
Abstract
Monosaccharide- and polysaccharide biopolymer-stabilized rhenium oxide nanoparticles (ReOxNPs) were effectively applied for deactivation of furazolidone (FRz) and chloramphenicol (ChRP) dissolved in solutions. The employed for that purpose monosaccharide- and polysaccharide biopolymer-stabilized ReOxNPs were synthesized in a two-step procedure. In the first step, the raw-ReOxNPs were produced using a cold atmospheric pressure plasma (CAPP)-based approach. In the second step, the raw-ReOxNPs were incorporated within either D-fructose or Arabic gum, acting as mono- and polysaccharide matrices. Optical and granulometric properties of the so-obtained stabilized ReOxNPs were revealed using several experimental techniques such as UV/Vis absorption spectrophotometry (UV/Vis), scanning electron microscopy (SEM), transmission electron microscopy (TEM), selected area electron diffraction (SAED), energy dispersive X-ray scattering (EDAX), attenuated total reflectance Fourier transform infrared spectroscopy (ATR FT-IR), X-Ray photoelectron spectroscopy (XPS), and dynamic light scattering (DLS). It was found that the produced raw-ReOxNPs, fructose-stabilized ReOxNPs, and Arabic gum-stabilized ReOxNPs were a blend of Re0 and different O-doped Re species, that were octahedral in shape and exhibited the size of 240.6, 461.8, and 7.54 nm, respectively, as was determined using DLS. The obtained ReOxNPs led to losses of FRz in 97, 91, and 62%, for raw-ReOxNPs, fructose-ReOxNPs, and Arabic-gum-stabilized ReOxNPs, respectively, as was estimated using high-performance liquid chromatography with a diode-array detector (HPLC–DAD). The applied approach led to the 100% conversion of ChRP by raw-ReOxNPs, while the saccharide-stabilized nanostructure turned out to be inefficient in reducing ChRP. The method described here might be applied as a tempting alternative to other methods used in the wastewaters purification processes.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Nowadays, utilization of antimicrobial agents for the treatment of bacterial and viral infections is a very common practice. As such, nitro-based antibiotics, i.e. furazolidone (FRz, [3-(5-nitrofurfurylidenamino)-2-oxazolidinone], CAS 67-45-8), and chloramphenicol (ChRP, D-(–)-threo-2-Dichloroacetamido-1-(4-nitrophenyl)-1,3-propanediol, CAS 56-75-7) play a particularly important role. FRz is widely used in veterinary for the treatment of fish diseases in addition to the treatment of poultry diseases [1]. ChRP in turn is defined as a broad-spectrum bacteriostatic antibiotic that is applied for the treatment of kidney and vaginal bacterial infections caused by vancomycin-resistant Enterococcus bacteria [2].
Due to a widespread and often unjustified use of antibiotics in veterinary and medicine, the natural environment is easily contaminated with them. Such a practice may lead to a dangerous increase of multidrug resistance among pathogenic microorganisms against which they were applied [3]. In this scenario, if the multidrug resistance reaches the virulent human pathogens, our community will have no effective drugs for the treatment of common infections. Bearing this in mind, special attention should be paid to deactivation of antimicrobial agents in liquids.
The conventional techniques used in wastewater treatment plants are often unable to remove polar micropollutants. Generally, there are two groups of advanced methods used in chemical degradation of antibacterial agents in liquid wastes. The first group of methods are advanced oxidation processes (AOPs) that include, e.g. Fenton [4], photo-Fenton [5], or ozonation [6] reactions. The second group of methods are based on the application of cold atmospheric pressure plasma (CAPP), in which the plasma-derived reactive species produced during its operation plays a crucial role in the processes of organic compounds degradation [3, 7]. However, both of these approaches exhibit some limitations, mainly associated with an incomplete degradation of the organic pollutants (in the case of AOPs) or a necessity to use specific plasma reactors (in the case of CAPP). Therefore, the application of nanomaterials (NMs) of defined catalytic properties for the treatment of antimicrobial agents could be a tempting alternative to the above-mentioned methods.
