade dysplasia, adenomas with high-grade dysplasia, CRC and metastatic-CRC, respectively [92]. Likely as a result of this observed hypermethylation in CRC, the SST mRNA level was decreased in CRC compared to normal tissue, as demonstrated with microarray data [93]. In a small pilot study with only 4 samples collected from patients with pre-neoplastic colorectal sessile serrated adenomas, SST hypermethylation was demonstrated in all examined patients [94]. These results indicate that, among others, the downregulation of SST may be involved in the development of CRC. In line with this, SST promotor methylation is increased in CRC, associating with reduced expression levels [69, 93, 95]. In agreement with the results observed in CRC samples, SST promotor methylation levels were also significantly increased when focused specifically on primary colon cancer samples compared to normal colonic mucosae, i.e. 88% versus 47%, respectively [96].
Limited information is available about the epigenetic regulation of SST in other types of cancer. SST promotor hypermethylation was shown in pancreatic PANC-1 cells. Here, CpG methylation rates of 96–98% were associated with extremely low SST mRNA levels [79]. Moreover, knockdown of DNMT1 increased SST expression, emphasizing the role of DNA methylation and thus the epigenetic machinery in the regulation of SST expression. Furthermore, analysis of glioblastoma multiforme tissue samples demonstrated both SST hypermethylation on CpG sites and a 80.5-fold downregulated SST expression level compared to control brain tissue [97]. SST hypermethylation has also been reported for human tissue derived from cervical cancer [98] and anal cancer [99], and both in cell lines and human tissue derived from esophageal carcinomas [100], gliomas [101] as well as HNSCC [70, 102, 103]. For esophageal carcinomas, gliomas and HNSCC, a negative correlation with mRNA expression was found. It has even been suggested that SST may be used as a methylation-based biomarker for the prognosis and/or diagnosis of HNSCC [102], CRC [69, 95], cervical [98] and anal cancer [99].
There is a possibility that the presence of endocrine cells in the examined tissues affect the outcome of these studies, for example the presence of enteroendocrine cells in control colorectal and CRC tissue. These endocrine cells are characterized by the expression of SST, and differences in the number of these cells in normal and tumor tissue, and thus the level of SST expression, may bias the conclusions focusing on downregulated SST expression levels in tumor tissue. However, SST hypermethylation, which is reported for several types of tumors, still suggests that SST, with its anti-proliferating and anti-secretory effects, is under the control of the epigenetic machinery. To the best of our knowledge, no data are available with respect to the role of histone acetylation within this process. In line with SSTR2 regulation, SST expression can therefore be modified by the use of epigenetic drug inhibitors targeting DNA methylation, i.e. DNMTis.
4.1 Modulation of SST expression in vitro and in vivo
In vitro studies have demonstrated that the DNMTi 5-AZA-dC modulates SST expression. 5-AZA-dC has been shown to induce demethylation of the SST gene and/or concomitantly increased SST protein expression levels in cell lines derived from colon cancer [96], renal cell carcinoma [90] and esophageal cancer [100]. Similar results were observed in the PANC-1 pancreatic cancer cell line upon 5-AZA treatment [79]. Results were also confirmed in vivo. Subcutaneous inoculation of 5-AZA pre-treated cells in athymic nude mice resulted in reduced tumor growth. Moreover, 5-AZA treatment of PANC-1 xenograft-bearing mice induced a significant reduction in tumor volume. Examination of the resected tumors showed that SST mRNA levels were significantly increased after 5-AZA treatment. Moreover, the SST promotor was demethylated at CpG sites upon epigenetic drug treatment.
