
Abstract
Abstract
Sepsis remains the most common cause of death in intensive care units in the USA, with a current estimate of at least 750,000 cases per year, and 215,000 deaths annually. Despite extensive research still we do not quite understand the cellular and molecular mechanisms that are involved in triggering and propagation of septic injury. Endotoxin (lipopolysaccharide from Gram-negative bacteria, or LPS) has been implicated as a major cause of this syndrome. Inflammatory shock as a consequence of LPS release remains a serious clinical concern. In humans, inflammatory responses to LPS result in the release of cytokines and other cell mediators from monocytes and macrophages, which can cause fever, shock, organ failure and death. A number of different approaches have been investigated to try to treat and/or prevent the septic shock associated with infections caused by Gram-negative bacteria, including blockage of one or more of the cytokines induced by LPS. Recently several novel amphipathic compounds have been developed as direct LPS antagonists at the LPS receptor, TLR4. This review article will outline the current knowledge on the TLR4-LPS synthesis and discuss the signaling, in vitro pre-clinical and in vivo clinical evaluation of TLR4 antagonists and their potential use in sepsis and a variety of diseases such as atherosclerosis as well as hepatic and renal malfunction.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
LPS AND TLR
Bacteria are classified into two groups based on a staining procedure (1). This staining response is a consequence of the composition of their membranes. Gram-positive bacteria present a multi-layered, cross-linked polymer of peptidoglycan surrounding their plasma membrane, whereas Gram-negative bacteria have essentially a monolayer (1). The Gram-negative outer membrane is an asymmetric lipid bilayer interspersed with proteins. The lipid of this outer leaflet is almost exclusively constituted by LPS molecules.
Bacterial infection can be life threatening, requiring the host organism to develop a system to respond to this insult. The innate immune response is the first line of defense against infectious agents and is devoted to recognize highly conserved pathogen motifs in lipopeptides, DNA, dsRNA, ssRNA, specific proteins and LPS. These motifs are known as pathogen-associated molecular patterns (PAMPs) (2).
Lipopolysaccharide is composed of three distinct domains, lipid A, a short core of oligosaccharide and the O-antigen polysaccharide (Fig. 1). The lipid A domain is the bioactive component and is recognized during human infection. The composition of the O-antigen varies between different Gram-negative bacterial strains. The presence or absence of O chains determines whether LPS is considered rough or smooth (3). Full length O chains would render the LPS smooth while the absence or reduction of O-chains would make the LPS rough (3,4).

The structure of LPS.
Lipopolysaccharide is a potential drug target since its presence is critical in membrane stability and also it plays a prominent role in raising an immune response (2). LPS triggers the release of many inflammatory cytokines, in particular TNFα, interleukin-1β and IL-6, and it has been implicated as the etiological agent of a variety of pathologies ranging from mild (fever) to lethal (septic shock, organ failure and death) (5). Thus the structure, function and biosynthesis of LPS have been areas of intense research in the last decade (6).
The receptors capable of recognizing the pathogen-associated molecular patterns are Toll-like receptors (TLR) and scavenger receptors. Ten members of the TLR family have been identified in humans (7). The Toll was originally described as a type I transmembrane receptor that controls the embryonic dorsal-ventral pattern of Drosophila (8). In fact this pioneering work identified a group of ten different genes which when deleted produced qualitatively similar phenotypes. Null mutations on any of these genes lead to a failure to differentiate patterns on the dorsoventral axis and resulted on embryonic lethality. The identification of the sequence of Toll led to the recognition that its carboxyl terminal domain was significantly related to that of the vertebrate interleukin-1 receptor (IL-1R) (8). IL-1R activation is part of a cascade of events linked to an acute phase response to infection. This suggested that TLRs could not only be involved in development but also in the initial responses to infection in vertebrates. This hypothesis received further support from the work of Lemaitre et al., who found that Toll and other genes from the dorsal group played a role in innate immune responses to pathogenic fungi and bacteria (9).
The TLRs belong to a cluster of molecules called the IL-1R/TLR super-family characterized by the presence of cytoplasmic Toll/IL-1R (TIR) domains (10). The three subgroups are: the IL-1R (which present extracellular immunoglobulin domains), the adapter subgroup (cytoplasmic proteins without extracellular region) and the TLRs (9). TLRs are type I transmembrane proteins with extracellular amino terminus and a carboxy terminal intracellular domain. The extracellular domain of the TLR4 contains over 600 amino acids and is highly polymorphic compared with the transmembrane and cytosolic domains (6). The TIR domain, composed of three highly conserved regions, contains 150 amino acids and modulates protein-protein interactions between the TLRs and the adaptor proteins involved in the signal transduction cascade (10). Unlike other receptors, TLRs do not have an enzymatic activity (6). Researchers have identified at least fifteen different negative regulators of the TLRs, including MyD88s (a short form of MyD88), IRAKM, suppressor of cell signaling-1 (SOCS1), nucleotide-binding oligomerization domain 2 (NOD2), phosphatidylinositol-3-kinase (PI3K) and Toll-interacting protein (TOLLIP) (11). The TLR activation leads to responses that involve the induction of new genes via transduction pathways such as NFκB and AP-1 (9). The discovery of TLR lead to the understanding that an adaptive response mediated by antibody responses and T cell activation is tightly coupled to a second unknown process that requires the presence of microbial extracts (2,7).
Toll-like receptor 4 (TLR4) is the central signaling receptor for LPS in mammals (12). The current knowledge on the structure and function of the TLR4 has opened the possibility to develop new drug targets to fight sepsis and other diseases associated with this signaling molecule. TLR4 was identified as the first human homologue of the Drosophila Toll (13). TLR4 not only engages LPS but it recognizes an envelope glycoprotein encoded by mouse mammary tumor virus (MMTV) (14). In addition, TLR4 recognizes ligands such as heat shock proteins and EDA (extracellular domain A) in fibronectin (15,16).
TLR4 SIGNALING
TLRs activate a potent immunostimulatory response which needs to be tightly controlled. TLRs homo o heterodimerize upon ligand binding whereas TLR4 and TLR9 homodimerize (6). TLR signaling involves a family of adaptor proteins which recruit downstream protein kinases which activate transcription factors such as nuclear factor-kB (NF-κB) and members of the interferon (IFN)-regulatory factor (IRF) family (10). LPS signaling involves the binding of the LPS-binding protein (LBP) to LPS; this interaction leads to a disruption of LPS aggregates (10) (Fig. 2 LPS signaling, modified from (10) with permission). Upon ligand binding there is the formation of a TLR4 complex with CD14. CD14 was the first molecule shown to enhance LPS signals (17). Interestingly TLR4 does not require CD14 to trigger epithelial signaling to uropathogenic E. coli since bladder cells do not express CD14 (18). In addition a small molecule, myeloid differentiation 2 receptor (MD-2), participates in this complex by associating with the TLR4 extracellular domain (19).

LPS signaling [modified from O’Neill and Bowie (10) with permission]. TLR4 requires four signaling adaptors to function upon activation by LPS. Similarly to TLR2 it uses MAL to recruit MyD88 and to activate the NFκB pathway and p38 and JNK MAPK pathways. A second signaling cascade triggered by the LPS-TLR4 interaction involves TRAM. TRAM recruits TRIF which activates pathways involving TBK1 to IRF3, TRAF6 to NFκB and RIP1 to apoptosis.