Overall, NMs exhibit unique catalytical, optical, chemical, biological, and mechanical properties as compared to the materials of macroscopic sizes [8,9,10]. Considering the catalytic properties of NMs of noble metals, e.g. platinum nanoparticles (PtNPs) [11, 12] and palladium nanoparticles (PdNPs) [12], rhenium nanoparticles (ReNPs) as well as rhenium oxide nanoparticles (ReOxNPs) [13, 14] have them the most desired and that is why they are recognized as novel and very prospective materials. Only in 2020, Re-based NMs were synthesized and used, e.g. as a drug delivery platform for HSP 90 inhibitor [15], as a component of solid oxide fuel cells [16], and as an element of DNA scaffolds with Re electrocatalysts for enhanced hydrogen evolution reaction (HER) [17], and in other applications [18,19,20]. Simultaneously, taking into account decomposition of nitro-based organic compounds, ReNPs and ReOxNPs appears to have excellent catalytic properties but there are only few studies, in which they were used for decomposition of selected nitrocompounds, including nitrobenzene, 2-nitroaniline, 4-nitroaniline, 4-nitrophenol, and 2,4-dinitrophenol [13, 14, 21]. Based on these works, the Re-based NMs certainly outperformed other nanocatalysts in terms of the reaction kinetics and yields (%) of –NO2 groups conversion. However, they either revealed some limitations similar as those reported for other nanocatalysts like PtNPs and PdNPs [22, 23]. These limitations are associated with stability of the Re nanostructures and limited selectivity towards the reduction reaction of –NO2 groups [24, 25]. Therefore, it is hypothesized that proper stabilization of the Re nanostructures could resolve both of these issues. As such, mono- and polysaccharides, i.e. fructose and Arabic gum, could be applied for the capping of the synthesized Re nanostructures. The proper stabilization of Re-based NMs could not only prevent them from their sedimentation but also could modify their structure to gain their selectivity to –NO2 groups [26]. Therefore, such modification could be perfect for the catalytic conversion of nitro-based antibiotics. In this context, to the best of our knowledge, there are no works, in which biopolymer-stabilization of the Re-based NMs was ever studied in reference to efficient deactivation of the nitro-antibiotics.
Therefore, in the present contribution, we have used for the first time the ReOxNPs stabilized with mono- and polysaccharides for the process of deactivation of two antibiotics with –NO2 group, i.e. FRz and ChRP. To reach this aim, we have produced at first the raw-ReOxNPs using an effective, environmental-friendly, and low-cost CAPP-based approach, i.e. applying as the CAPP source a direct current atmospheric pressure glow discharge (dc-APGD) system, operated in contact with a flowing liquid cathode (FLC). Afterwards, to be able to assess the role of the applied stabilizer on the catalytic activity of the obtained raw-ReOxNPs, we have incorporated them within the mono- and polysaccharide matrices, made of either fructose or Arabic Gum, respectively. Then, we have characterized the optical, granulometric, and catalytic properties of the so-produced Re-based NMs and employed them in the deactivation process of FRz and ChRP. Finally, we have assessed the yield of antibiotic degradation achieved with the aid of the synthesized, biopolymer-based Re nanocatalysts.
Materials and Methods
Reagents and Solutions
A raw-ReOxNPs precursor solution was consisted of 1000 mg L−1 of Re(VII) ions (NH4ReO4, Merck, Poland), and applied as a working solution. Arabic gum (pure powder, Avantor Performance Materials, Poland) and D-(–)-fructose (> 99%, Sigma-Aldrich, Germany) were used as stabilizers of ReOxNPs. FRz, belonging to the nitrofurans group, was from Sigma-Aldrich while ChRP, belonging to the chloramphenicols group, was purchased from A&A Biotechnology (Poland). The concentration of both antibiotics in their final water solutions was 0.1 mmol L−1. Sodium borohydride (NaBH4) was used as a reducing agent in the catalytic reduction of –NO2 groups. The reagent was purchased from Merck (Poland), the reducing agent solutions contained 0.1 mol L−1 of NaBH4. The reagents for detection of –NH2 groups, i.e. phenol (> 99%), pyridine (99%), KCN (> 96%), and ninhydrin were acquired also from Merck, and used as received. De-ionized water (0.05 µs/cm) was used throughout.
