
Abstract
Cinnamomum camphora linalool type (C. camphora linalool type) is an economically important plant that contains abundant linalool. To explore the optimal concentration of exogenously applied indole-3-butyric acid (IBA) for root formation and development, morphological analyses were performed with plants after a 1-min soaking treatment with five different concentrations of IBA. When grown on the medium (laterite with plant ash) for 60 days after treatment with 3000 mg/L IBA, the root ratio and the number of roots were significantly higher than those obtained with 0, 1000, 2000, or 4000 mg/L IBA treatment. After soaking with 3000 mg/L IBA, the stem bases at five key stages of root development (0, 14, 21, 28, and 42 days) were selected for global transcriptome analyses. IBA treatment modified the expression of genes related to the auxin, brassinosteroid, and abscisic acid signalling transduction pathways. The rooting process reduced the accumulation of starch and the synthesis of linalool but promoted the metabolism of D-glucose. Our study identified the optimum concentration of IBA for a short treatment of C. camphora linalool type stem segments to regulate root formation and development at the transcriptional level.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cinnamomum camphora (Linn.) Presl (C. camphora) is one of the oldest herbal medicines used as in traditional medicine (Chen et al. 2020). It can be divided into the isoborneol, camphora, cineole, linalool, and borneol types according to the composition of the essential oils of the branches and leaves (Guo et al. 2017). Most of these types have been used in biomedicines, spices, cosmetics, and forest planting (Chen et al. 2018; Liu et al. 2019; Wu et al. 2019).
Cinnamomum camphora var. linaloolifera Fujita is the linalool-type C. camphora and is an evergreen broadleaved tree with the advantages of rapid growth, dense branches, and straight trunks. It is also an excellent urban green-tree species in southern China and is cultivated as an economically important tree for its medicinal and aromatic properties (Chen et al. 2017; Li et al. 2018). The volatile oil, based on the GC–MS analysis, in C. camphora linalool type leaves is mainly composed of linalool (60.97%, the highest in all C. camphora), camphor (7.13%), caryophyllene (5.17%), and eucalyptus oleol (3.8%) (Guo et al. 2016; Xinyu Fu et al. 2016; Wang et al. 2019). Linalool extracted from the leaves, twigs, and seeds shows insecticidal and repellent activities against cotton aphid (Jiang et al. 2016), Schistosomiasis japonicum (Yang et al. 2014), Pseudomonas aeruginosa, and Escherichia coli (Wang et al. 2019). With the increasing interest in naturally occurring essential oils with insecticidal and insect repellent activity, including linalool, C. camphora linalool type has gained the attention of researchers.
At present, cultivation of C. camphora plants is mainly through cutting, grafting, and tissue culture seedlings (Wu Youmei et al. 2009). Previous research found that the growth and biomass of tissue culture seedlings were higher than that of cutting seedlings and grafting seedlings, with more adventitious roots with high quality. And the number of thick roots of cuttings was less and the quality of adventitious roots was lower than that of tissue culture seedlings (Yue-Qiu and Wang 2015). Cutting seedlings also has the disadvantages of low survival rates, slow growth rates and other limitations to rapid propagation without any treatment with exogenous phytohormones (over 50 days for the annual branches on May) (Guorong Huang 2020). Auxin, abscisic acid (ABA), brassinosteroid (BRs), and other phytohormones play important roles in the rooting process of C. camphora linalool type and show a dynamic equilibrium (Lal et al. 2007; Du et al. 2012; Gonzalez et al. 2015; Jamro et al. 2018). ABA plays a crucial role in root architecture (Dong et al. 2018), while auxin plays a key role in lateral root formation (Xie et al. 2000) and BRs determine the root hair specification by regulating the fate of the root epidermal cell (Cheng et al. 2014). Considering that the cost and it is difficult to popularize tissue culture on a large scale, exogeneous application of plant hormone (such as IBA) is the most suitable way to address disadvantages of traditional cutting method.
In this study, stem segments of the annual shoot were used for morphological analysis to identify the optimal exogenous concentration (3000 mg/L) of β-oxidation of indole-3-butyric acid (IBA) to maximize the rooting potential of C. camphora linalool type. The rooting process of stems after the treatment of 3000 mg/L IBA was observed for 6 weeks, and five root development key nodes (0, 14, 21, 28, and 42 days) were selected for the determination of the transcriptome. According to the transcriptome analysis results, auxin and BR signalling were found to be involved in the regulation of root development, linalool biosynthesis, and carbohydrate metabolism. RT-PCR of core DEGs and the content determination of linalool, starch, and soluble sugar were also performed to provide further evidences. We propose that a combination of the metabolic regulation of core substances and the signal transduction of plant hormones provides the molecular basis for the development of C. camphora linalool type roots and rapid seeding.
Results
The Optimum Concentration of Exogenous IBA in Stem Segment Culture
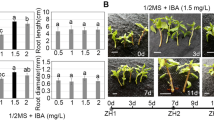
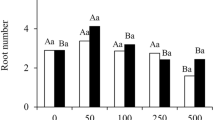
To explore the optimum concentration of exogenous IBA in the stem segment culture of C. camphora linalool type, different concentrations of IBA solutions (1000, 2000, 3000, and 4000 mg/L) were used to soak the stem bases for 1 min, which were then grown on culture medium (laterite with plant ash) for 60 days (Fig. 1A). The root ratio of the stem segments treated with IBA was significantly higher than that of the control group (0 mg/L). The highest rooting rates were found after 3000 mg/L IBA treatment (67.67 ± 3.52%). The root number of stem segments after the treatment with 3000 mg/L IBA was also the highest (3.27 ± 0.08), followed by 2000 mg/L IBA, and all of them were significantly higher than that of control. The root length of stem segments after treatment with 4000 mg/L IBA was the longest (4.20 ± 0.87 cm) but showed no significant difference from that of 3000 mg/L IBA (3.89 ± 0.43 cm). There were no significant differences in root diameters among the treatments (Fig. 1B). In summary, 3000 mg/L IBA is the optimum concentration to promote the rooting of C. camphora linalool type stem segments.