MD-2 binds to the LPS monomer and is sensitive to the acylation pattern of the lipid A moiety. Association of the MD-2:LPS complex to the ectodomain of the TLR4 finally transduces the signal through the association of intracellular TIR domain, recruiting the adapter proteins triggering the signaling cascade (20). In a similar way to TLR2, TLR4 uses the myeloid differentiation primary-response gene 88 adapter like protein (MAL) as a bridging adaptor to recruit the myeloid differentiation primary-response gene 88 (MyD88) to activate the NF-κB, p38 and JNK/MAPK pathways via TRAF6 (9). MAL is recruited to plasma membrane microdomains containing the phospholipid PtdIns (4,5)P2 (phosphatidylinositol-4,5-bisphosphate). MAL subsequently recruits MyD88 (20). Another pathway activated by TLR4 involves TRIF-related adaptor molecule (TRAM). Similar to MAL, TRAM is also membrane proximal and requires myristoylation to lodge into the membrane. TRAM recruits the Toll/interleukin-1 receptor (TIR)-domain-containing adaptor protein inducing interferon-β (TRIF) which activates the tumor-necrosis factor-receptor-associated factor 3 (TRAF3), TRAF6 and receptor interacting protein 1(RIP1). Recent work with CD14 knockout mice suggested that TRL4 can function in two ways: one where full signaling occurs in the presence of CD14 and one limited to MyD88-dependent signaling (21).
In addition to blocking the intracellular LPS signaling there are other means to modulate the endotoxin response. Approaches to alleviate the morbidity and mortality of patients associated with severe sepsis and septic shock include: (a) neutralizing LPS or blocking initial LPS-signaling events by preventing the generation of cell-surface signals, (b) blocking the intracellular signals induced by endotoxin or the synthesis of cytokines and other cellular mediators, (c) inhibiting the release of cytokines (Il-1, IL-6 and IL-8) and cellular mediators, (d) blocking the TNF-α and IL-1 receptors to the cellular mediators on their responsive target cell and (e) inhibiting downstream pathophysiological events such as acute respiratory distress or aberrant blood clotting (22).
POTENTIAL CLINICAL USES OF TLR4 ANTAGONISTS
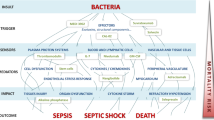
TLR mediated innate and/or adaptive immune responses play an important role in a variety of diseases, including sepsis, infectious disease, atherosclerosis, kidney failure, liver disease, pulmonary disease and myocardial ischemia/reperfusion injury (5,23–28). TLRs are expressed in a variety of cell types including immune and non-immune cells. In addition, the capability of these receptors to recognize PAMPs is indicative of their distinct roles in infection, inflammation and tissue damage (29) (Fig. 3).

TLRs are involved in protective immunity in many infectious diseases, cancer, allergies and in the pathogenesis of sepsis, autoimmune diseases or atherosclerosis. An efficient TLR antagonist may be of benefit to block or reduce exaggerated TLR stimulation [modified with permission from Ishii et al. (78)].
TLR4 and Sepsis
According to the definition made by ACCP/SCCM Consensus Conference in 1992, sepsis is known to be an early syndrome that may progress to a pathologic state manifested by hypotension and hypoperfusion known as septic shock. LPS has been associated with sepsis and the high mortality rate seen in septic shock (5). However, it is the exaggerated host response to the systemic release of endotoxin that accounts for septic shock from Gram-negative bacteria (23).
TLR4 up-regulation in non-immune cells after initial TLR mediated immune response may trigger secondary responses such as activation of endothelial cells that promotes the production of adhesion molecules, followed by macrophage infiltration and vascular permeability during infection (30). This cascade may result in a systemic septic syndrome including tissue perfusions, an imbalanced coagulation cascade and organ failure (31).
TLR4 and Atherosclerosis
Atherosclerosis is an inflammatory disease where activated cells are involved in its initiation and progression. Guha and Mackman (32) have shown that activated TLR4 elicits the production of inflammatory cytokines and chemokines. Edfeldt et al. (33) have also found that TLR4 is prominently expressed in endothelial cells of human atherosclerotic lesion, but poorly expressed in normal human arteries. In the early atherosclerotic lesion, LPS and other ligands can stimulate the TLR4 expression on macrophages. The activated receptors can then initiate the signaling cascade that induces the expression of inflammatory cytokines, proteases, and cytotoxic oxygen and nitrogen radicals. These entities further speed up the progression of the atherosclerotic lesion (34). In advanced atherosclerotic lesion, LPS can induce the proliferation of vascular smooth muscle cells, as well as the expression of elastin-degrading enzyme via TLR4 (35). Besides that, in response to chemokines, more smooth muscle cells will also migrate to the sites of the lesions (36). These predominant changes cause the accumulation of cells, extracellular matrix components, thickening of the intima, as well as the deformity of the arterial wall.
Furthermore, TLR4 signaling might also be involved in atherosclerotic plaque destabilization. Grenier and Grignon have demonstrated that LPS induces the expression of matrix metalloproteinase-9 (MMP-9) by TLR4 in macrophages; MMP-9 has been shown to degrade collagen fibrous cap, thus predisposing plaque to rupture (37).
TLR4 and Liver Injury
In many forms of liver diseases such as alcoholic or non-alcoholic liver disease, liver failure and inflammation are the result of a cascade of insults which result in hyper-activation of inflammatory pathways and liver injury (26,27). Velayudham and colleagues (26) have shown that there is an up-regulation of TLR4 receptors in liver granulomas and LPS induced liver injury. Pathogen-induced TLR4 activation also activates reactive oxygen species (ROS), which is a major source of acute hepatocyte injury and death in the liver. Up-regulation of peripheral blood monocyte expression of TLR4 also occurs in patients with chronic hepatitis C (38). In addition, endogenous gut-derived bacterial LPS have also been implicated as important cofactors in the pathogenesis of liver injury.
Within the liver, LPS binds to LPS-binding protein (LBP), which then facilitates its transfer to membrane CD14 on the surface of Kupffer cells in the liver (39). Moreover, TLR4 can also interact with a protein ligand released from damaged hepatocytes to extend an existing injury in the liver (40).
In other studies, there is evidence that high-mobility group box 1 (HMGB1) can interact with both TLR2 and TLR4 to induce an inflammatory response during liver ischemia/reperfusion (IR) injury similar to that initiated by LPS (41,42). HMGB1 is an intracellular protein present in many species that functions in regulation and modulation of gene transcription (42). HMGB1 is released readily from necrotic or damaged cells, which may signal through TLR4 the presence of advancing tissue injury, initiating an inflammatory response that further damages viable cells (42).
TLR4 and Ischemia/Reperfusion Injury
The restoration of blood flow to the ischemic heart has often caused myocardial ischemic/reperfusion (MI/R) injury. An inflammatory response triggered by MI/R injury can irreversibly cause damage to the viable tissue surrounding the infarct, thereby further extending the injury. It is still unclear how innate immune signaling pathways are initiated during MI/R injury (43). However, TLR4, which is also present in cardiomyocytes, has been thought to play a role in mediating MI/R injury. Schuster and Nelson (28) have shown that TLR4 receptor is up-regulated in response to myocardial injury. Furthermore, Shimamoto and colleagues have shown that TLR4 activates NF-κB-dependent transcription of inflammatory cytokine genes in MI/R injury. The TLR4-mediated injury appears to occur through activation of c-Jun NH2-terminal kinase (JNK) and translocation of NF-κB (41). It is also believed that TLR4 recognizes endogenous molecules that are exposed during cellular injury and extracellular matrix remodeling, independent of pathogen invasion (44). Thus, inhibition of TLR4 signaling pathway may be a potential therapeutic target to treat the myocardium damage in the ischemia/reperfusion setting.