Synthesis of Raw- and Saccharide-Stabilized ReOxNPs
The synthesis of biopolymer-stabilized ReOxNPs was performed in two steps. In the first step (Fig. 1), the raw-ReOxNPs were produced using a method patented in our research group and described in details in ref [27]. Briefly, a raw-ReOxNPs precursor solution was consistently introduced to a highly-throughput CAPP-based system at a flow rate of 3.0 mL min−1, using a four-channel peristaltic pump (Masterflex L/S, Core Palmer, USA), through a quartz-graphite tube (OD = 6.00 mm, ID = 4.00 mm). Here, dc-APGD, operated in a 2.5 mm distance between a metallic tungsten anode (OD = 4.00 mm) and the ReOxNPs precursor solution acting as the FLC of the dc-APGD system), was used as the CAPP source. The discharge current was 45 mA, while the discharge voltage was 1200 V. The mean power of this the CAPP-based system was 54 Watts. After the ReOxNPs precursor solution was treated by dc-APGD, the resulting product, i.e. the raw-ReOxNPs, was collected to 30 mL glass vials.

The graphical representation of the system used for the raw-ReOxNPs synthesis. (1) (2) High voltage inputs; (3) four-channel peristaltic pump; (4) inlet of a Re(VII) solution; (5) molybdenum electrode; (6) dc-APGD; (7) quartz-graphite tube; (8) sample outflow; (9) sample container
In the next step, the synthesized raw-ReOxNPs were incorporated within the saccharide-based matrices as follows. Firstly, a 13.5 mL of a 0.5% (m/v) solution of Arabic gum or D-(–)-fructose were prepared in a glass vial and heated to 100 °C, using a heating plate equipped with a magnetic stirrer (IKA, C-MAG HS7, Germany). Secondly, a 1.5 mL of a raw-ReOxNPs colloidal solution was mixed with the properly heated up solution of Arabic gum or fructose. After mixing both solutions, the N2 atmosphere (99.9999%, SIAD, POLAND) was provided in order to protect the ReOxNPs against any uncontrolled oxidation. The obtained reaction mixture was maintained in 100 °C for 20 min. Finally, the both saccharide-stabilized ReOxNPs were collected and subjected to further analyses, aimed at assessing their optical, granulometric, and catalytic properties.
Instrumentation
In order to reveal the optical, structural, and catalytic properties of the CAPP-synthesized ReOxNPs, several experimental techniques were employed.
At the beginning, the UV/Vis absorption spectra of all studied ReOxNPs were acquired using a Specord 210 Plus (Analytik Jena, Germany) to determine the optical properties of the studied Re-based NMs. The spectra were recorded with resolution 1 nm, in the range of 200–1000 nm.
Then, the granulometric properties of the ReOxNPs and their morphology were assessed using scanning and transmission electron microscopy (SEM, TEM). The corresponding photomicrographs were acquired using a JEOL JSM-6610A SEM instrument (Japan), and a Hitachi (Tokyo, Japan) H-800 TEM instrument, respectively. The elemental composition of both the raw and the stabilized ReOxNPs were revealed by energy-dispersive X-ray (EDX) microanalysis, using the JEOL JED-2300 EDX analyzer attached to the SEM instrument. The crystallinity of the tested NPs was verified by means of selected area energy diffraction (SAED), which facility was attached to the TEM instrument (Hitachi). The samples for the TEM analyses were placed onto Cu grids and evaporated to dryness under ambient temperature. The average size of the resultant ReOxNPs was assessed with the aid of dynamic light scattering (DLS) using a Litasizer 500 instrument (Anton Paar GmbH, Austria). The DLS measurements were carried out using a detector angle of 173° in an optically homogenous square polystyrene cuvette (Sardsted, Germany). Afterwards, the ReOxNPs stabilization by mono- and polysachcride applied was assessed using attenuated total reflectance Fourier-transformation infrared spectroscopy (ATR FT-IR). The ATR FT-IR spectra were recorded with the aid of a Jasco (MD, USA) FT-IR 4700 spectrometer. The corresponding spectra were acquired with resolution of 4 cm−1, taking 64 scans for each tested sample and zeroing the instrument with water.