Root development under the different concentration of IBA. A Phenotypes of the stem sections before and 60 days after a 1-min soaking treatment of 0, 1000, 2000, 3000, and 4000 mg/L IBA solutions. White bar = 2 cm. B Root ratio, number of root, root length, and root diameter after a 60-day stem section cultures after a 1-min soaking treatment of 0, 1000, 2000, 3000, and 4000 mg/L IBA. C Phenotypes of the stem sections with a 0-day, 7-day, 14-day, 21-day, 28-day, 35-day, and 42-day stem section cultures after the treatment of 3000 mg/L IBA. The 0-day, 14-day, 21-day, 28-day, and 42-day plants were chosen and named as F1 to F5 respectively. White bar = 2 cm. All bars represent means ± s.d, and those labelled with a letter are significantly different at P < 0.05 by Duncan’s test. Three biological replicates in the experiment were performed, and each biological replicate consists of 450 stems
Phenotypes of the Stem Segment Base
The phenotypes of the stem sections after 0, 7, 14, 21, 28, 35, and 42 days of stem section culture after treatment with 3000 mg/L IBA are shown in Fig. 1C. There were no obvious morphological changes at the base of the cuttings from the first week, and the cuttings healed slowly in the second week. The incision swelling was more obvious during the third week and more callus appeared at the base. The callus area was enlarged in the fourth week. A large number of adventitious roots appeared after 6 weeks of culture accompanied by fibrous roots. Considering that the base of the stem is the part of direct contact and absorption of exogenous IBA, and is the active and core area of cell differentiation during rooting, the base of the cuttings at five time points (0, 14, 21, 28, and 42 days) representing the major stages in root development was chosen for global transcriptome analysis and were respectively named F1, F2, F3, F4, and F5.
Differentially Expressed Genes During Root Development
To understand the molecular mechanism and regulation networks of C. camphora linalool type during its root developmental periods, differentially expressed genes (DEGs; log2 (relative expression level) ≥ 1 and ≤ − 1, p ≤ 0.05) in F2, F3, F4, and F5 compared with F1 were identified by RNA-Seq (Supplemental Tables S1, S2, S3 and S4). Principal component analysis (PCA) of the DEGs revealed the separation of gene expression profiles among F1 to F5 was different from each other. The distribution of the three biological replicates was aggregated (Fig. 2A). As summarised in Fig. 2B, compared with F1, 1523 up- and 1530 down-regulated genes were observed in F2, 3011 up- and 2119 down-regulated genes were observed in F3 compared with F1, 5049 up- and 3614 down-regulated genes were observed in F4 against F1, and 5316 up- and 3733 down-regulated genes were observed in F5 compared with F1 (Fig. 2B). When comparing the DEGs of different groups, 1208 genes were consistently up- or down-regulated during the developmental periods and had a large number of specifically expressed genes (Fig. 2C). The clustering analysis of the relative expression levels of all of the DEGs (union of 10 comparative combinations in Fig. 2B) is shown in Fig. 2D.

Overall differentially expressed genes (DEGs) during the stem segment culturing. A Principal component analysis (PCA) of differentially expressed genes (log2 (relative expression level) ≥ 1 and ≤ − 1, P < 0.05, DEGs) in F1, F2, F3, F4, and F5. B The histogram of the statistical results for the DEGs in 5 groups. The blue for down-expressed genes and the gray for up-expressed genes. C Venn diagram of the overlap between the DEGs between F2 and F1, F3 and F2, F4 and F3, F5 and F4. D The clustering analysis results of the relative expression levels of DEGs between five different groups. The abscissa is the sample name, and the ordinate is the normalized value of the differential gene FPKM. The redder the colour, the higher the expression level. The more green the colour, the lower the expression
Gene ontology (GO) enrichment analyses were performed with the DEGs in F2 to F5 compared with F1 (Fig. 3A). The up-regulated genes were enriched in the metabolism of cellular glucan and polysaccharides, as well as the biosynthesis of cellular carbohydrate and ATP, especially in response to external stimuli (such as pathogenesis-related protein CKAN_01791200 /CKAN_01791000) and oxidative stress (such as peroxidase CKAN_00579200/CKAN_00756800) (Fig. 3A, red dotted box). Down-regulated genes were enriched in the metabolism of coenzymes, cofactors and nucleotides (Fig. 3A, green dotted box), glycolysis, and the response to chemicals. Each pair of genes (red and green points in Fig. 3A) with significant changes in the expression of pathogenesis-related protein and peroxidase families were selected for real-time RT-PCR analysis. The time-specific expression assay results (Fig. 3B) are in agreement with the RNA-seq results. The activity of peroxidase enzyme (POD) was consistent with the expression trend (Fig. 3C). The KEGG pathway enrichment assay was also performed with these DEGs, and both the up- and down-regulated genes were enriched in starch and sucrose metabolism, while the up-regulated genes were specifically enriched in plant hormone signal transduction and the down-regulated genes were specifically enriched in linalool biosynthesis (Fig. 3C, orange or blue squares, respectively, for the up- and down-regulated genes).

GO and KEGG enrichment of the DEGs during the stem segment culturing. A GO (biological process) enrichment analysis (P-value for significance test between GeneRatio and BgRatio) clustering results and the interactive graph analysis results of the up-expressed (red) and down-expressed (green) gene of F2, F3, F4, and F5 in contrast to F1. The clustering analysis results of genes, which belonged to the response to external stimulus and oxidative stress, the metabolic of trehalose, relative expression levels of the different groups based on the results of RNA-seq analysis. B Expression profiles of genes (real-time RT-PCR results) selected in A (red and green points). C The POD enzyme activity determination results. D KEGG pathway enrichment analysis clustering results of the up-expressed (orange) and down-expressed (blue) gene of F2, F3, F4, and F5 in contrast to F1. All bars represent means ± s.d. (three biological replicates, each biological replicate consists of 5 stems) and those labelled with a letter are significantly different at P < 0.05 by Duncan’s test
Transcriptome Differences in Plant Hormone Signal Transduction Pathways During Root Development
Both the GO and KEGG pathway of DEGs were significantly enriched in the response to extremal stimuli and plant hormone signal transduction (Figs. 4A and C). By comparison, auxin and brassinosteroid (BR) were the two most altered hormone signalling cascades, as demonstrated by the following: down-regulated expression of eight AUX/IAA (auxin-responsive protein) genes; increased expression of auxin response genes, including one ARF (Arf GTPase activating protein), two GH3 (beta-glucosidase), and six SAUR-like auxin-responsive family genes; increased expression of five BRI1 (BRASSINOSTEROID INSENSITIVE 1) genes, three BZR1/2 (BRASSINAZOLE-RESISTANT 1/2) genes, one CYCD3 (cyclin D-type protein 3) gene, and five TCH4 (xyloglucan:xyloglucosyl transferase 4) genes. Concomitantly, each pair of genes with significant changes in the expression of AUX/IAA and SAUR, BRI1, BZR1/2, and TCH4 family members were selected for real-time RT-PCR analysis. The time-specific expression assay results (Fig. 4B) were consistent with the RNA-seq results.