TLR4 and Kidney Disease
Acute renal failure (ARF) occurs in close to 5% of hospital admissions, and is a leading cause of morbidity and mortality. A common cause of ARF is sepsis, which results from overwhelming infection (25). Cunningham and colleagues (25) have shown that LPS insult leads to renal cell apoptosis and renal neutrophil infiltration.
Tubular epithelial cells of the kidney are among the non-immune cells that express TLR1, TLR1-2, TLR1-3, TLR1-4, and TLR1-6, suggesting that these TLR might contribute to the activation of immune responses in tubulointerstitial injury (45). In addition, receptors such as TLR4, TLR2, CD91 and the receptor for the advanced glycation end-products (RAGE), allow leukocytes and renal cells to recognize molecules released by injured cells. These receptors are sentinels for tissue necrosis (46). Upon stimulation with LPS in renal infection or other endogenous ligands from necrotic tubular cells, the activated TLR4 has been shown to specifically stimulate the NF-κB pathway in response to oxidative stress (47). Furthermore, TLR4 activation on tubular epithelial cells and circulating immune cells leads to secretion of cytokines and chemokines that either directly or indirectly contributes to renal injury.
TLR4 in Inflammatory Bowel Disease
Inflammatory bowel disease (IBD) is a medical condition that predominantly affects the gastrointestinal tract (48). De Jager et al. showed that TLR4 and its signaling molecule TIRAP affect susceptibility to IBD (49). Recent studies have shown that TLR4−/− and MyD88−/− knockout mice tend to be more prone to severe dextran sulfate sodium-induced colitis than their wild-type littermates (50). Interestingly, CRX-526 a TLR4 antagonist has been shown to prevent an inflammatory disease in the dextran sulfate sodium and mdr1a−/−/1b−/− deficient mice models (51). To explain these contradictory results we have to consider that constitutive signaling through TLR4 may result in the production of tissue protective factors such as IL-6 and TNF-α (49). This is the scenario in the MyD88−/− knockout mice, while in the case of the CRX-526 we may have selective downregulation of one of the TLR-4/LPS signaling pathways.
TLR4 and Pulmonary Disease
Simpson and colleagues observed an increased expression of TLR2, TLR4 and CD14, as well as the pro-inflammatory cytokines IL-8 and IL-1β in neutrophilic asthma and bronchiectasis patients compared to controls. These groups also had higher airway endotoxin levels than the control group (23). In another study, there was also an increased pulmonary expression of inflammatory cytokines occurring in the lung during experimental endotoxemia. The cytokine production further contributes to acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) (52,53). Baumgarten and colleagues showed that LPS induces pro-inflammatory cytokines in the lung via the TLR4/CD14 signaling cascade, suggesting a role of the innate immune response in the pathogenesis of ALI/ARDS (52).
SYNTHESIS OF NOVEL ANALOGUES
LPS biosynthesis occurs by two distinct, yet convergent pathways: one for the lipid A core and another for the polysaccharide O antigen. After independent synthesis, the two parts are ligated together to complete the LPS molecule (4). Amongst the most important TLR4 antagonists developed so far we have CRX-526, E5531 and E5564.
CRX-526
Aminoalkyl glucosaminide 4-phosphates or AGPs are a class of lipid A mimetics in which the reducing sugar of lipid A has been replaced with an N-acylated aminoalkyl aglycon unit (54). The AGPs contain an L-serine-based aglycon unit as well as three (R)-3-n-alkanoyloxytetradecanoyl residues comprised of even-numbered normal fatty acyl chains between 6 and 14 carbon atoms in length. All members of this family have 14 carbon atoms in the “primary” fatty acid, which is the -hydroxy fatty acid attached directly to the AGP backbone. -Hydroxymyristic acid is the most common of the primary fatty acids present in lipid A. These compounds were used in a variety of cell-based and an in vivo model to determine structure-activity relationships related to AGP acyl chain length and stimulation via TLR4 (54). Figure 4 shows the chemical structure of two AGPs: MPL and CRX-526. The structure of CRX-526 differs significantly from monophosphoryl lipid A (MPL) and other TLR4-agonist AGP in the length of its secondary fatty acyl chains (SAC): for instance CRX-526 contains 3 SAC of 6 carbons in length, whereas MPL and other AGP, which signal through the TLR4 complex, contain SAC of >10 carbons in length (55).

The chemical structures of MPL and CRX-526 [modified from (51) with permission].
The synthesis of AGPs has been described elsewhere (55). Briefly, the AGPs were prepared by a highly convergent method, which allowed chemical differentiation of the hydroxyl and amino groups and sequential introduction of the (R)-3-n-alkanoyloxytetrahexanoyl residues. The AGPs were purified by flash chromatography on silica gel (to >95% purity) and analyzed as a triethylammonium salt by standard analytical methods. For stimulation in vitro, the AGPs were formulated in water containing 216 μg/ml dipalmitoylphosphatidyl choline [aqueous formulation (AF)], 0.2% triethanolamine (pH 7.4), or in 2% glycerol (i.v. formulation).
E5531 and E5564
E5531 (6-O-{2-deoxy-6-O-methyl-4-O-phosphono-3-O-[(R)-3-Z-dodec-5-endoyloxydecl]-2-[3-oxo-tetradecanoylamino]-O-phosphono-d-glucopyranose tetrasodium salt) and E5564 (Eritoran ™) [-d-glucopyranose,3-O-decyl-2-deoxy-6-O-[2-deoxy-3-O-[(3R)-3-methoxydecyl]-6-O-methyl-2-[[(11Z)-1-oxo-11-octadecenyl]amino]-4-O-phosphono-d-glucopyranosyl]-2-[(1,3-dioxotetradecyl)amino]-1-(dihydrogen phosphate), tetrasodium salt], formula weight 1401.60) (molecular formula: C66H122N2Na4O19P2) were synthesized by Eisai Research Institute of Boston (Andover, MA, USA) (53). The structure of E5531, an analog of the lipid A of Rhodobacter capsulatus, is presented elsewhere (56). The structure of E5564 is depicted in Fig. 5. The crystal structure of the TLR4-MD2 complex with bound E5564 was recently published (57) suggesting that the mechanism of action of E5564 is binding through a large internal pocket in MD-2.

The chemical structure of E5564.
PRE-CLINICAL EVALUATION
An important consideration in the development of assays is to determine the efficacy and toxicity of the compounds and the potency required to obtain a biological effect. A high potency is a key factor that would limit the high cost of synthesis and purification of such a compound. To illustrate the challenges in developing a synthetic endotoxin receptor antagonist we present the case of E5564.