To verify the oxidation state of Re species the ReOxNPs consists of, the X-Ray photoelectron spectroscopy (XPS) was applied. The XPS analyses were carried out in a PHI VersaProbeII Scanning XPS system using monochromatic Al Kα (1486.6 eV) X-rays focused to a 100 µm spot and scanned over the area of 400 × 400 µm. The photoelectron take-off angle was 45° and the pass energy in the analyzer was set to 117.50 eV (0.5 eV step) for survey scans and 46.95 eV (0.1 eV step) to obtain high energy resolution spectra for the C 1s, O 1s, Si 2p, Au 4f and Re 4f regions. A dual beam charge compensation with 7 eV Ar+ ions and 1 eV electrons were used to maintain a constant sample surface potential regardless of the sample conductivity. All XPS spectra were charge referenced to the unfunctionalized, saturated carbon (C–C) C 1 s peak at 285.0 eV. The operating pressure in the analytical chamber was less than 4 × 10−9 mbar. Deconvolution of spectra was carried out using PHI MultiPak software (v.9.9.2). Spectrum background was subtracted using the Shirley method. Samples were prepared by successive drop-casting and evaporation of the solvent from nanoparticles dispersion deposited onto a substrate consisting of a thin layer of gold (80 nm) evaporated on silicon wafer.Lastly, the UV/Vis absorption spectrophotometry was used to control the progress of the catalytic reaction. The corresponding UV/Vis spectra were recorded using a Jasco (MD, USA) V570 spectrophotometer with resolution of 1 nm, and in the range of 200–1000 nm. Finally, to verify the yield of conversion of FRz and ChRP in the presence of Re-based NMs, high-performance liquid chromatography with a diode-array detector (HPLC–DAD) was employed. The HPLC–DAD analyses were performed by using a Nexera XR (Shimadzu) system, equipped with: an autosampler SIL-20AC, a pump LC-20AD, a column oven CTO-20AC, and a detector SPD-M20A. The HPLC column was Gemini-NX (5 µm, C18 110A, 150 × 4.6 mm). The injection volume was 25 μL. The autosampler oven temperature was set to 10 °C. The mobile phase component A was 10 mmol L−1 ammonium formate (pH 3.2), while the component B was acetonitrile (ACN). The column oven was set to 27 °C. The elution started with 10% of B but during 8 min the B component increased to 90% and was maintained through next 2 min.. The mobile phase flow rate was 1 mL min−1. The DAD was set to measure the wavelengths from 190 to 800 nm. The analytical wavelength was set to 365 nm for FRz, and 280 nm for ChRP. The retention time of FRz and ChRP was 5.55 min and 5.98 min, respectively. The calibration curve spanned the concentration range from 0.01 to 50 mg L−1. The removal efficiency of FRz and ChRP was calculated by comparing the concentration of the antibiotic found before and after the treatment with NaBH4 in the presence of the raw-ReOxNPs or the saccharide-stabilized ReOxNPs.
Catalytic Reduction of Antibiotics Triggered by the ReOxNPs Application
To deactivate the antimicrobial agents containing –NO2 groups, i.e. FRz and ChRP, the nitro-moieties thereof (see Fig. 2 for details) were reduced to –NH2 groups with NaBH4 via the catalytic process.

Chemical structures of A furazolidone (FRz) and B chloramphenicol (ChRP)
For the tests on catalytic deactivation of both antibiotics, water-based solutions of FRz and ChRP (0.1 mmol L−1), NaBH4 (0.1 mol L−1), and colloidal suspensions of proper ReOxNPs (raw- or saccharide-stabilized) were used. The catalytic reaction was carried out in the following way. First, 2.7 mL of the FRz or ChRP solution was introduced to a quartz cuvette. Afterwards, 0.2 mL of the NaBH4 solution was introduced. Finally, 0.2 mL of the so-prepared Re-based nanocatalyst suspension was added, which initiated the catalytic reduction reaction of –NO2 groups. The course of both reactions was constantly monitored using UV/Vis absorption spectrophotometry, i.e. the corresponding UV/Vis absorption spectra were recorded, and the absorbance readings at λmax assigned to –NO2 groups (λmax ~ 364 nm for FRz and 278 nm for ChRP), and at λmax assigned –NH2 groups (λmax 213–235 nm) were carried out. The use of the excessive amount of NaBH4 in respect to FRz and ChRP enabled to model of the undergoing catalytic reduction reaction using the pseudo-first order kinetics. Because the decrease of the absorbance associated with reduction of –NO2 groups is proportional to the decrease of their concentration, the pseudo-first order kinetics constant (k1, min−1) was acquired based on the slope of the ln(At/A0) = k1t plot, where A0 and At are the initial absorbance, and the absorbance at time t. To enable the direct comparison between the catalysts the obtained pseudo-first order constants (k1, min−1) were re-calculated to mass-normalized rate constants (km, min−1 mg−1) that reveal the catalytic activity in the context of the amount of active sites applied. After completing the catalytic reduction reaction, samples were also collected and analyzed using HPLC–DAD in order to determine whether and to what extent FRz and ChRP were converted. Finally, the presence of –NH2 groups in the resultant products were confirmed using the Kaiser test, that is a color reaction of free –NH2 groups in the presence of KCN and ninhydrin [28]. The test was carried in the samples of FRz and ChRP after the catalytic process, and the obtained result was compared to the same test carried out for the unprocessed antibiotics as well as the catalysts used for the research.