Additionally, in the AGS gastric cancer cell line, the effect of 5-AZA-dC was dose-dependent. A lower dose (1.6 μM; 3 days treatment) had no effect on SST mRNA levels in the AGS cell line [89], whereas a higher dose (5 μM; 3 days treatment) reduced SST DNA promotor methylation levels and restored mRNA expression to detectable levels. Combination treatment of 5-AZA-dC and TSA even further increased SST mRNA levels [87]. Studies with other gastric cancer cell lines showed higher SST mRNA levels upon 5-AZA-dC in a subset of the tested cell lines [89], suggesting cell line-specific responses. Cell line-specific responses were also reported for gliomas [101], only demonstrating SST upregulation upon 5-AZA-dC treatment in cell lines characterized by promotor hypermethylation. The glioma cell lines U251 and SF767 are characterized by 51.6% and 77.1% methylation of the SST promotor, respectively. Upon 5-AZA-dC treatment, SST mRNA levels were significantly increased. Of note, these effects were not observed in SF126 cells, characterized by only 14.2% methylation. This suggests that DNMTi are only effective in cell lines with high promotor methylation levels. Upon inducing SST expression, there was enrichment of activating histone marks H3Ac and H3K4me3, and reduction of inhibiting mark H2K9me3, suggesting an interplay between the promotor region and chromatin structure.
Altogether, several lines of evidence suggest that DNA methylation as well as histone modifications are involved in deregulated SST expression in various types of cancer. Moreover, SST expression can be modulated by the use of epigenetic drugs, thereby further supporting the involvement of the epigenetic machinery.
5 Conclusion
In conclusion, understanding of the epigenetic mechanisms involved in the regulation of the expression of the SST-system is important for both NETs and other tumor types. A detailed analysis of this system potentially opens up new possibilities to develop or improve treatment options for different types of SSTR-expressing tumors, including NETs. Although studies clearly prove the involvement of epigenetics in the regulation of SSTRs and SST expression in vitro, more in-depth studies are required to confirm the ability to upregulate SSTR2 by using epigenetic drugs in vivo. Proper analysis to confirm the mechanism of action of epigenetic drugs are often lacking, e.g. examining histones profiles, immunohistochemistry, RT-qPCR and/or autoradiography to confirm increased SSTR expression.
Moreover, receptor-specificity should be determined after epigenetic drug treatment. Since most of the knowledge on the epigenetic regulation of the SST-system is derived from in vitro studies in cell lines and experimental tumor models, future studies should also focus on the role of epigenetic marks in determining SSTR expression in primary NET tissues from patients. Moreover, the safety profile of epigenetic drugs on healthy tissue should be assessed as these drugs may potentially upregulate physiological SSTR2 expression which possibly results in enhanced (radio)toxicity in non-targeted organs. Known and future insights in the epigenetic regulation of SST and SSTR in NETs may result in the development of epidrug-based treatment modalities aiming to increase SST and SSTR2 expression. Increased intra-tumoral SST expression may in turn lead to anti-secretory and anti-proliferative effects, whereas increased SSTR2 expression could improve tumor visibility with SSTR-scintigraphy and enhance tumor response to (radiolabeled) SSAs. This may in particular be beneficial for patients with low or insufficient SSTR expression. Moreover, future studies, also including safety, are required to define the optimal dose and treatment duration for mono- and combination therapy with DNMTis and/or HDACis.
References
Olias G, Viollet C, Kusserow H, Epelbaum J, Meyerhof W. Regulation and function of somatostatin receptors. J Neurochem. 2004;89:1057–91.
Barnett P. Somatostatin and somatostatin receptor physiology. Endocrine. 2003;20:255–64.
Patel YC. Somatostatin and its receptor family. Front Neuroendocrinol. 1999;20:157–98.
Günther T, Tulipano G, Dournaud P, Bousquet C, Csaba Z, Kreienkamp H-J, et al. International Union of Basic and Clinical Pharmacology. CV. Somatostatin receptors: structure, function, ligands, and new nomenclature. Pharmacol Rev. 2018;70:763–835.
Gatto F, Barbieri F, Arvigo M, Thellung S, Amaru J, Albertelli M, et al. Biological and biochemical basis of the differential efficacy of first and second generation Somatostatin receptor ligands in neuroendocrine neoplasms. Int J Mol Sci. 2019;20:3940.
Barbieri F, Bajetto A, Pattarozzi A, Gatti M, Würth R, Thellung S, et al. Peptide receptor targeting in cancer: the somatostatin paradigm. Int J Pept. 2013;2013:926295.
Qian ZR, Li T, Ter-Minassian M, Yang J, Chan JA, Brais LK, et al. Association between Somatostatin receptor expression and clinical outcomes in neuroendocrine tumors. Pancreas. 2016;45:1386–93.