In vitro, E5564 dose-dependently inhibited LPS-mediated activation of primary cultures of human myeloid cells and mouse tissue culture macrophage cell lines as well as human or animal whole blood at nanomolar concentrations as measured by production of tumor TNF- and other cytokines. E5564 also blocked the ability of Gram negative bacteria to stimulate human cytokine production in whole blood. In vivo, E5564 blocked induction of LPS-induced cytokines and LPS or bacterial-induced lethality in primed mice. E5564 was devoid of agonistic activity when tested both in vitro and in vivo and had no antagonistic activity against Gram positive-mediated cellular activation at concentrations up to 1 μM. E5564 blocked LPS-mediated activation of nuclear factor-B in TLR4/MD-2-transfected cells. In a mouse macrophage cell line, activity of E5564 was independent of serum, suggesting that E5564 exerts its activity through the cell surface receptor(s) for LPS, without the need for serum LPS transfer proteins. Similar to E5531, another lipid A-like antagonist, E5564 associates with plasma lipoproteins, causing low concentrations of E5564 to be quantitatively inactivated in a dose- and time-dependent manner. However, compared with E5531, E5564 is a more potent inhibitor of cytokine generation, and higher doses retain activity for a period of time likely sufficient to permit clinical application. These results indicate that E5564 is a potent LPS antagonist and lacks agonistic activity in human and animal model systems, making it a potentially effective therapeutic agent for treatment of disease states caused by LPS.
Compared with E5531, E5564 is structurally and synthetically less complex, yet seems to possess superior activity and pharmacological characteristics. Although E5531 demonstrated potent inhibition of LPS when added to blood in vitro and in vivo, activity decreased as a function of time. This reaction has been shown to be due to interaction of E5531 with plasma lipoproteins (58–60). E5564 is an inhibitor of LPS-mediated stimulation of responsive cells in vitro and in vivo as measured by production of cytokines, as well as morbidity and mortality associated with LPS poisoning in animal models. Because E5564 is a structural analog of the lipid A portion of LPS, it is logical to hypothesize that the antagonist interacts with the same signaling components that bind to LPS such as the soluble serum proteins LBP and sCD14, as well as membrane-associated CD14 and perhaps the TLR4/MD-2 receptor complex. E5564 blocked LPS/sCD14-induced reporter activity in TLR4/MD-2-expressing HEK293 (61), but not TLR2-mediated signaling by heat-killed S. aureus. These findings indicate that E5564 selectively inhibits LPS signaling via TLR4/MD-2. However, a limitation to this model system is that LPS requires the presence of sCD14 for cellular activation, making it difficult to determine whether E5564 blocks LPS binding to sCD14 or TLR4/MD-2. Results from experiments indicated that serum components did not affect the potency of E5564, indicating that they are not critical to E5564 antagonistic activity (61).
Further support of the hypothesis that interaction of E5564 at CD14 does not play a key role in its activity comes from a previous study by Lien et al. describing the activity of novel synthetic acyclic lipid A-like agonists that activate TLR4/MD-2 in the absence of CD14 (62). E5564 inhibited the actions of these agonists under serum-free conditions. Taken together, these lines of evidence make it tempting to speculate that E5564 binds to TLR4/MD-2 complex, thereby blocking LPS binding or transmembrane signaling. The downstream effect of inhibiting the initial signaling by LPS seems to be an inhibition of all LPS-induced cytokines measured, including TNF-, IL-1, IL-6, IL-8, IL-10, and nitric oxide, which was measured in cultured cells, whole blood, and in vivo. Recently the crystal structure of the TLR4/MD-2 and E5564 has been described confirming the physical interaction of these molecules (57).
Comparisons of antagonistic potency in cells cultured in 10% serum versus whole blood allow us to determine whether the high concentration of proteins/lipoproteins present in serum inhibit E5564 activity. In all systems but the rat, antagonistic activity of E5564 in cultured cells was within fourfold that measured in high serum (blood) compared with assays done in low-serum conditions (cultured cells or monocytes) (60,63). This indicates that serum has little or no inhibitory effect on antagonistic activity under these in vitro conditions. However, extended incubations in whole blood demonstrated that activity of E5564 was measurably reduced. Other studies indicate that like E5531, E5564 is not rapidly metabolized, but binds to lipoproteins, and time dependently loses antagonistic activity (64). The observation that lipoproteins reduce drug activity may explain the poor activity of E5564 in rat blood that has relatively high lipoprotein content.
During extended incubation in whole blood, E5564 retained activity better than similar concentrations of the first-generation antagonist E5531. Based on the proposed mechanism of action as a cell surface antagonist, it is likely that E5564 can completely block cellular activation by LPS. This block is achieved by concentrations of E5564 as low as 10 nM (14 ng/ml) in vitro, and at doses of 1 mg/kg or less in animal models challenged with lethal LPS doses.
Both LPS-challenge model and infection model use animals that have been sensitized or primed to LPS by previous infection with BCG, increasing cytokine response and lowering the threshold lethal dose of endotoxin (61). All animal models of sepsis and infection have been criticized for their inability to closely mimic human sepsis. The primed model is the most relevant to the study of endotoxin antagonists such as E5564. It is well known that compared with humans, unprimed rodents such as rats and mice and primates demonstrate a profound insensitivity to endotoxin, requiring endotoxin doses as high as milligrams per kilogram, whereas humans demonstrate reproducible response to endotoxin at doses as low as 2 ng/kg. This argues that either LPS contributes only weakly to the inflammatory process in animal models, or that response to infection occurs only after the level of infection is very high, representing a process different from that in more LPS-sensitive species such as humans (61).
Even in primed animal models, lethal doses of LPS are high, approximately 100 μg/kg, generating estimated plasma concentrations of ∼1 μg/ml. These plasma levels are still >100-fold that found in even the most extreme cases of human sepsis (65). Because the dose of E5564 required to protect against LPS is proportional to the LPS challenge dose, studying E5564 in these animal models indicates that E5564 can be a safe and effective antagonist even under these extraordinary conditions. E5564 is approximately tenfold better in human blood than mouse blood (IC50 = 1.6 nM in human whole blood; Table I versus ∼20 nM in mouse whole blood; Table II).
Complete block of cytokine response by 10 nM E5564 in blood extrapolates to a human dose of approximately 100 μg in a 70-kg individual. Recent studies have supported this extrapolation by finding that a dose of 100 μg of E5564 given to normal volunteers over 30 min completely blocks response (signs, symptoms, and cytokines) to a dose of 4 ng/kg endotoxin administered at the midpoint of the E5564 infusion (66).
In vitro and ex vivo assays have found that low concentrations of E5564 time dependently lose ability to inhibit LPS response. In light of these observations, it is perhaps not surprising that low doses of E5564 demonstrate a time-dependent loss of activity after administration into normal volunteers. This loss in activity is overcome when E5564 doses are increased (67). Phase I clinical safety and tolerability assays indicate that E5564 is safe and except for the occurrence of phlebitis, well tolerated at doses up to 252 mg administered over 72 h. At this dose, in vivo antagonistic activity is retained for at least an additional 72 h after discontinuing infusion. This leads us to believe that sufficient therapeutic activity can readily be administered to patients (67). The safety and efficacy of E5564 are currently been analyzed in a phase III randomized controlled study.
TAK-242
TAK-242 has been demonstrated to suppress LPS-induced inflammation (68,69). Recently, TAK-242 has been shown to almost completely suppress production of nitric oxide or TNFα induced by LPS in mouse RAW264.7, human U937 and P31/FUJ cells (70). In a HEK293 cell model where TLR4, MD-2 and CD14 were co-expressed, this antagonist showed specificity to TLR4 as other TLRs, TLR1/2, TLR2/6, TLR3, TLR5, TLR7 and TLT9 were not affected by this drug (70).