Results and Discussion
Characterization of the ReOxNPs Obtained Usig a CAPP-Based Approach
It is well known that in the case of CAPP operated in contact with liquids, different long-lived and short-lived reactive oxygen and nitrogen species (RONS) of certain red-ox potentials in addition to hydrated electrons (e−aq) are present in these liquids and are involved in the nucleation and growth of metallic NPs [29]. Bearing this in mind, we have employed dc-APGD, operated in the surrounding atmospheric air (no additional noble or non-noble gas was required) and in contact with the continuously flowing solution of Re(VII) ions and, to produce ReOx of nanometric size through the CAPP-liquid interactions, e.g. reduction reactions of Re(VII) ions with RONS and e−aq.
To verify our hypothesis about the stabilization effect on the catalytic properties of the Re-based NMs, the resulting CAPP-synthesized ReOxNPs were incorporated into saccharide matrices, and we have characterized the obtained products. The optical properties of both the raw- and the saccharide-stabilized ReOxNPs were assessed using UV/Vis absorption spectroscopy (Fig. 3).

UV/Vis absorption spectra of A the raw-ReOxNPs, B the fructose-stabilized ReOxNPs, and C the Arabic gum-stabilized ReOxNPs
Generally, Re-based NMs, due to the low dielectric constant value for Re, do not have any characteristic fingerprinting regions presented in the UV/Vis absorption spectra (the UV/Vis spectra displayed in a full range are provided for the reference in Figures S1 and S2, Supplementary Materials) [13, 30]. However, as displayed in the UV/Vis absorption spectrum of the raw-ReOxNPs (Fig. 3A), a series of small bumps at λmax 350–380 nm were found, likely due to the interferences caused by the scattering of light by the nanostructures present in the colloidal solution. These bumps significantly faded after stabilization of the ReOxNPs (Fig. 3B, C), suggesting that the monosaccharide-stabilization and polysaccharide biopolymer-stabilization of ReOxNPs could be indeed occur. Similar conclusion can be made when considering the saccharides themselves. The spectrum of fructose (Fig. 3B and S1) displays a series of bands at λmax ~ 250 nm. In contrast, the spectrum of fructose-stabilized ReOxNPs (Fig. 3B) reveal no additional interferences. In turn, the spectrum of the Arabic gum biopolymer shows a maximum at λmax 281 nm (fingerprint of Arabic gum [31]). This maximum shifted to λmax 277 nm (Figs. 3C, S1 and S2) after ReOxNPs stabilization. All of these observations suggest that the saccharide stabilization actually took place [31, 32]
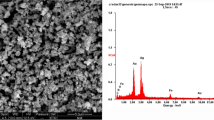
To verify the elemental composition of the synthesized Re-based NMs, the raw-ReOxNPs were subjected to the SEM-EDAX analysis. Based on the acquired SEM photomicrograph (Fig. 4A) and the registered EDAX spectrum (Fig. 4B), it was established that the synthesized ReOxNPs indeed based on Re. Simultaneously, some small peaks in the range of 0.065–0.130 keV were assigned to the presence of C and O (Fig. 4B), which elements originated from saccharides and the ReOxNPs, respectively. However, Re(VII) in the form of ReO4− ions is difficult to be reduced. This is attributed to their relatively low reducing potential of 0.510 V (for instance, the reduction potential of PtCl42− ions is 0.755 V, and of AuCl4− ions is 1.002 V) [33,34,35]. Simultaneously, Re can form a variety of species at 0, +3, +4, +6 and +7 oxidation states. Therefore, to determine the nature of the CAPP-synthesized ReOxNPs in more details, the TEM analysis was also employed. Figure 5 displays the photomicrographs of the raw- and saccharide-stabilized ReOxNPs. Additionally, the SAED pattern of the raw-ReOxNPs is also provided.