Differential expression of representative genes involved in plant hormone signal transduction. A DEGs in auxin and brassinosteroid signal transduction pathway. B Expression profiles of genes involved in auxin and brassinosteroid signal transduction pathway from F1 to F5. C DEGs in abscisic acid signal transduction pathway. The absolute values of log2 (F2, 3, 4, 5/F1) ≥ 1 and FDR < 0.001 were used as the criteria for DEGs. The colour of the box represents up (red) and down (green)-regulated genes. All bars represent means ± s.d (three biological replicates, each biological replicate consists of 5 stems) and those labelled with a letter are significantly different at P < 0.05 by Duncan’s test
During the formation of adventitious roots, there are contrasting changes in the amount of endogenous IAA and ABA in the roots. The expression of the ABA signalling pathway transduction genes in the transcriptome data conforms to this, as demonstrated by the down-regulated expressions of four PYR/PYL (REGULATORY COMPONENT OF ABA RECEPTOR) genes and four ABF (ABA responsive element binding factor) genes, as well as the up-regulated expressions of nine PP2C (type 2C protein phosphatases) genes (Fig. 4C).
Expression Profiles of Carbohydrate Metabolism Pathway Genes
DEGs were significantly enriched in the linalool biosynthesis, as well as starch and sucrose metabolic pathways, and we, therefore, compared the expression trends of core genes in these pathways. Significant repression of genes involved in the linalool biosynthesis process was observed, including three (3S)-linalool synthases and five linalool 8-monooxygenases (Fig. 5A). Genes in the starch and sucrose metabolic pathway showed significant changes in expression during the culture (Fig. 5B). Genes encoding four sucrose synthases, three trehalose phosphatases, five glycosyl hydrolases, five β-glucosidases, four β-amylases, two starch synthase, one 4-α-glucanotransferase, and five glycosyltransferases were significantly up-regulated in F2 to F5 compared with F1 (Fig. 5B). Genes encoding four sucrose synthases, one cell wall invertase, two fructokinases, two glucose-6-phosphate isomerases, and two phosphoglucomutases were repressed in F2 to F5 compared with F1 (Fig. 5B). Genes with significant changes in expression of (3S)-linalool synthase and linalool 8-monooxygenase, glycosyl hydrolase, β-glucosidase, 4-α-glucanotransferase, and glycosyltransferase families were also selected for real-time RT-PCR analysis to confirm the RNA-Seq assay results (Fig. 5C). The linalool, starch, and soluble sugar contents in stems during the five periods were determined after 12-h light exposure. The content of soluble sugars was increased, while the content of starch and linalool were decreased with the time of culture (Fig. 5D).

Differential expression of representative genes involved in linalool biosynthesis and starch and sucrose metabolism. A DEGs in linalool biosynthesis. B DEGs in starch and sucrose metabolism. C Expression profiles of representative genes involved in linalool biosynthesis and starch and sucrose metabolism. D Changes of starch, soluble sugar and linalool contents in F1, F2, F3, F4, and F5. The absolute values of log2 (F2, 3, 4, 5/F1) ≥ 1 and FDR < 0.001 were used as the criteria for DEGs. The colour of the box represents up (red)- and down (green)-regulated genes. All bars represent means ± s.d (three biological replicates, each biological replicate consists of 5 stems) and those labelled with a letter are significantly different at P < 0.05 by Duncan’s test
Discussion
Accuracy and Representativeness of Transcriptome Data
Indole-3-butyric acid plays a role in the root regeneration of different plant species (Pan and Tian 1999; Rout 2006; Ali et al. 2009), and its physiological role is two-fold: a low concentration of IBA promotes plant growth, while a high concentration retards development through regulating the proliferation and expansion of cells (Björkman 2004; Iqbal et al. 2016). Based on the root ratios, the number of roots, and root length and diameter, 3000 mg/L IBA was considered to be the optimum concentration for further transcriptome measurements. Plant tissue cultivation always begins with the process of the stress response. Based on the GO enrichment results (Fig. 3A), the expression of genes involved in reactive oxygen stress peroxidases (Fu 2014), salicylic acid, and auxin response pathogenesis-related protein (Mitsuhara et al. 2008) were significantly induced. The peroxidase enzyme activity measured during the five stages was similar with the trend in the level of transcription (Fig. 3C). This result illustrates the accuracy of transcriptome sampling and the reliability of transcriptome data from the side.