CLINICAL EVALUATION
Sepsis is a major cause of high mortality rate in intensive care units in the USA (71). Severe sepsis usually leads to organ failure. Currently, over 30 pharmaceutical products have been in the development stage to treat this condition, yet only few have reached the market (72). Many of these target specific inflammatory mediators have been unsuccessful because of the complex nature of sepsis. For the treatment of sepsis, there are a few products that are being investigated in clinical studies via blocking different mechanisms of the body’s innate immune system. Eli Lilly’s Xigris® was one of the few drugs currently available on the market to treat sepsis. Xigris® is a recombinant human activated protein C that has anti-inflammatory, anti-thrombotic and pro-fibrinolytic properties to block the coagulation cascade which plays a critical role in the development of organ failure due to sepsis (73). In addition, simvastatin and atorvastatin had also shown to have some non-specific anti-inflammatory effects contributing to their clinical benefits in treating sepsis (63). However, statins are currently not been considered as a treatment for sepsis. To find a more specific target, scientists have identified TLR4 as one of the candidates in blocking the innate immune system. Only two TLR4 antagonists, E5564 and TAK-242, have made far into the clinical phase (Table III).
E5564
In Wong, et al., determined the safety and tolerability of E5564 following a 30-min intravenous infusion in healthy male volunteers (74). This was a single-center, randomized, double-blind, placebo-controlled, sequential-group, single-dose study of E5564. The drug dose levels used were 350, 1,000, 2,000 or 3,500 μg. All doses of E5564 presented a long pharmacokinetic half-life and short in vivo pharmacodynamic half life which generally less than several hours when it is co-administered with LPS in healthy volunteers (74). The C max and AUC (area under the curve) of E5564 increased in a dose-dependent manner. E5564 pharmacokinetics was characterized by a slow clearance (0.67–0.95 ml h−1 kg−1), a small volume of distribution (41–54 ml/kg), and a relatively long elimination half life (42–51 h) in healthy male volunteers. Thus, to overcome this low PD, the doses of E5564 given to the volunteers needed to be adjusted. In summary, all doses were demonstrated to be safe and well tolerated. Safety and tolerability assessments included monitoring and questioning of the subjects about adverse events, physical examinations, clinical laboratory tests (including hematology, blood chemistry, and urinalysis), and vital sign measurements (including supine and standing pulse rate and blood pressure), and 12-lead electrocardiograms (ECGs) and cytokine concentration testing. In this study, E5564 inhibited LPS-induced tumor necrosis factor-α in a dose-dependent manner, and at the higher doses (2 and 3.5 mg), antagonistic activity was measurable up to 8 h post-infusion. E5564 lacked LPS-like agonist activity at doses up to 3.5 mg (74).
In another study of healthy volunteers with experimental endotoxemia, Lynn et al. (66) found phlebitis was only associated with 72 h continuous intravenous infusion of E5564 but not with four hour infusion of E5564 into a peripheral vein. In this study the authors explored the possibility of extended pharmacokinetic activity of E5564. The infusion period was changed from bulk dosing to a 4- and 72-h infusions of E5564 into normal volunteers. They observed that at 4 h infusion of E5564, 3 mg/h completely blocked endotoxin administered 8 h post-dosing. Additionally, they observed that administration of 3.4 mg of E5564/hX72 h completely blocked the effects of endotoxin challenge at the end of dosing (72 h), and at 48 and 72 h post dosing. A lower dose of E5564 of 2 mg was also studied, and they found that 0.5 mg/h × 4 h, ameliorated but did not block most effects of endotoxin 8 h post-dosing. This work also studied the effect of varying plasma lipoprotein content on E5564 activity in subjects who have high or low cholesterol levels (>180 or <140 mg/dl) after a 72 h infusion of 252 mg of E5564. The distribution of E5564 into the lipoprotein fractions was not significantly different between the low- and high-cholesterol groups (66).
In another study by Rossignol et al., a 72 h intravenous infusion and higher doses (500, 2,000 or 3,500 μg/h) of E5564 were administered into healthy volunteers (67). E5564 has a slow plasma clearance (0.679 to 0.930 ml h−1 kg−1 of body weight), a small volume of distribution (45.6 to 49.8 ml/kg), and a relatively long half-life (50.4 to 62.7 h). All these pharmacokinetic parameters obtained are comparable to the study done by Wong et al. (74). The association of E5564 with plasma lipoproteins was also investigated and it was found that the majority (∼55%) of the drug was bound specifically to high-density lipoprotein (HDL), but not low-density lipoproteins, very-low-density lipoproteins, or albumin (67).
A Phase II multi-site, double-blind, randomized, ascending-dose, placebo-controlled safety study on E5564 was conducted in cardiac surgery patients (75). Patients undergoing coronary artery bypass graft and/or cardiac valvular surgery with cardiopulmonary bypass were enrolled. Patients received a four hour infusion of 2, 12 or 28 mg of E5564 before cardiopulmonary bypass. No significant safety concerns were identified. No significant difference was observed in most variables related to systemic inflammation or organ dysfunction/injury. This phase II safety study suggests that the administration of E5564 is not associated with toxicity in cardiac surgical patients. However, the relatively small sample size used in this study limits the conclusion regarding rare adverse events or the potential clinical benefits of this drug (75).
The potential of E5564 as a sepsis treatment was addressed by Kaneko et al. (76), Surface Plasmon resonance (SPR) analysis indicated that E5564 binds to LPS binding protein (LBP), in a manner similar to LPS. Blood withdrawn from healthy volunteers was treated with heparin to prevent clotting. At doses of E5564 relevant to its clinical use (i.e. 6 μg/ml), antibodies against LBP did not influence either the distribution of E5564 to non-HDL lipoprotein fractions or the transfer of E5564 from non-HDLs to HDL. LBP binds E5564 in a manner similar to LPS, but does not play a role in E5564 redistribution/binding to lipoprotein and plasma clearance.
Czeslick et al. (77) carried out an ex vivo study on the effect of E5564 on production of LPS-induced pro-inflammatory cytokines, particularly IL-6 and TNF-α, in LPS-induced human monocytes. In this study, they recruited 10 healthy volunteers and obtained their whole blood samples and pre-incubated with 0.001, 0.003, 0.01, 0.03, 0.1, 1 and 10 ng/ml E5564 for 45 min and after stimulated with 0.2 ng/ml of LPS. They found that E5564 (0.003 up to 10 ng/ml) caused a dose-dependent inhibitory effect on IL-6 and TNF-α production in LPS-stimulated human monocytes. They concluded that E5564 has a significant LPS inhibitory effect via down regulation of the intracellular generation of pro-inflammatory cytokines IL-6 and TNF-α in human monocytes (77).
The association of E5564 with plasma protein and lipoprotein was studied in plasma obtained from fasted human subjects with various lipid concentrations (64). It was reported that the majority of E5564 was recovered in the high-density lipoprotein (HDL) fraction. Additionally, they had shown increasing levels of TG-rich lipoprotein (TRL) lipid (TC and TG) concentrations resulted in a significant increase in the percentage of E5564 recovered in the TRL fraction. Furthermore, their findings had suggested that E5564 does not influence CETP-mediated transfer activity (64).