A The SEM photomicrograph and B the EDAX spectrum of the raw-ReOxNPs

TEM photomicrographs of A the raw-ReOxNPs, B the SAED pattern of the raw-ReOxNPs C The fructose-stabilized ReOxNPs, and D the Arabic gum-stabilized ReOxNPs
The SAED pattern displayed in Fig. 5B confirms that the synthesis of the ReOxNPs led to the formation of NMs that were a blend of different Re species. Based on the calculated d-spacings, the ReOxNPs were possibly the blend of Re3O10 (d-spacing 3.38 Å), Re2O7 (d-spacing 1.91 Å) [16], ReO2 (d-spacing 2.87 Å) [16, 36], ReO3 (d-spacings 1.51, 3.80 Å) [37], and Re(0) (d-spacings 2.21, 2.72 Å) [36, 38].
As can be seen in Fig. 5A, the CAPP-synthesized raw-ReOxNPs were octahedral in shape with the size ranging from 80 to 130 nm. Meanwhile, the fructose- and the Arabic gum-stabilized ReOxNPs (Fig. 5C, D) were much smaller and clearly occluded in the organic matrix (the effect visible as the dark shades around NPs). The diameters of the saccharide-stabilized ReOxNPs ranged from 2 to 20 nm and simultaneously, the shapes thereof seemed to be spherical instead of octahedral. This suggested that the monosaccharide and the polysacchcride biopolymer played a significant role not only in the stabilization of the resultant Re-based NMs but also could affect the reduction of Re(VII) ions used for the synthesis. This phenomenon will be discussed in the next section. Besides the size revealed by the TEM analysis, DLS was additionally applied to get this size of the CAPP-synthesized ReOxNPs. It was established that the average size by number of the ReOxNPs was 240, 460, and 7.5 nm for the raw-ReOxNPs, the fructose-stabilized ReOxNPs, and the Arabic gum-stabilized ReOxNPs, respectively. This stands in contrast with the TEM-determined diameters of the ReOxNPs, which were considerably smaller as compared to those determined by the DLS technique. The differences between these diameters were probably caused by the nature of the DLS measurement itself (the technique is based on the light scattering) [39]. Therefore, it is likely that the DLS analysis could be interfered by the hydration radius of the ReOxNPs, resulting from their capping by fructose and Arabic gum. This conclusion seemed to be supported by the determined hydrodynamic diameters (the average size by intensity) that were respectively: 850, 400, and 270 nm. Because the ReOxNPs were sparingly described in the literature, the assessment of their morphology and composition is challenging. The above-mentioned results indicate on the significant differences between raw- and saccharide stabilized ReOxNPs. For this reason, to ultimately verify the oxidation state of the Re in ReOxNPs, the XPS analysis was carried out. The Fig. 6 displays Re 4f spectra of the raw- and saccharide-stabilized ReOxNPs. Additionally, the C 1s, O 1s, Si 2p, Au 4f spectra are provided in the supplementary materials in Figure S3. The XPS survey spectra are also displayed in the supplementary materials in Figure S4. In turn, Table 1 shows surface concentrations of chemical bonds obtained from fitting XPS data for all analyzed samples.

Re 4f XPS spectra of the A raw-ReOxNPs, B fructose-ReOxNPs, and C Arabic gum-ReOxNPs
The Re 4f spectra were fitted with up to three doublet structures (doublet separation f7/2–f5/2 equals 2.43 eV) with first main 4f7/2 line centered at 42.5 eV which indicate Re4+ oxidation state like in ReO2, second 4f7/2 line centered at 44.2 eV points out the existence of Re6+ oxidation state like in ReO3 and last 4f7/2 line found at 46.3 eV indicate Re7+ oxidation state like in Re2O7 (Fig. 6) [40, 41]. The latter one could also originate from Re7+ in ReO4− used as ReOxNPs precursor. These results are consistent with the TEM/SAED analysis, where similar structures were detected. However, in that case, the lattice fingers of Re0 (Fig. 5) were also found. Although the XPS analysis does not confirm this, the formation of Re0 cannot be excluded, as this species may be at very low concentration, be greatly dispersed or buried in a nanoparticle (as information depth in XPS is about 5 nm i.e. the analyzed part of nanopraticles is only the very surface not bulk). What should be also noticed is the atomic composition (%) of ReOxNPs surface. As can be seen in Table 1, the raw-ReOxNPs contains much greater concentration of Re7+ atoms as compared to the saccharide stabilized samples. Because the Re7+ could originate from i.a. unreacted precursor (ReO4−), much smaller concentrations thereof detected in the stabilized samples may suggest that the fructose and Arabic gum indeed participated in the formation of ReOxNPs. The support for this conclusion can be further found in accessing the O 1s spectra (Figure S3), that were fitted with three components: first line centered at 531.6 eV which points out the existence of oxygen in metal oxides (Re–O, Si–O), second line centered at 533.1 indicate presence of O–C type bonds and third line at 534.7 eV indicate –OH type compounds and/or adsorbed water [40]. Furthermore, the C 1s spectra can be fitted with three components: first line at 285.0 eV indicate presence of aliphatic carbon, third line C–O groups (286.7 eV) and C=O and/or O–C=O groups evidenced with the line centered at 288.3 eV (Figure S3) [42]. All of these observations may suggest that the saccharides not only stabilized the ReOxNPs but also they could participate in the reduction of Re(VII).