Papotti M, Bongiovanni M, Volante M, Allia E, Landolfi S, Helboe L, et al. Expression of somatostatin receptor types 1-5 in 81 cases of gastrointestinal and pancreatic endocrine tumors. A correlative immunohistochemical and reverse-transcriptase polymerase chain reaction analysis. Virchows Arch. 2002;440:461–75.
Evans AA, Crook T, Laws SA, Gough AC, Royle GT, Primrose JN. Analysis of somatostatin receptor subtype mRNA expression in human breast cancer. Br J Cancer. 1997;75:798–803.
Qiu C-Z, Wang C, Huang Z-X, Zhu S-Z, Wu Y-Y, Qiu J-L. Relationship between somatostatin receptor subtype expression and clinicopathology, Ki-67, Bcl-2 and p53 in colorectal cancer. World J Gastroenterol. 2006;12:2011–5.
Hennigs JK, Müller J, Adam M, Spin JM, Riedel E, Graefen M, et al. Loss of somatostatin receptor subtype 2 in prostate cancer is linked to an aggressive cancer phenotype, high tumor cell proliferation and predicts early metastatic and biochemical relapse. PLoS One. 2014;9:e100469.
Werner C, Dirsch O, Dahmen U, Grimm M-O, Schulz S, Lupp A. Evaluation of Somatostatin and CXCR4 Receptor Expression in a Large Set of Prostate Cancer Samples Using Tissue Microarrays and Well-Characterized Monoclonal Antibodies. Transl Oncol. 2020;13:100801.
Stafford ND, Condon LT, Rogers MJ, MacDonald AW, Atkin SL. The expression of somatostatin receptors 1 and 2 in benign, pre-malignant and malignant laryngeal lesions. Clin Otolaryngol Allied Sci. 2003;28:314–9.
Zou Y, Tan H, Zhao Y, Zhou Y, Cao L. Expression and selective activation of somatostatin receptor subtypes induces cell cycle arrest in cancer cells. Oncol Lett. 2019;17:1723–31.
Singh S, Granberg D, Wolin E, Warner R, Sissons M, Kolarova T, et al. Patient-reported burden of a neuroendocrine tumor (NET) diagnosis: results from the first global survey of patients with NETs. J Glob Oncol. 2016;3:43–53.
Zandee WT, de Herder WW. The evolution of neuroendocrine tumor treatment reflected by ENETS guidelines. Neuroendocrinology. 2018;106:357–65.
Hofland J, Kaltsas G, de Herder WW. Advances in the diagnosis and Management of Well-Differentiated Neuroendocrine Neoplasms. Endocr Rev. 2020;41:371–403.
Mizutani G, Nakanishi Y, Watanabe N, Honma T, Obana Y, Seki T, et al. Expression of Somatostatin receptor (SSTR) subtypes (SSTR-1, 2A, 3, 4 and 5) in neuroendocrine tumors using real-time RT-PCR method and immunohistochemistry. Acta Histochem Cytochem. 2012;45:167–76.
Stueven AK, Kayser A, Wetz C, Amthauer H, Wree A, Tacke F, et al. Somatostatin analogues in the treatment of neuroendocrine tumors: past. Present and Future Int J Mol Sci. 2019;20:3049.
Bergsma H, van Vliet EI, Teunissen JJ, Kam BL, de Herder WW, Peeters RP, et al. Peptide receptor radionuclide therapy (PRRT) for GEP-NETs. Best Pract Res Clin Gastroenterol. 2012;26:867–81.
Van Essen M, Krenning EP, De Jong M, Valkema R, Kwekkeboom DJ. Peptide receptor radionuclide therapy with radiolabelled somatostatin analogues in patients with somatostatin receptor positive tumours. Acta Oncol. 2007;46:723–34.
Ruszniewski P, Valle JW, Lombard-Bohas C, Cuthbertson DJ, Perros P, Holubec L, et al. Patient-reported outcomes with lanreotide autogel/depot for carcinoid syndrome: an international observational study. Dig Liver Dis. 2016;48:552–8.
O'Toole D, Ducreux M, Bommelaer G, Wemeau JL, Bouche O, Catus F, et al. Treatment of carcinoid syndrome: a prospective crossover evaluation of lanreotide versus octreotide in terms of efficacy, patient acceptability, and tolerance. Cancer. 2000;88:770–6.