Dynamic Homeostasis of Plant Hormones During Root Development
By comparing the expression of key genes in various plant hormone signal transduction pathways, it was found that the auxin, BR, and ABA signal transduction pathway genes were significantly altered during root development (Figs. 4A and C). Auxin plays a crucial role in root growth and developmental regulation by regulating the expression of AUX/IAA, GH3, and SAUR et al. Two Indole-3-pyruvate monooxygenase (CKAN_01183000 and CKAN_00588900), which involved in auxin biosynthesis, were repressed in F2 to F5 when compared with F1. That is consistent with previous studies that exogenous IBA inhibits endogenous auxin biosynthesis. The AUX/IAAs are also repressed by treatment with exogenous auxin and encode short-lived nuclear proteins (Park et al. 2002). In our study, CKAN_01958500, CKAN_01302600, and another six genes were significantly repressed in F2 to F5 compared with F1. The AtIAA2/3 orthologs in Arabidopsis regulate the development of lateral roots through activating the expression of LBD16/18 (Lee et al. 2015a, b). As the downstream regulating gene of AUX/IAA, CKAN_01177000 encodes AUXIN RESPONSE FACTOR (ARF) and was up-regulated in the later stages of tissue culture when AUX/IAA may retard the expression of ARF TFs in the absence of auxin (Korasick et al. 2014). CKAN_02009600 and CKAN_02242800 are orthologs of AtGH3.17 in C. camphora linalool type and encode IAA-amino synthases (group II family) (Pierdonati et al. 2019). There was increased expression of CKAN_02519500, CKAN_01995300, and another four genes, which are the orthologs of SAUR41 (SMALL AUXIN UPREGULATED 41) in C. camphora linalool type and encode clade III SAUR proteins that contribute to radial root pattern formation (Qiu et al. 2013, 2019). Transcriptome data also revealed that a large number of auxin efflux carriers (PIN, involved in the root-specific auxin transport, such as CKAN_01601500, CKAN_00099200, and CKAN_00765500) and auxin-induced proteins 6B (CKAN_00774200) and X15 (CKAN_00761400) were significantly up-regulated after IBA treatment.
In other species, the content of IAA and ABA show a contrasting trend; i.e., the content of ABA is lowest when the IAA content is highest, during the development of seeds and roots (García-Martín et al. 2005; Albacete et al. 2008; Bosco et al. 2014). In this study, the expression of the core genes in the ABA signal transduction pathway, such as the PYR/PYL and ABF, were just repressed gradually (Choi et al. 2000; Kim et al. 2004; Fujii et al. 2007; Ma et al. 2009). PYR/PYLs are ABA-receptors that function at the apex of a negative regulatory pathway by inhibiting PP2Cs (Park et al. 2009; Hao et al. 2011), which is similar to the gradually increasing expression of CKAN_02175500 and another eight PP2Cs. The increased expression of PP2C inactivates SnRK2 by direct dephosphorylation to retard the expression of ABF indirectly (Umezawa et al. 2010).
Brassinosteroid is a plant growth-promoting steroid that was first isolated from the pollen of Brassica napus (Grove et al. 1979). BRI1 is an important component of the BR receptor that is required for systemin-induced root elongation (Holton et al. 2007). BZR1/2 controls root meristem development by regulating the cell division (Hak-Soo et al. 2015; Espinosa-Ruiz et al. 2017) and increasing the expression of CYCD3 and TCH4. CYCD3 positively affects root development and cell division (Li et al. 2008; Randall et al. 2015), while TCH4 encodes one of the xyloglucan endo-transglycosylase (XET) enzymes to increase cell elongation and induced expressed by the auxin (Xu et al. 1996; Koka et al. 2000; Friedrichsen and Chory 2001).
In summary, the expression trend of auxin signalling transduction pathway genes positively correlated with that of BRs and negatively correlated with that of ABA. Together, these indicate a trend towards the occurrence of root primordia and the development of roots.
Changes in Carbon Metabolism and Secondary Metabolism During Rooting
As a crucial secondary metabolite of C. camphora linalool type, linalool has insecticidal and insect repellent activity. (3S)-Linalool synthase (TPS14) synthesises linalool from geranyl diphosphate (Cseke et al. 1998; Aubourg et al. 2002). Geranyl diphosphate is an intermediate product of the terpenoid backbone biosynthesis process, whose substrate is glyceraldehyde 3-phosphate (G3P, intermediate product of the glycolysis pathway), and G3P forms acetyl-CoA via glycolysis (Lehninger et al. 2008; Hu et al. 2017). Linalool is then metabolised by 8-monooxygenase (CYP111A) into 8-hydroxylinalool and 8-oxolinalool (Bell et al. 2010; Ilc et al. 2017). Both TPS14 and CYP111A were repressed during the rooting process, which suggests that the decline in linalool levels might be caused by a decrease in biosynthesis rates. It is worth noting that the KEGG pathway genes were enriched in the starch and sucrose metabolism process (Fig. 3C). By comparing the expression of key genes based on both the RNA-seq and real-time RT-PCR data, a large amount of starch and sucrose can be converted into D-glucose (Figs. 5B and C). We also found that genes encoding two glyceraldehyde-3-phosphate dehydrogenase (CKAN_01457400 and CKAN_01754000, 2.67- and 1.13-fold, respectively), one pyruvate kinase (CKAN_01349800, 2.59-fold) and one enolase (CKAN_01387600, 0.36-fold expression level in F5 compared with F1), were up-regulated. All of these genes encode key rate-limiting enzymes in the glycolysis pathway that converts glucose into pyruvate and acetyl-CoA (Chuang et al. 2005; Piast et al. 2005). Therefore, we speculate that during rooting, C. camphora linalool type plants accumulated glucose as a substrate to produce a large amount of acetyl-CoA to provide energy (such as ATP) through the tricarboxylic acid cycle for the rooting and stress response processes. At the same time, the reduction of the G3P content retarded the synthesis of linalool.
Conclusion
Based on transcriptome data and physical and chemical indicators, a proposed model for the stem segment cultures after the treatment of exogenous IBA can be proposed: exogenous IBA modifies carbohydrate, secondary metabolism and the auxin, and BR and ABA signalling transduction pathways (Fig. 6). Auxin and BRs play a positive and synergistic role in the rooting process, but this is in contrast to the expression trend of ABA signalling transduction pathway-related genes. On the other hand, the accumulation of starch gradually decreases and a large amount of D-glucose is used to synthesise pyruvate and acetyl-CoA through the glycolysis pathway to provide more energy for the rooting and stress responses. In consequence, the synthesis of linalool was slowed down by retarding the expression of terpenoid backbone biosynthesis pathway genes.