TAK-242
Human peripheral blood mononuclear cells (PBMCs) were isolated from peripheral blood obtained from healthy human volunteers by density gradient centrifugation (68). TAK-242 was effective in human cells and inhibited the production of TNF-α, IL-6, and IL-1b from PBMCs stimulated with LPS and IFN-gamma, with IC50 values of TAK-242 ranging from 5.3 to 58 nM. There were four donors used for this study. There was no marked difference in the IC50 values of TAK-242 amongst them. TAK-242 showed suppressive effects on the production of various inflammatory mediators from human monocytes and macrophages stimulated with LPS. TAK-242 also suppressed the production of these cytokines from LPS-stimulated human peripheral blood mononuclear cells (PBMCs) at IC50 values from 11 to 33 nM. In addition, the inhibitory effects on the LPS-induced IL-6 and IL-12 production were similar in human PBMCs, monocytes, and macrophages. TAK-242 suppressed the cytokine production induced by Toll-like receptor (TLR) 4 ligands, but not by ligands for TLR2, TLR3, and TLR9. TAK-242 suppresses the production of multiple cytokines by selectively inhibiting TLR4 intracellular signaling (68).
CONCLUSIONS
The manipulation or intervention of TLR-mediated immune responses is a potential approach to treat and prevent the septic shock and variety of associated diseases. However, blocking TLR may lead to ‘inappropriate’ immune responses such allergic Th2 responses, or immunological tolerance (78). Thus, it seems clear that the risks and benefits of manipulation of TLR mediated immune responses need to be balanced and require further investigation.
References
T. J. Beveridge. Use of the gram stain in microbiology. Biotech. Histochem. 76:111–118 (2001).
C. A. Janeway Jr, and R. Medzhitov. Innate immune recognition. Annu. Rev. Immunol. 20:197–216 (2002).
J. C. Chow, D. W. Young, D. T. Golenbock, W. J. Christ, and F. Gusovsky. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J. Biol. Chem. 274:10689–10692 (1999).
J. A. Yethon, and C. Whitfield. Lipopolysaccharide as a target for the development of novel therapeutics in gram-negative bacteria. Curr. Drug Targets Infect. Disord. 1:91–106 (2001).
J. E. Parrillo, M. M. Parker, C. Natanson, A. F. Suffredini, R. L. Danner, R. E. Cunnion, and F. P. Ognibene. Septic shock in humans. Advances in the understanding of pathogenesis, cardiovascular dysfunction, and therapy. Ann. Intern. Med. 113:227–242 (1990).
N. J. Gay, and M. Gangloff. Structure and function of toll receptors and their ligands. Annu. Rev. Biochem. 76:141–165 (2007).
T. K. Means, D. T. Golenbock, and M. J. Fenton. Structure and function of Toll-like receptor proteins. Life Sci. 68:241–258 (2000).
C. Hashimoto, K. L. Hudson, and K. V. Anderson. The Toll gene of Drosophila, required for dorsal-ventral embryonic polarity, appears to encode a transmembrane protein. Cell. 52:269–279 (1988).
S. L. Doyle, and L. A. O’Neill. Toll-like receptors: from the discovery of NFkappaB to new insights into transcriptional regulations in innate immunity. Biochem. Pharmacol. 72:1102–1113 (2006).
L. A. O’Neill, and A. G. Bowie. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 7:353–364 (2007).
F. Y. Liew, D. Xu, E. K. Brint, and L. A. O’Neill. Negative regulation of toll-like receptor-mediated immune responses. Nat. Rev. Immunol. 5:446–458 (2005).
L. L. Stoll, G. M. Denning, and N. L. Weintraub. Endotoxin, TLR4 signaling and vascular inflammation: potential therapeutic targets in cardiovascular disease. Curr. Pharm. Des. 12:4229–4245 (2006).
R. Medzhitov, P. Preston-Hurlburt, and C. A. Janeway Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 388:394–397 (1997).
B. A. Jude, Y. Pobezinskaya, J. Bishop, S. Parke, R. M. Medzhitov, A. V. Chervonsky, and T. V. Golovkina. Subversion of the innate immune system by a retrovirus. Nat. Immunol. 4:573–578 (2003).
Y. Okamura, M. Watari, E. S. Jerud, D. W. Young, S. T. Ishizaka, J. Rose, J. C. Chow, and J. F. Strauss 3rd. The extra domain A of fibronectin activates Toll-like receptor 4. J. Biol. Chem. 276:10229–10233 (2001).
S. P. Gondokaryono, H. Ushio, F. Niyonsaba, M. Hara, H. Takenaka, S. T. Jayawardana, S. Ikeda, K. Okumura, and H. Ogawa. The extra domain A of fibronectin stimulates murine mast cells via toll-like receptor 4. J. Leukoc. Biol. 82:657–665 (2007).
S. D. Wright, R. A. Ramos, P. S. Tobias, R. J. Ulevitch, and J. C. Mathison. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science. 249:1431–1433 (1990).
J. C. Sirard, M. Bayardo, and A. Didierlaurent. Pathogen-specific TLR signaling in mucosa: mutual contribution of microbial TLR agonists and virulence factors. Eur. J. Immunol. 36:260–263 (2006).
R. Shimazu, S. Akashi, H. Ogata, Y. Nagai, K. Fukudome, K. Miyake, and M. Kimoto. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J. Exp. Med. 189:1777–1782 (1999).
R. Jerala. Structural biology of the LPS recognition. Int. J. Med. Microbiol. 297:353–363 (2007).
Z. Jiang, P. Georgel, X. Du, L. Shamel, S. Sovath, S. Mudd, M. Huber, C. Kalis, S. Keck, C. Galanos, M. Freudenberg, and B. Beutler. CD14 is required for MyD88-independent LPS signaling. Nat. Immunol. 6:565–570 (2005).
L. D. Hawkins, W. J. Christ, and D. P. Rossignol. Inhibition of endotoxin response by synthetic TLR4 antagonists. Curr. Top Med. Chem. 4:1147–1171 (2004).
J. L. Simpson, T. V. Grissell, J. Douwes, R. J. Scott, M. J. Boyle, and P. G. Gibson. Innate immune activation in neutrophilic asthma and bronchiectasis. Thorax. 62:211–218 (2007).
K. S. Michelsen, T. M. Doherty, P. K. Shah, and M. Arditi. Role of Toll-like receptors in atherosclerosis. Circ. Res. 95:e96–e97 (2004).
P. N. Cunningham, Y. Wang, R. Guo, G. He, and R. J. Quigg. Role of Toll-like receptor 4 in endotoxin-induced acute renal failure. J. Immunol. 172:2629–2635 (2004).
A. Velayudham, I. Hritz, A. Dolganiuc, P. Mandrekar, E. Kurt-Jones, and G. Szabo. Critical role of toll-like receptors and the common TLR adaptor, MyD88, in induction of granulomas and liver injury. J. Hepatol. 45:813–824 (2006).
A. M. Diehl. Cytokine regulation of liver injury and repair. Immunol. Rev. 174:160–171 (2000).
J. M. Schuster, and P. S. Nelson. Toll receptors: an expanding role in our understanding of human disease. J. Leukoc. Biol. 67:767–773 (2000).
H. Li, Y. He, J. Zhang, S. Sun, and B. Sun. Lipopolysaccharide regulates toll-like receptor 4 expression in human aortic smooth muscle cells. Cell Biol. Int. 31:831–835 (2007).
H. Bosshart, and M. Heinzelmann. Targeting bacterial endotoxin: two sides of a coin. Ann. N. Y. Acad. Sci. 1096:1–17 (2007).