Appetecchia M, Baldelli R. Somatostatin analogues in the treatment of gastroenteropancreatic neuroendocrine tumours, current aspects and new perspectives. J Exp Clin Cancer Res. 2010;29:19.
Rinke A, Muller HH, Schade-Brittinger C, Klose KJ, Barth P, Wied M, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID study group. J Clin Oncol. 2009;27:4656–63.
Caplin ME, Pavel M, Cwikla JB, Phan AT, Raderer M, Sedlackova E, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371:224–33.
Brabander T, van der Zwan WA, Teunissen JJM, Kam BLR, Feelders RA, de Herder WW, et al. Long-Term Efficacy, Survival, and Safety of [177Lu-DOTA0,Tyr3]octreotate in Patients with Gastroenteropancreatic and Bronchial Neuroendocrine Tumors. Clin Cancer Res. 2017;23:4617–24.
Kwekkeboom DJ, de Herder WW, Kam BL, van Eijck CH, van Essen M, Kooij PP, et al. Treatment With the Radiolabeled Somatostatin Analog [177Lu-DOTA0,Tyr3]Octreotate: Toxicity, Efficacy, and Survival. J Clin Oncol. 2008;26:2124–30.
Strosberg J, El-Haddad G, Wolin E, Hendifar A, Yao J, Chasen B, et al. Phase 3 trial of (177)Lu-Dotatate for Midgut neuroendocrine tumors. N Engl J Med. 2017;376:125–35.
van Vliet EI, van Eijck CH, de Krijger RR, Nieveen van Dijkum EJ, Teunissen JJ, Kam BL, et al. Neoadjuvant Treatment of Nonfunctioning Pancreatic Neuroendocrine Tumors with [177Lu-DOTA0,Tyr3]Octreotate. J Nucl Med. 2015;56:1647–53.
Kwekkeboom DJ, Teunissen JJ, Bakker WH, Kooij PP, de Herder WW, Feelders RA, et al. Radiolabeled Somatostatin Analog [177Lu-DOTA0,Tyr3]Octreotate in Patients With Endocrine Gastroenteropancreatic Tumors. J Clin Oncol. 2005;23:2754–62.
Righi L, Volante M, Tavaglione V, Bille A, Daniele L, Angusti T, et al. Somatostatin receptor tissue distribution in lung neuroendocrine tumours: a clinicopathologic and immunohistochemical study of 218 'clinically aggressive' cases. Ann Oncol. 2010;21:548–55.
Diakatou E, Kaltsas G, Tzivras M, Kanakis G, Papaliodi E, Kontogeorgos G. Somatostatin and dopamine receptor profile of gastroenteropancreatic neuroendocrine tumors: an immunohistochemical study. Endocr Pathol. 2011;22:24–30.
Di Domenico A, Wiedmer T, Marinoni I, Perren A. Genetic and epigenetic drivers of neuroendocrine tumours (NET). Endocr Relat Cancer. 2017;24:R315–R34.
Kanwal R, Gupta S. Epigenetic modifications in cancer. Clin Genet. 2012;81:303–11.
Rodenhiser D, Mann M. Epigenetics and human disease: translating basic biology into clinical applications. CMAJ. 2006;174:341–8.
Skinner MK. Role of epigenetics in developmental biology and transgenerational inheritance. Birth Defects Res C Embryo Today. 2011;93:51–5.
Lewis EM, Kroll KL. Development and disease in a dish: the epigenetics of neurodevelopmental disorders. Epigenomics. 2018;10:219–31.
Jeffries MA, Sawalha AH. Autoimmune disease in the epigenetic era: how has epigenetics changed our understanding of disease and how can we expect the field to evolve? Expert Rev Clin Immunol. 2015;11:45–58.
Finnerty BM, Gray KD, Moore MD, Zarnegar R, Fahey Iii TJ. Epigenetics of gastroenteropancreatic neuroendocrine tumors: a clinicopathologic perspective. World J Gastrointest Oncol. 2017;9:341–53.
Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36.
Maleszewska M, Kaminska B. Is glioblastoma an epigenetic malignancy? Cancers (Basel). 2013;5:1120–39.