The proposed model for the stem segment cultures after the treatment of exogenous IBA. Exogenous IBA promotes the rooting of Cinnamomum camphora linalool type cuttings by regulating carbohydrate, secondary metabolism and the auxin, BR, and ABA signalling transduction pathways
Materials and Methods
Plant Materials
The branches were from cuttings of 4 years old Cinnamomum camphora linalool type ‘Ganfang No. 1’ (Gan-S-SC-CC-001–2020). The average oil yield of fresh weight, dry weight, and L-linalool content of leaves were 2.53%, 6.03%, and 93.50% respectively. The mother tree of Ganfang No. 1 is from Xikou Village, Datong Town, Jiande City, Zhejiang Province, with a diameter at breast height of 25 cm, a height of 12 m, and a north–south crown width of 10 m. In the experiment, 1-year semi-lignified softwood twigs with one leaf and one bud were selected as cuttings. Stem segment cutting were performed by randomizing block arrangement after the 1-min soaking treatment of 0, 1000, 2000, 3000, and 4000 mg/L IBA solutions on the medium (Laterite: plant ash, v:v, 4:1). Use plastic film to maintain humidity above 80%, the temperature to 15 ~ 20 °C (night)/25 ~ 30 °C (day), and to avoid direct light (shading 70 ~ 75%). Before cutting, 0.1% carbendazim solution was used to sterilize the mediums and cuttings, and once a week after cutting. Root ratio, number of root, root length, and root diameter were calculated after a 60-day culturing. Three biological replicates in the experiment were performed, and each biological replicate consists of 450 stems. For RNA extraction and content measurement, fresh tissues of the stem base (15 mm, 5 plants per sample) taken 0 day, 14 days, 21 days, 28 days, and 42 days after planting on the media were collected.
Library Construction
All samples were sent to Novogene (Beijing, China) for extraction, sequencing, and bioinformatics analyses. The total RNA was extracted using Trizol reagent (Takara, Japan) and treated with 1U DNAse I (Life Technologies). Total RNA integrity, RIN = 7.4–8.8, 28S/18S = 1.3–1.4; total RNA purity, OD260/OD280 = 1.88–1.97, OD 260/0D230 = 1.93–2.04; total RNA amount = 89.6–162.4 μg were quantified by Agilent 2100 bioanalyzer and Nanodrop spectrophotometer (Thermo Scientific). mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. Fragmentation was carried out using divalent cations under elevated temperature in NEBNext First Strand Synthesis Reaction Buffer (5X). First-strand cDNA was synthesized using random hexamer primer and M-MuLV Reverse Transcriptase (RNase H-). Second strand cDNA synthesis was subsequently performed using DNA Polymerase I and RNase H. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of the 3’ ends of the DNA fragments, NEBNext Adaptor with a hairpin loop structure was ligated to prepare for hybridization. In order to select cDNA fragments that were preferentially 250–300 bp in length, the library fragments were purified with an AMPure XP system (Beckman Coulter, Beverly, USA). Then, 3 μl USER Enzyme (NEB, USA) was used with size-selected, adaptor-ligated cDNA at 37 °C for 15 min, followed by 5 min at 95 °C before PCR analysis. Then, PCR was performed with Phusion High-fidelity DNA polymerase, Universal PCR primers, and Index (X) Primer (14 cycles). Finally, PCR products were purified (AMPure XP system) and library quality was assessed on the Agilent Bioanalyzer 2100 system (OD260/280 = 1.79–1.84, OD260/230 = 1.71–1.78).
Clustering and Sequencing
Clustering of the index-coded samples was performed on a cBot Cluster Generation System using a TruSeq PE Cluster Kit v3-cBot-HS (Illumina) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina Novaseq platform and 150-bp paired-end reads were generated. The range of raw reads of these 15 samples was from 46,066,820 to 53,058.852, with the error rates from 0.2 to 0.3, Q30 from 92.39 to 94.51. Reads with adapter, N (undetermined base) and reads with low quality (over 50% bases showed Qphred ≤ 20) were filtered out, and the clean reads were from 43,920,844 to 51,980,346. Exactly 83.55 to 87.24% of the clean reads were mapped on the genome of a total of 83.55 to 87.24% of the sequences that were associated with the reference genome of Cinnamomum kanehirae (Asbrc_ckan_0, BioProject: PRJNA477266), and 73.51–75.12%, 2.73–6.21%, and 22.15–20.25% of the mapped reads were mapped on the exon, intron, and intergenic respectively.
Differential Expression Analysis
Differential expression analysis of five groups (three biological replicates per condition) was performed using the R (3.5.1) package DESeq2_1.20.0 (Love et al. 2014). DESeq2 provides statistical routines for determining differential expressions in digital gene expression data using a model based on the negative binomial distribution. The resulting P-values were adjusted using Benjamini and Hochberg’s approach for controlling the false discovery rate. Genes with an adjusted P < 0.05 found by DESeq2 were assigned as differentially expressed. The selection of differentially expressed genes was based on FDR ≤ 0.001 and an absolute value of Log2 (relative expression level, compared with WT) ≥ 1.
GO and KEGG Enrichment Analysis of Differentially Expressed Genes
Case special deep analysis based on DEGs, including GO and KEGG pathway enrichment analysis, was done by our group. Gene ontology (GO) enrichment analysis of differentially expressed genes was implemented by the cluster Profiler R(3.5.1) package (Young et al. 2010), in which gene length bias was corrected. GO terms with corrected P < 0.05 were considered significantly enriched by differential expressed genes. KEGG is a database for understanding the high-level functions and utilities of biological systems, such as cells, organisms, and ecosystems, from molecular-level information, especially large-scale molecular datasets generated by genome sequencing and other high-throughput experimental technologies (http://www.genome.jp/kegg/). We used the cluster Profiler R(3.5.1) package to test the statistical enrichment of differentially expressed genes in KEGG pathways (Kanehisa et al. 2008).