M. Wendel, R. Paul, and A. R. Heller. Lipoproteins in inflammation and sepsis. II. Clinical aspects. Intensive. Care Med. 33:25–35 (2007).
M. Guha, and N. Mackman. LPS induction of gene expression in human monocytes. Cell Signal. 13:85–94 (2001).
K. Edfeldt, J. Swedenborg, G. K. Hansson, and Z. Q. Yan. Expression of toll-like receptors in human atherosclerotic lesions: a possible pathway for plaque activation. Circulation. 105:1158–1161 (2002).
S. T. Smiley, J. A. King, and W. W. Hancock. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J. Immunol. 167:2887–2894 (2001).
M. Watari, H. Watari, I. Nachamkin, and J. F. Strauss. Lipopolysaccharide induces expression of genes encoding pro-inflammatory cytokines and the elastin-degrading enzyme, cathepsin S, in human cervical smooth-muscle cells. J. Soc. Gynecol. Investig. 7:190–198 (2000).
S. Sasu, D. LaVerda, N. Qureshi, D. T. Golenbock, and D. Beasley. Chlamydia pneumoniae and chlamydial heat shock protein 60 stimulate proliferation of human vascular smooth muscle cells via toll-like receptor 4 and p44/p42 mitogen-activated protein kinase activation. Circ. Res. 89:244–250 (2001).
D. Grenier, and L. Grignon. Response of human macrophage-like cells to stimulation by Fusobacterium nucleatum ssp. nucleatum lipopolysaccharide. Oral Microbiol Immunol. 21:190–196 (2006).
S. M. Riordan, N. A. Skinner, J. Kurtovic, S. Locarnini, C. J. McIver, R. Williams, and K. Visvanathan. Toll-like receptor expression in chronic hepatitis C: correlation with pro-inflammatory cytokine levels and liver injury. Inflamm. Res. 55:279–285 (2006).
G. L. Su. Lipopolysaccharides in liver injury: molecular mechanisms of Kupffer cell activation. Am. J. Physiol. Gastrointest. Liver Physiol. 283:G256–G265 (2002).
A. Tsung, R. A. Hoffman, K. Izuishi, N. D. Critchlow, A. Nakao, M. H. Chan, M. T. Lotze, D. A. Geller, and T. R. Billiar. Hepatic ischemia/reperfusion injury involves functional TLR4 signaling in nonparenchymal cells. J. Immunol. 175:7661–7668 (2005).
A. Shimamoto, A. J. Chong, M. Yada, S. Shomura, H. Takayama, A. J. Fleisig, M. L. Agnew, C. R. Hampton, C. L. Rothnie, D. J. Spring, T. H. Pohlman, H. Shimpo, and E. D. Verrier. Inhibition of Toll-like receptor 4 with eritoran attenuates myocardial ischemia-reperfusion injury. Circulation. 114:I270–4 (2006).
J. S. Park, F. Gamboni-Robertson, Q. He, D. Svetkauskaite, J. Y. Kim, D. Strassheim, J. W. Sohn, S. Yamada, I. Maruyama, A. Banerjee, A. Ishizaka, and E. Abraham. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am. J. Physiol. Cell. Physiol. 290:C917–C924 (2006).
D. J. Kaczorowski, A. Nakao, K. P. Mollen, R. Vallabhaneni, R. Sugimoto, J. Kohmoto, K. Tobita, B. S. Zuckerbraun, K. R. McCurry, N. Murase, and T. R. Billiar. Toll-like receptor 4 mediates the early inflammatory response after cold ischemia/reperfusion. Transplantation. 84:1279–1287 (2007).
F. Hua, T. Ha, J. Ma, Y. Li, J. Kelley, X. Gao, I. W. Browder, R. L. Kao, D. L. Williams, and C. Li. Protection against myocardial ischemia/reperfusion injury in TLR4-deficient mice is mediated through a phosphoinositide 3-kinase-dependent mechanism. J. Immunol. 178:7317–7324 (2007).
H. J. Anders, B. Banas, and D. Schlondorff. Signaling danger: toll-like receptors and their potential roles in kidney disease. J. Am. Soc. Nephrol. 15:854–867 (2004).
C. Y. Lu, J. Hartono, M. Senitko, and J. Chen. The inflammatory response to ischemic acute kidney injury: a result of the ‘right stuff’ in the ‘wrong place’? Curr. Opin. Nephrol. Hypertens. 16:83–89 (2007).
M. Yu, D. Shao, J. Liu, J. Zhu, Z. Zhang, and J. Xu. Effects of ketamine on levels of cytokines, NF-kappaB and TLRs in rat intestine during CLP-induced sepsis. Int. Immunopharmacol. 7:1076–1082 (2007).
R. K. Weersma, H. M. van Dullemen, G. van der Steege, I. M. Nolte, J. H. Kleibeuker, and G. Dijkstra. Review article: Inflammatory bowel disease and genetics. Aliment. Pharmacol. Ther. 26(Suppl 2):57–65 (2007).
P. L. De Jager, D. Franchimont, A. Waliszewska, A. Bitton, A. Cohen, D. Langelier, J. Belaiche, S. Vermeire, L. Farwell, A. Goris, C. Libioulle, N. Jani, T. Dassopoulos, G. P. Bromfield, B. Dubois, J. H. Cho, S. R. Brant, R. H. Duerr, H. Yang, J. I. Rotter, M. S. Silverberg, A. H. Steinhart, M. J. Daly, D. K. Podolsky, E. Louis, D. A. Hafler, and J. D. Rioux. The role of the Toll receptor pathway in susceptibility to inflammatory bowel diseases. Genes Immun. 8:387–397 (2007).
A. Araki, T. Kanai, T. Ishikura, S. Makita, K. Uraushihara, R. Iiyama, T. Totsuka, K. Takeda, S. Akira, and M. Watanabe. MyD88-deficient mice develop severe intestinal inflammation in dextran sodium sulfate colitis. J. Gastroenterol. 40:16–23 (2005).
M. M. Fort, A. Mozaffarian, A. G. Stover, S. Correia Jda, D. A. Johnson, R. T. Crane, R. J. Ulevitch, D. H. Persing, H. Bielefeldt-Ohmann, P. Probst, E. Jeffery, S. P. Fling, and R. M. Hershberg. A synthetic TLR4 antagonist has anti-inflammatory effects in two murine models of inflammatory bowel disease. J. Immunol. 174:6416–6423 (2005).
G. Baumgarten, P. Knuefermann, H. Wrigge, C. Putensen, H. Stapel, K. Fink, R. Meyer, A. Hoeft, and C. Grohe. Role of Toll-like receptor 4 for the pathogenesis of acute lung injury in Gram-negative sepsis. Eur. J. Anaesthesiol. 23:1041–1048 (2006).
J. Qu, J. Zhang, J. Pan, L. He, Z. Ou, X. Zhang, and X. Chen. Endotoxin tlerance inhibits lipopolysaccharide-initiated acute pulmonary inflammation and lung injury in rats by the mechanism of nuclear factor-kappaB. Scand. J. Immunol. 58:613–619 (2003).
D. A. Johnson. Synthetic TLR4-active Glycolipids as Vaccine Adjuvants and Stand-alone Immunotherapeutics. Curr. Top. Med. Chem. 8:64–79 (2008).