Alaskhar Alhamwe B, Khalaila R, Wolf J, von Bülow V, Harb H, Alhamdan F, et al. Histone modifications and their role in epigenetics of atopy and allergic diseases. Allergy Asthma Clin Immunol. 2018;14:39.
Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38:23–38.
Hervouet E, Peixoto P, Delage-Mourroux R, Boyer-Guittaut M, Cartron P-F. Specific or not specific recruitment of DNMTs for DNA methylation, an epigenetic dilemma. Clin Epigenetics. 2018;10:17.
Hessmann E, Johnsen S, Siveke J, Ellenrieder V. Epigenetic treatment of pancreatic cancer: is there a therapeutic perspective on the horizon? Gut. 2016;66:168–79.
Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol. 2014;6:a018713.
Klieser E, Urbas R, Stattner S, Primavesi F, Jager T, Dinnewitzer A, et al. Comprehensive immunohistochemical analysis of histone deacetylases in pancreatic neuroendocrine tumors: HDAC5 as a predictor of poor clinical outcome. Hum Pathol. 2017;65:41–52.
Alvarez MJ, Subramaniam PS, Tang LH, Grunn A, Aburi M, Rieckhof G, et al. A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nat Genet. 2018;50:979–89.
Mafficini A, Scarpa A. Genetics and epigenetics of Gastroenteropancreatic neuroendocrine neoplasms. Endocr Rev. 2019;40:506–36.
Petersenn S, Rasch AC, Presch S, Beil FU, Schulte HM. Genomic structure and transcriptional regulation of the human somatostatin receptor type 2. Mol Cell Endocrinol. 1999;157:75–85.
Xu Y, Berelowitz M, Bruno JF. Characterization of the promoter region of the human somatostatin receptor subtype 2 gene and localization of sequences required for estrogen-responsiveness. Mol Cell Endocrinol. 1998;139:71–7.
Pscherer A, Dorflinger U, Kirfel J, Gawlas K, Ruschoff J, Buettner R, et al. The helix-loop-helix transcription factor SEF-2 regulates the activity of a novel initiator element in the promoter of the human somatostatin receptor II gene. EMBO J. 1996;15:6680–90.
Torrisani J, Hanoun N, Laurell H, Lopez F, Maoret JJ, Souque A, et al. Identification of an upstream promoter of the human somatostatin receptor, hSSTR2, which is controlled by epigenetic modifications. Endocrinology. 2008;149:3137–47.
Veenstra MJ, van Koetsveld PM, Dogan F, Farrell WE, Feelders RA, Lamberts SWJ, et al. Epidrug-induced upregulation of functional somatostatin type 2 receptors in human pancreatic neuroendocrine tumor cells. Oncotarget. 2018;9:14791–802.
Wanek J, Gaisberger M, Beyreis M, Mayr C, Helm K, Primavesi F, et al. Pharmacological inhibition of class IIA HDACs by LMK-235 in pancreatic neuroendocrine tumor cells. Int J Mol Sci. 2018;19:3128.
Taelman VF, Radojewski P, Marincek N, Ben-Shlomo A, Grotzky A, Olariu CI, et al. Upregulation of key molecules for targeted imaging and therapy. J Nucl Med. 2016;57:1805–10.
Jin XF, Auernhammer CJ, Ilhan H, Lindner S, Nölting S, Maurer J, et al. Combination of 5-fluorouracil with epigenetic modifiers induces radiosensitization, somatostatin receptor 2 expression, and radioligand binding in neuroendocrine tumor cells in vitro. J Nucl Med. 2019;60:1240–6.
Guenter R, Aweda T, Carmona Matos DM, Jang S, Whitt J, Cheng Y-Q, et al. Overexpression of somatostatin receptor type 2 in neuroendocrine tumors for improved Ga68-DOTATATE imaging and treatment. Surgery. 2019;167:189–96.
Sun L, Qian Q, Sun G, Mackey LV, Fuselier JA, Coy DH, et al. Valproic acid induces NET cell growth arrest and enhances tumor suppression of the receptor-targeted peptide-drug conjugate via activating somatostatin receptor type II. J Drug Target. 2016;24:169–77.