Plant RNA Extraction and Real-Time RT-PCR
The total RNA was extracted using Trizol reagent (Takara, Japan) and treated with 1U DNAse I (Life Technologies). Total RNA integrity, RIN = 7.1–8.2, 28S/18S = 1.2–1.4; total RNA purity, OD260/OD280 = 1.82–1.98, OD 260/0D230 = 1.89–2.01; and total RNA amount = 102.13–245.80 μg were quantified by Agilent 2100 bioanalyzer and Nanodrop spectrophotometer (Thermo Scientific). An amount of 500 ng total RNA was used for inverse transcription with a Transcriptor First Strand cDNA Synthesis Kit (Takara, Japan). We diluted the cDNA 10 times and then used it as the template for real-time PCR (RT-PCR). The RT-PCR reaction system comprised SYBR® Premix Ex TaqTM (2 ×) 5 μl (Takara, Japan), PCR Forward Primer (10 μM) 0.2 μl, PCR Reverse Primer (10 μM) 0.2 μl, cDNA template 1 μl, and distilled deionized water up to 10 μl. The PCR process used the following conditions: 95 °C for 15 s, 58.2–66.8 °C for 30 s, and 72 °C for 30 s for 40 cycles. CKAN_02474600 which encoded Actin was selected as the stably expressed reference genes for the normalization of the RT-qPCR data. Transcriptome data indicated that the expression level of CKAN_02474600 showed no significant difference between these five groups (|log2 FC|< 0.01), and the amplification efficiency of its primer was 103.97% with R2 = 0.9867. Melt curves of 22 genes were all unimodal, which indicates that the primer specificity is high. Details of primers sequences, melting temperature, primer efficiency (E, %), and R2 were showed in Supplemental Table S5.
Determination of POD Enzyme Activity and the Content of Soluble Sugars, Starch, and Linalool
The activity of peroxidase enzyme (POD) were determined according to the method of Vetter (Vetter et al. 1958). Starch and soluble sugars were determined with Anthrone reagent (Hansen and Moller 1975; Taylor et al. 1982) with the stems (1.5 cm to the end) as the materials (three biological replications, 5 plants for each biological replication). Linalool were determined with gas chromatography (Wang et al. 2006).
References
Albacete A, Ghanem ME, Martínez-Andújar C, Acosta M, Pérez-Alfocea F (2008) Hormonal changes in relation to biomass partitioning and shoot growth impairment in salinized tomato (Solanum lycopersicum L.) plants. J Exp Bot 59:4119–4131
Ali A, Ahmad T, Abbasi NA, Hafiz IA (2009) Effect of different concentrations of auxins on in vitro rooting of olive cultivar ‘Moraiolo.’ Pakistan J Bot 41:1223–1231
Aubourg S, Lecharny A, Bohlmann J (2002) Genomic analysis of the terpenoid synthase (AtTPS) gene family of Arabidopsis thaliana. Mol Genet Genomics 267:730–745
Bell SG, Dale A, Rees NH, Wong LL (2010) A cytochrome P450 class i electron transfer system from Novosphingobium aromaticivorans. Appl Microbiol Biotechnol 86:163–175
Björkman T (2004) Effect of Trichoderma colonization on auxin-mediated regulation of root elongation. Plant Growth Regul 43:89–92
Bosco R, Caser M, Ghione GG, Mansuino A, Giovannini A, Scariot V (2014) Dynamics of abscisic acid and indole-3-acetic acid during the early-middle stage of seed development in Rosa hybrida. Plant Growth Regul 75:265–270
Chen C, Zheng Y, Liu S, Zhong Y, Meng X (2017) The complete chloroplast genome of Cinnamomum camphora and its comparison with related Lauraceae species. PeerJ 5(2):e3820
Chen C, Zheng Y, Zhong Y, Wu Y, Meng X (2018) Transcriptome analysis and identification of genes related to terpenoid biosynthesis in Cinnamomum camphora. BMC Genomics 19:550
Chen J, Tang C, Zhang R, Ye S, Yang D (2020) Metabolomics analysis to evaluate the antibacterial activity of the essential oil from the leaves of Cinnamomum camphora (Linn.) Presl. J Ethnopharmacol. 253:112652
Cheng Y, Zhu W, Chen Y, Ito S, Asami T, Wang X (2014) Brassinosteroids control root epidermal cell fate via direct regulation of a MYB-bHLH-WD40 complex by GSK3-like kinases. Elife. 3:e02525
Choi HI, Hong JH, Ha JO, Kang JY, Kim SY (2000) ABFs, a family of ABA-responsive element binding factors. J Biol Chem 275:1723–1730
Chuang DM, Hough C, Senatorov VV (2005) Glyceraldehyde-3-phosphate dehydrogenase, apoptosis, and neurodegenerative diseases. Annu Rev Pharmacol Toxicol 45:269–290
Cseke L, Dudareva N, Pichersky E (1998) Structure and evolution of linalool synthase. Mol Biol Evol 15:1491–1498
Dong H, Bai L, Chang J, Song CP (2018) Chloroplast protein PLGG1 is involved in abscisic acid-regulated lateral root development and stomatal movement in Arabidopsis. Biochem Biophys Res Commun 495:280–285
Du J, Yin H, Zhang S, Wei Z, Zhao B, Zhang J, Gou X, Lin H, Li J (2012) Somatic embryogenesis receptor kinases control root development mainly via brassinosteroid-independent actions in Arabidopsis thaliana. J Integr Plant Biol 54:388–399
Espinosa-Ruiz A, Martínez C, de Lucas M, Fàbregas N, Bosch N, Caño-Delgado AI, Prat S (2017) TOPLESS mediates brassinosteroid control of shoot boundaries and root meristem development in Arabidopsis thaliana. Dev 144:1619–1628
Friedrichsen D, Chory J (2001) Steroid signaling in plants: from the cell surface to the nucleus. Bioessays 23:1028–1036
Fu JY (2014) Cloning of a new glutathione peroxidase gene from tea plant (Camellia sinensis) and expression analysis under biotic and abiotic stresses. Bot Stud 55:7
Fu YX, Jiang XM, Luo LP, Zhang T, Guo XL, He YC (2016) A GC-MS analysis of volatile oil from different types of Cinnamomum camphora leaves. J For Eng. 72–76