D. A. Johnson, C. G. Sowell, C. L. Johnson, M. T. Livesay, D. S. Keegan, M. J. Rhodes, J. T. Ulrich, J. R. Ward, J. L. Cantrell, and V. G. Brookshire. Synthesis and biological evaluation of a new class of vaccine adjuvants: aminoalkyl glucosaminide 4-phosphates (AGPs). Bioorg. Med. Chem. Lett. 9:2273–2278 (1999).
W. J. Christ, O. Asano, A. L. Robidoux, M. Perez, Y. Wang, G. R. Dubuc, W. E. Gavin, L. D. Hawkins, P. D. McGuinness, and M. A. Mullarkey. E5531, a pure endotoxin antagonist of high potency. Science. 268:80–83 (1995).
H. M. Kim, B. S. Park, J. I. Kim, S. E. Kim, J. Lee, S. C. Oh, P. Enkhbayar, N. Matsushima, H. Lee, O. J. Yoo, and J. O. Lee. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 130:906–917 (2007).
T. Kawata, J. R. Bristol, J. R. Rose, D. P. Rossignol, W. J. Christ, O. Asano, G. R. Dubuc, W. E. Gavin, L. D. Hawkins, and Y. Kishi. Anti-endotoxin activity of a novel synthetic lipid A analog. Prog. Clin. Biol. Res. 392:499–509 (1995).
K. M. Wasan, F. W. Strobel, S. C. Parrott, M. Lynn, W. J. Christ, L. D. Hawkins, and D. P. Rossignol. Lipoprotein distribution of a novel endotoxin antagonist, E5531, in plasma from human subjects with various lipid levels. Antimicrob. Agents Chemother. 43:2562–2564 (1999).
J. R. Rose, M. A. Mullarkey, W. J. Christ, L. D. Hawkins, M. Lynn, Y. Kishi, K. M. Wasan, K. Peteherych, and D. P. Rossignol. Consequences of interaction of a lipophilic endotoxin antagonist with plasma lipoproteins. Antimicrob. Agents Chemother. 44:504–510 (2000).
M. Mullarkey, J. R. Rose, J. Bristol, T. Kawata, A. Kimura, S. Kobayashi, M. Przetak, J. Chow, F. Gusovsky, W. J. Christ, and D. P. Rossignol. Inhibition of endotoxin response by e5564, a novel Toll-like receptor 4-directed endotoxin antagonist. J. Pharmacol. Exp. Ther. 304:1093–1102 (2003).
E. Lien, J. C. Chow, L. D. Hawkins, P. D. McGuinness, K. Miyake, T. Espevik, F. Gusovsky, and D. T. Golenbock. A novel synthetic acyclic lipid A-like agonist activates cells via the lipopolysaccharide/toll-like receptor 4 signaling pathway. J. Biol. Chem. 276:1873–1880 (2001).
H. Methe et al. Statins decrease toll-like receptor 4 expression and downstream signaling in human CD14 monocytes. Arterioscler. Thromb. Vasc. Biol. 25:1439 (2005).
K. M. Wasan, O. Sivak, R. A. Cote, A. I. MacInnes, K. D. Boulanger, M. Lynn, W. J. Christ, L. D. Hawkins, and D. P. Rossignol. Association of the endotoxin antagonist E5564 with high-density lipoproteins in vitro: dependence on low-density and triglyceride-rich lipoprotein concentrations. Antimicrob. Agents Chemother. 47:2796–2803 (2003).
S. M. Opal, P. J. Scannon, J. L. Vincent, M. White, S. F. Carroll, J. E. Palardy, N. A. Parejo, J. P. Pribble, and J. H. Lemke. Relationship between plasma levels of lipopolysaccharide (LPS) and LPS-binding protein in patients with severe sepsis and septic shock. J. Infect. Dis. 180:1584–1589 (1999).
M. Lynn, D. P. Rossignol, J. L. Wheeler, R. J. Kao, C. A. Pardomo, R. Noveck, R. Vargas, T. D’Angelo, S. Gotzkowsky, and F. G. McMahon. Blocking of responses to endotoxin by E5564 in healthy volunteers with experimental endotoxemia. J. Infect. Dis. 187:631–639 (2003).
D. P. Rossignol, K. M. Wasan, E. Choo, E. Yau, N. Wong, J. Rose, J. Moran, and M. Lynn. Safety, pharmacokinetics, pharmacodynamics, and plasma lipoprotein distribution of eritoran (E5564) during continuous intravenous infusion into healthy volunteers. Antimicrob. Agents Chemother. 48:3233–3240 (2004).
M. Ii et al. A novel cyclohexene derivative, ethyl (6 R)-6-[N-(2-chloro-4-fluorophenyl) sulfamoyl] cyclohex-1-ene-1-carboxylate (TAK-242), selectively inhibits toll-like receptor 4-mediated cytokine production through suppression of intracellular signaling. Mol. Pharmacol. 69:1288 (2006).
T. Sha, M. Sunamoto, T. Kitazaki, J. Sato, M. Ii, and Y. Iizawa. Therapeutic effects of TAK-242, a novel selective Toll-like receptor 4 signal transduction inhibitor, in mouse endotoxin shock model. Eur. J. Pharmacol. 571:231–239 (2007).
T. Kawamoto, M. Ii, T. Kitazaki, Y. Iizawa, and H. Kimura. TAK-242 selectively suppresses Toll-like receptor 4-signaling mediated by the intracellular domain. Eur. J. Pharmacol. 584(1):40–8 (2008).
D. C. Angus, and R. S. Wax. Epidemiology of sepsis: an update. Crit. Care Med. 29:S109–S116 (2001).
R. S. Hotchkiss, and I. E. Karl. The Pathophysiology and Treatment of Sepsis. N. Engl. J. Med. 348:138 (2003).
G. Bernard. Drotrecogin alfa (activated) (recombinant human activated protein C) for the treatment of severe sepsis. Crit. Care Med. 31:85–93 (2003).
Y. N. Wong, D. Rossignol, J. R. Rose, R. Kao, A. Carter, and M. Lynn. Safety, pharmacokinetics, and pharmacodynamics of E5564, a lipid A antagonist, during an ascending single-dose clinical study. J. Clin. Pharmacol. 43:735–742 (2003).
E. Bennett-Guerrero. A phase II, double-blind, placebo-controlled, ascending-dose study of Eritoran (E5564), a lipid A antagonist, in patients undergoing cardiac surgery with cardiopulmonary bypass. Anesth. Analg. 104:378 (2007).
K. Kaneko, R. Ueda, T. Kawata, S. Ishizaka, and T. Yoshimura. LPS binding protein does not participate in the pharmacokinetics of E5564. J. Endotoxin Res. 10:185 (2004).
E. Czeslick, A. Struppert, A. Simm, and A. Sablotzki. E5564 (Eritoran) inhibits lipopolysaccharide-induced cytokine production in human blood monocytes. Inflamm. Res. 55:511 (2006).
K. J. Ishii, S. Uematsu, and S. Akira. ‘Toll’ gates for future immunotherapy. Curr. Pharm. Des. 12:4135–4142 (2006).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Leon, C.G., Tory, R., Jia, J. et al. Discovery and Development of Toll-Like Receptor 4 (TLR4) Antagonists: A New Paradigm for Treating Sepsis and Other Diseases. Pharm Res 25, 1751–1761 (2008). https://doi.org/10.1007/s11095-008-9571-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-008-9571-x