Arvidsson Y, Johanson V, Pfragner R, Wangberg B, Nilsson O. Cytotoxic effects of Valproic acid on neuroendocrine tumour cells. Neuroendocrinology. 2016;103:578–91.
Li SC, Martijn C, Cui T, Essaghir A, Luque RM, Demoulin JB, et al. The somatostatin analogue octreotide inhibits growth of small intestine neuroendocrine tumour cells. PLoS One. 2012;7:e48411.
Hofving T, Arvidsson Y, Almobarak B, Inge L, Pfragner R, Persson M, et al. The neuroendocrine phenotype, genomic profile and therapeutic sensitivity of GEPNET cell lines. Endocr Relat Cancer. 2018;25:367–80.
Alvarez MJ, Yan P, Alpaugh ML, Bowden M, Sicinska E, Zhou CW, et al. Reply to ‘H-STS, L-STS and KRJ-I are not authentic GEPNET cell lines’. Nat Genet. 2019;51:1427–8.
Guenter RE, Aweda T, Carmona Matos DM, Whitt J, Chang AW, Cheng EY, et al. Pulmonary Carcinoid Surface Receptor Modulation Using Histone Deacetylase Inhibitors. Cancers (Basel). 2019;11:767.
Chen G, Jaskula-Sztul R, Harrison A, Dammalapati A, Xu W, Cheng Y, et al. KE108-conjugated unimolecular micelles loaded with a novel HDAC inhibitor thailandepsin-a for targeted neuroendocrine cancer therapy. Biomaterials. 2016;97:22–33.
Jaskula-Sztul R, Chen G, Dammalapati A, Harrison A, Tang W, Gong S, et al. AB3-loaded and tumor-targeted Unimolecular micelles for medullary thyroid Cancer treatment. J Mater Chem B. 2017;5:151–9.
Jaskula-Sztul R, Xu W, Chen G, Harrison A, Dammalapati A, Nair R, et al. Thailandepsin A-loaded and octreotide-functionalized unimolecular micelles for targeted neuroendocrine cancer therapy. Biomaterials. 2016;91:1–10.
Liu J, Li H, Sun L, Wang Z, Xing C, Yuan Y. Aberrantly methylated-differentially expressed genes and pathways in colorectal cancer. Cancer Cell Int. 2017;17:75.
Misawa K, Misawa Y, Kondo H, Mochizuki D, Imai A, Fukushima H, et al. Aberrant methylation inactivates somatostatin and somatostatin receptor type 1 in head and neck squamous cell carcinoma. PLoS One. 2015;10:e0118588.
Shen Z, Chen X, Li Q, Zhou C, Li J, Ye H, et al. SSTR2 promoter hypermethylation is associated with the risk and progression of laryngeal squamous cell carcinoma in males. Diagn Pathol. 2016;11:10.
Zhao J, Liang Q, Cheung KF, Kang W, Lung RWM, Tong JHM, et al. Genome-wide identification of Epstein-Barr virus-driven promoter methylation profiles of human genes in gastric cancer cells. Cancer. 2013;119:304–12.
Shi X, Li X, Chen L, Wang C. Analysis of somatostatin receptors and somatostatin promoter methylation in human gastric cancer. Oncol Lett. 2013;6:1794–8.
Kim HJ, Kang TW, Haam K, Kim M, Kim SK, Kim SY, et al. Whole genome MBD-seq and RRBS analyses reveal that hypermethylation of gastrointestinal hormone receptors is associated with gastric carcinogenesis. Exp Mol Med. 2018;50:1–14.
Sun G, Mackey LV, Coy DH, Yu CY, Sun L. The histone deacetylase inhibitor vaproic acid induces cell growth arrest in hepatocellular carcinoma cells via suppressing notch signaling. J Cancer. 2015;6:996–1004.
Sun L, He Q, Tsai C, Lei J, Chen J, Makcey LV, et al. HDAC inhibitors suppressed small cell lung cancer cell growth and enhanced the suppressive effects of receptor-targeting cytotoxins via upregulating somatostatin receptor II. Am J Transl Res. 2018;10:545–53.
Franko-Tobin LG, Mackey LV, Huang W, Song X, Jin B, Luo J, et al. Notch1-mediated tumor suppression in cervical cancer with the involvement of SST signaling and its application in enhanced SSTR-targeted therapeutics. Oncologist. 2012;17:220–32.