Fujii H, Verslues PE, Zhu JK (2007) Identification of two protein kinases required for abscisic acid regulation of seed germination, root growth, and gene expression in Arabidopsis. Plant Cell 19:485–494
García-Martín G, Manzanera JA, González-Benito ME (2005) Effect of exogenous ABA on embryo maturation and quantification of endogenous levels of ABA and IAA in Quercus subersomatic embryos. Plant Cell Tissue Organ Cult 80:171–177
Gonzalez AA, Agbévénou K, Herrbach V, Gough C, Bensmihen S (2015) Abscisic acid promotes pre-emergence stages of lateral root development in Medicago truncatula. Plant Signal Behav 10:e977741
Grove MD, Spencer GF, Rohwedder WK, Mandava N, Cook J (1979) Brassinolide, a plant growth-promoting steroid isolated from Brassica napus pollen. Nature 281:216–217
Guo S, Geng Z, Zhang W, Liang J, Wang C, Deng Z, Du S (2016) The chemical composition of essential oils from Cinnamomum camphora and their insecticidal activity against the stored product pests. Int J Mol Sci 17:1836
Guo X, Cui M, Deng M, Liu X, Luo L (2017) Molecular differentiation of five Cinnamomum camphora chemotypes using desorption atmospheric pressure chemical ionization mass spectrometry of raw leaves. Sci Rep 7:46579
Hansen J, Moller IB (1975) Analysis of starch and soluble sugars with Anthrone reagent. Anal Biochem 68(1):87–94
Hao Q, Yin P, Li W, Wang L, Yan C, Lin Z, Wu JZ, Wang J, Yan SF, Yan N (2011) The molecular basis of ABA-independent inhibition of PP2Cs by a subclass of PYL proteins. Mol Cell 42:662–672
Holton N, Caño-Delgado A, Harrison K, Montoya T, Chory J, Bishop GJ (2007) Tomato BRASSINOSTEROID INSENSITIVE1 is required for systemin-induced root elongation in Solanum pimpinellifolium but is not essential for wound signaling. Plant Cell 19:1709–1717
Hu Z, Tang B, Wu Q, Zheng J, Leng P, Zhang K (2017) Transcriptome sequencing analysis reveals a difference in monoterpene biosynthesis between scented Lilium ‘Siberia’ and unscented Lilium ‘Novano.’ Front Plant Sci 8:1351
Huang G (2020) The cutting propagation Technique of Cinnamomum camphora. Fujian for 210:48–50
Ilc T, Halter D, Miesch L, Lauvoisard F, Kriegshauser L, Ilg A, Baltenweck R, Hugueney P, Werck-Reichhart D, Duchêne E, Navrot N (2017) A grapevine cytochrome P450 generates the precursor of wine lactone, a key odorant in wine. New Phytol 213:264–274
Iqbal MA, Khalid M, Zahir ZA, Ahmad R (2016) Auxin producing plant growth promoting rhizobacteria improve growth, physiology and yield of maize under saline field conditions. Int J Agric Biol 18:37–45
Jamro M, Shah AN, Nizamani FK (2018) Effects of IBA and NAA on integrated root development in aerial offshoots of Phoenix dactylifera L. Bangladesh J Bot 47:287–292
Jiang H, Wang J, Song L, Cao X, Yao X, Tang F, Yue Y (2016) Gc×Gc-tofms analysis of essential oils composition from leaves, twigs and seeds of cinnamomum camphora l. presl and their insecticidal and repellent activities. Molecules. 21:423
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y (2008) KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36 (Database issue):D480–4
Kim S, Kang JY, Cho DI, Park JH, Kim SY (2004) ABF2, an ABRE-binding bZIP factor, is an essential component of glucose signaling and its overexpression affects multiple stress tolerance. Plant J 40:75–87
Koka CV, Cerny RE, Gardner RG, Noguchi T, Fujioka S, Takatsuto S, Yoshida S, Clouse SD (2000) A putative role for the tomato genes DUMPY and CURL-3 in brassinosteroid biosynthesis and response. Plant Physiol 122:85–98
Korasick DA, Westfall CS, Lee SG, Nanao MH, Dumas R, Hagen G, Guilfoyle TJ, Jez JM, Strader LC (2014) Molecular basis for AUXIN RESPONSE FACTOR protein interaction and the control of auxin response repression. Proc Natl Acad Sci USA 111:5427–5432
Lal S, Tiwari JP, Awasthi P, Singh G (2007) Effect of IBA and NAA on rooting potential of stooled shoots of guava (Psidium guajava L.) cv. sardar. ACTA Horticulturae 735:193–196
Lee HS, Kim Y, Pham G, Kim JW, Song JH, Lee Y, Hwang YS, Roux SJ, Kim SH (2015a) Brassinazole resistant 1 (BZR1)-dependent brassinosteroid signalling pathway leads to ectopic activation of quiescent cell division and suppresses columella stem cell differentiation. J Exp Bot 66:4835–4849
Lee HW, Cho C, Kim J (2015b) LBD16 and LBD18 Act downstream of the AUX1 and LAX3 auxin influx carriers to control lateral root development in Arabidopsis thaliana. Plant Physiol 168:1792–1806
Lehninger A, Nelson DL, Cox MM (2008) Glycolysis, gluconeogenesis, and the pentose phosphate pathway. Lehninger Princ Biochem Fourth Ed 556–557
Li F, Xu Y, Zhang J, Yin S, Yu FG (2008) Overexpression of D-type cyclin CYCD3;1 affects root development in Arabidopsis thaliana L. Plant Physiol Commun 44:431–435
Li YR, Fu CS, Yang WJ, Wang XL, Feng D, Wang XN, Ren DM, Lou HX, Shen T (2018) Investigation of constituents from Cinnamomum camphora (L.) J. Presl and evaluation of their anti-inflammatory properties in lipopolysaccharide-stimulated RAW 264.7 macrophages. J Ethnopharmacol 221:37–47
Liu X, Meng Y, Zhang Z, Wang Y, Zhang D (2019) Functional nano-catalyzed pyrolyzates from branch of Cinnamomum camphora. Saudi J Biol Sci 26:1227–1246
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550
Ma Y, Szostkiewicz I, Korte A, Moes D, Yang Y, Christmann A, Grill E (2009) Regulators of PP2C phosphatase activity function as abscisic acid sensors. Science 324:1064–1068
Mitsuhara I, Iwai T, Seo S, Yanagawa Y, Kawahigasi H, Hirose S, Ohkawa Y, Ohashi Y (2008) Characteristic expression of twelve rice PR1 family genes in response to pathogen infection, wounding, and defense-related signal compounds (121/180). Mol Genet Genomics 279:415–427