Tsai C, Leslie JS, Franko-Tobin LG, Prasnal MC, Yang T, Vienna Mackey L, et al. Valproic acid suppresses cervical cancer tumor progression possibly via activating Notch1 signaling and enhances receptor-targeted cancer chemotherapeutic via activating somatostatin receptor type II. Arch Gynecol Obstet. 2013;288:393–400.
Gailhouste L, Liew LC, Hatada I, Nakagama H, Ochiya T. Epigenetic reprogramming using 5-azacytidine promotes an anti-cancer response in pancreatic adenocarcinoma cells. Cell Death Dis. 2018;9:468.
Wang B, Zhao L, Chi W, Cao H, Cui W, Meng W. Aberrant methylation-mediated downregulation of lncRNA SSTR5-AS1 promotes progression and metastasis of laryngeal squamous cell carcinoma. Epigenetics Chromatin. 2019;12:35.
Liu Z, Marquez M, Nilsson S, Holmberg AR. Incubation with somatostatin, 5-aza decitabine and trichostatin up-regulates somatostatin receptor expression in prostate cancer cells. Oncol Rep. 2008;20:151–4.
White-Al Habeeb NMA, Ho LT, Olkhov-Mitsel E, Kron K, Pethe V, Lehman M, et al. Integrated analysis of epigenomic and genomic changes by DNA methylation dependent mechanisms provides potential novel biomarkers for prostate cancer. Oncotarget. 2014;5:7858–69.
Zhao J, Liang Q, Cheung KF, Kang W, Dong Y, Lung RWM, et al. Somatostatin receptor 1, a novel EBV-associated CpG hypermethylated gene, contributes to the pathogenesis of EBV-associated gastric cancer. Br J Cancer. 2013;108:2557–64.
Chen W, Ding R, Tang J, Li H, Chen C, Zhang Y, et al. Knocking out SST gene of BGC823 gastric Cancer cell by CRISPR/Cas9 enhances migration, invasion and expression of SEMA5A and KLF2. Cancer Manag Res. 2020;12:1313–21.
Pyronnet S, Bousquet C, Najib S, Azar R, Laklai H, Susini C. Antitumor effects of somatostatin. Mol Cell Endocrinol. 2008;286:230–7.
Wu J, Gu Y, Xiao Y, Xia C, Li H, Kang Y, et al. Characterization of DNA methylation associated gene regulatory networks during stomach Cancer progression. Front Genet. 2018;9:711.
Jackson K, Soutto M, Peng D, Hu T, Marshal D, El-Rifai W. Epigenetic silencing of somatostatin in gastric cancer. Dig Dis Sci. 2011;56:125–30.
Li H, Liu JW, Liu S, Yuan Y, Sun LP. Bioinformatics-based identification of methylated-differentially expressed genes and related pathways in gastric Cancer. Dig Dis Sci. 2017;62:3029–39.
Zhang X, Yang JJ, Kim YS, Kim KY, Ahn WS, Yang S. An 8-gene signature, including methylated and down-regulated glutathione peroxidase 3, of gastric cancer. Int J Oncol. 2010;36:405–14.
Ricketts CJ, Morris MR, Gentle D, Brown M, Wake N, Woodward ER, et al. Genome-wide CpG island methylation analysis implicates novel genes in the pathogenesis of renal cell carcinoma. Epigenetics. 2012;7:278–90.
Morris MR, Gentle D, Abdulrahman M, Clarke N, Brown M, Kishida T, et al. Functional epigenomics approach to identify methylated candidate tumour suppressor genes in renal cell carcinoma. Br J Cancer. 2008;98:496–501.
Patai ÁV, Valcz G, Hollósi P, Kalmár A, Péterfia B, Patai Á, et al. Comprehensive DNA methylation analysis reveals a common ten-gene methylation signature in colorectal adenomas and carcinomas. PLoS One. 2015;10:e0133836.
Leiszter K, Sipos F, Galamb O, Krenács T, Veres G, Wichmann B, et al. Promoter hypermethylation-related reduced somatostatin production promotes uncontrolled cell proliferation in colorectal cancer. PLoS One. 2015;10:e0118332.