Pan R, Tian X (1999) Comparative effect of IBA, BSAA and 5,6-Cl2-IAA-Me on the rooting of hypocotyl in mung bean. Plant Growth Regul 27:91–98
Park JY, Kim HJ, Kim J (2002) Mutation in domain II of IAA1 confers diverse auxin-related phenotypes and represses auxin-activated expression of Aux/IAA genes in steroid regulator-inducible system. Plant J 32:669–683
Park SY, Fung P, Nishimura N, Jensen DR, Fujii H, Zhao Y, Lumba S, Santiago J, Rodrigues A, Chow TF, Alfred SE, Bonetta D, Finkelstein R, Provart NJ, Desveaux D, Rodriguez PL, McCourt P, Zhu JK, Schroeder JI, Volkman BF, Cutler SR (2009) Abscisic acid inhibits PP2Cs via the PYR/PYL family of ABA-binding START proteins. Science 324:1068–1071
Piast M, Kustrzeba-Wójcicka I, Matusiewicz M, Banaś T (2005) Molecular evolution of enolase. Acta Biochim Pol 52:507–513
Pierdonati E, Unterholzner SJ, Salvi E, Svolacchia N, Bertolotti G, Dello Ioio R, Sabatini S, Di Mambro R (2019) Cytokinin-dependent control of GH3 group II family genes in the Arabidopsis root. Plants 8:94
Qiu T, Chen Y, Li M, Kong Y, Zhu Y, Han N, Bian H, Zhu M, Wang J (2013) The tissue-specific and developmentally regulated expression patterns of the SAUR41 subfamily of SMALL AUXIN UP RNA genes: Potential implications. Plant Signal Behav 8:e25283
Qiu T, Qi M, Ding X, Zheng Y, Zhou T, Chen Y, Han N, Zhu M, Bian H, Jl W (2019) The SAUR41 subfamily of SMALL AUXIN UP RNA genes is abscisic acid-inducible to modulate cell expansion and salt tolerance in Arabidopsis thaliana seedlings. Ann Bot 125:805–819
Randall RS, Miyashima S, Blomster T, Zhang J, Elo A, Karlberg A, Immanen J, Nieminen K, Lee JY, Kakimoto T, Blajecka K, Melnyk CW, Alcasabas A, Forzani C, Matsumoto-Kitano M, Mähönen AP, Bhalerao R, Dewitte W, Helariutta Y, Murray JA (2015) AINTEGUMENTA and the D-type cyclin CYCD3; 1 regulate root secondary growth and respond to cytokinins. Biol Open 4:1229–1236
Rout GR (2006) Effect of auxins on adventitious root development from single node cuttings of Camellia sinensis (L.) Kuntze and associated biochemical changes. Plant Growth Regul 48:111–117
Taylor JS, Blake TJ, Pharis RP (1982) The role of plant hormones and carbohydrates in the growth and survival of coppiced Eucalyptus seedlings. Physiol Plant 48:111–117
Umezawa T, Nakashima K, Miyakawa T, Kuromori T, Tanokura M, Shinozaki K, Yamaguchi-Shinozaki K (2010) Molecular basis of the core regulatory network in ABA responses: sensing, signaling and transport. Plant Cell Physiol 51:1821–1839
Vetter JL, Steinberg MP, Nelson AI (1958) Enzyme assay, quantitative determination of peroxidase in sweet corn. J Agric Food Chem 6:39–41
Wang FM, Mao XH, Shen JI (2006) Determination of linalool in Rhizoma Homalomenae by GC. Chinese Tradit Pat Med 7:88–89
Wang W, Li D, Huang X, Yang H, Qiu Z, Zou L, Liang Q, Shi Y, Wu Y, Wu S, Yang C, Li Y (2019) Study on antibacterial and quorum-sensing inhibition activities of Cinnamomum camphora leaf essential oil. Molecules 24:3792
Wu K, Lin Y, Chai X, Duan X, Zhao X, Chun C (2019) Mechanisms of vapor-phase antibacterial action of essential oil from Cinnamomum camphora var. linaloofera Fujita against Escherichia coli. Food Sci Nutr 7:2546–2555
Wu YM, Wang PL, Wang YH, Wei YW, Li JF (2009) Analysis of growth traits on young camphor tree stands in different propagation types. China for Sci Technol 23:23–26
Xie Q, Frugis G, Colgan D, Chua NH (2000) Arabidopsis NAC1 transduces auxin signal downstream of TIR1 to promote lateral root development. Genes Dev 14:3024–3036
Xu W, Campbell P, Vargheese AK, Braam J (1996) The Arabidopsis XET-related gene family: environmental and hormonal regulation of expression. Plant J 9:879–889
Yang F, Long E, Wen J, Cao L, Zhu C, Hu H, Ruan Y, Okanurak K, Hu H, Wei X, Yang X, Wang C, Zhang L, Wang X, Ji P, Zheng H, Wu Z, Lv Z (2014) Linalool, derived from Cinnamomum camphora (L.) Presl leaf extracts, possesses molluscicidal activity against Oncomelania hupensis and inhibits infection of Schistosoma japonicum. Parasites and Vectors 7:407
Young MD, Wakefield MJ, Smyth GK, Oshlack A (2010) Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol 11:R14
Yue-Qiu HE, Wang JJ (2015) Tissue culture and plant regeneration of Cinnamomum camphora Yongjin. J South Agric 46(1):96–100
Funding
This study was supported by the Science and Technology Department of Jiangxi Education, China (GJJ151114), the Key Research and Development project of Science and Technology Department of Jiangxi province, China (20192BBFL60012), Forestry science and technology innovation project of Jiangxi province department forestry(Innovation project[2019]04), and National Natural Science Foundation of China (31600497).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Key Message
• Exogenous IBA modifies carbohydrate secondary metabolism and the auxin and BR and ABA signalling transduction pathways to assist the regulation of the rooting and stress response of Cinnamomum camphora linalool type
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xiao, Z., Zhang, B., Wang, Y. et al. Transcriptomic Analysis Reveals That Exogenous Indole-3-Butyric Acid Affects the Rooting Process During Stem Segment Culturing of Cinnamomum camphora Linalool Type. Plant Mol Biol Rep 40, 661–673 (2022). https://doi.org/10.1007/s11105-021-01304-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-021-01304-8