Patai ÁV, Barták BK, Péterfia B, Micsik T, Horváth R, Sumánszki C, et al. Comprehensive DNA methylation and mutation analyses reveal a methylation signature in colorectal sessile serrated adenomas. Pathol Oncol Res. 2017;23:589–94.
Kok-Sin T, Mokhtar NM, Ali Hassan NZ, Sagap I, Mohamed Rose I, Harun R, et al. Identification of diagnostic markers in colorectal cancer via integrative epigenomics and genomics data. Oncol Rep. 2015;34:22–32.
Mori Y, Cai K, Cheng Y, Wang S, Paun B, Hamilton JP, et al. A genome-wide search identifies epigenetic silencing of Somatostatin, Tachykinin-1, and 5 other genes in Colon Cancer. Gastroenterology. 2006;131:797–808.
Zhang M, Lv X, Jiang Y, Li G, Qiao Q. Identification of aberrantly methylated differentially expressed genes in glioblastoma multiforme and their association with patient survival. Exp Ther Med. 2019;18:2140–52.
Ongenaert M, Wisman GB, Volders HH, Koning AJ, Zee AG, van Criekinge W, et al. Discovery of DNA methylation markers in cervical cancer using relaxation ranking. BMC Med Genet. 2008;1:57.
van der Zee RP, Richel O, van Noesel CJM, Novianti PW, Ciocanea-Teodorescu I, van Splunter AP, et al. Host cell deoxyribonucleic acid methylation markers for the detection of high-grade anal intraepithelial Neoplasia and anal Cancer. Clin Infect Dis. 2019;68:1110–7.
Jin Z, Mori Y, Hamilton JP, Olaru A, Sato F, Yang J, et al. Hypermethylation of the somatostatin promoter is a common, early event in human esophageal carcinogenesis. Cancer. 2008;112:43–9.
Zhang Z, Tang H, Wang Z, Zhang B, Liu W, Lu H, et al. MiR-185 targets the DNA methyltransferases 1 and regulates global DNA methylation in human glioma. Mol Cancer. 2011;10:124.
Misawa K, Mima M, Imai A, Mochizuki D, Misawa Y, Endo S, et al. The neuropeptide genes SST, TAC1, HCRT, NPY, and GAL are powerful epigenetic biomarkers in head and neck cancer: a site-specific analysis. Clin Epigenetics. 2018;10:52.
Misawa K, Mochizuki D, Imai A, Endo S, Mima M, Misawa Y, et al. Prognostic value of aberrant promoter hypermethylation of tumor-related genes in early-stage head and neck cancer. Oncotarget. 2016;7:26087–98.
Marek L, Hamacher A, Hansen FK, Kuna K, Gohlke H, Kassack MU, et al. Histone deacetylase (HDAC) inhibitors with a novel connecting unit linker region reveal a selectivity profile for HDAC4 and HDAC5 with improved activity against chemoresistant cancer cells. J Med Chem. 2013;56:427–36.
Jang S, Yu XM, Odorico S, Clark M, Jaskula-Sztul R, Schienebeck CM, et al. Novel analogs targeting histone deacetylase suppress aggressive thyroid cancer cell growth and induce re-differentiation. Cancer Gene Ther. 2015;22:410–6.
Wang C, Henkes LM, Doughty LB, He M, Wang D, Meyer-Almes FJ, et al. Thailandepsins: bacterial products with potent histone deacetylase inhibitory activities and broad-spectrum antiproliferative activities. J Nat Prod. 2011;74:2031–8.
Acknowledgements
We thank the Medical Library of the Erasmus MC for assisting with the literature search.
Disclosures
L.J. Hofland: research grants from Novartis, Ipsen and Strongbridge; J. Hofland: speaker and travel fee from Ipsen, advisory board Novartis; R.A. Feelders: speaker fee Ipsen; no disclosures for S.U. Dalm, M. de Jong and M.J. Klomp.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Klomp, M., Dalm, S., de Jong, M. et al. Epigenetic regulation of somatostatin and somatostatin receptors in neuroendocrine tumors and other types of cancer. Rev Endocr Metab Disord 22, 495–510 (2021). https://doi.org/10.1007/s11154-020-09607-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11154-020-09607-z