Stabilization of ReOxNPs
All the observations made above suggested the primary role of the monosaccharide and the monosaccharide biopolymer in the stabilization of the resultant ReOxNPs. Therefore, the ATR FT-IR analyses were performed to assess the mechanism of this stabilization. The ATR FT-IR spectra were recorded for both the solutions containing the fructose- and the Arabic gum-stabilized ReOxNPs, as well as the solutions containing the stabilizers themselves. The recorded spectra are displayed in Fig. 7.

ATR FT-IR spectra of the raw-ReOxNPs, and A the fructose solution without the ReOxNPs and the solution containing the fructose-stabilized ReOxNPs; B the Arabic gum solution and the solution containing the Arabic gum-stabilized ReOxNPs
In the case of the ATR FT-IR spectra recorded for the solutions containing fructose (Fig. 7A), a series of bands associated with the O–H and C–H stretching vibrations were observed in the range of 3600–3200 cm−1. After the ReOxNPs stabilization, these bands sig changed their intensity and slightly shifted towards the smaller wavenumbers (1–2 cm−1 shifts). On the one hand, this could result from the formation of the ReOxNPs–OH bonds, but on the other hand, it could be related to the rearrangement of H–bonds due to the metal complexation [43]. Meanwhile, the bands observed in the region ~ 2880 cm−1, assigned to the C–H stretching vibrations in the monosaccharide (Fig. 7A) [44], also shifted to the lower wavenumbers (~ 20 cm−1 shift) and increased their intensity, which could be also linked with the coordination of the ReOxNPs by fructose. The lower region of the ATR FT-IR spectra (Fig. 7A) displayed a series of fingerprints associated with the saccharide ring. This included the bands situated at 1648 cm−1 (the OH bending of absorbed water), 1475, 1455 and 1419 cm−1 (the in-plane HCH and OCH bending), 1393 cm−1 (the in-plane CH bending), and 1065 cm−1 (the C–O–C symmetric stretching) [43, 44]. These bands changed their intensity and shifted towards the lower wavenumbers as well, suggesting the metal-sugar complexation. Similar phenomena were observed in the case of the ATR FT-IR spectra recorded for the solutions containing Arabic gum (Fig. 7B). The bands found in the region of 3600–3100 cm−1 (the O–H and C–H stretching) were shifted and changed their intensity as the result of the ReOxNPs stabilization. The band at 2914 cm−1 (the C–H stretching) was shifted about 20 cm−1 towards the lower wavenumbers similarly as in the case of the spectra recorded for fructose (Fig. 6). The same was observed for the bands at ~ 1652 and 1435 cm−1, attributed to the COO– asymmetric stretching [43,44,45]. Additionally, the wide band at ~ 1080 cm−1 that was assigned to the polyol moieties [46] in Arabic gum was also shifted about ~ 2 cm−1 from 1076 to 1074 cm−1 (Fig. 7B). All of these observations are consistent with the XPS analysis, that suggested, on the one hand, more efficient reduction of ReO4− oxyanion in the presence of saccharides, and on the other hand, formation of the above-mentioned bonds (Fig. 6, Table 1). This further allows to conclude that the primary mechanism of the ReOxNPs stabilization was possibly the metal–oxygen interactions. Figure 8 displays a simplified complexation mechanism of the ReOxNPs by fructose. The mechanism was likely similar in the case of Arabic gum-stabilized ReOxNPs, however, due to a complex structure of this polymer it was not displayed here.

The graphical representation of the ReOxNPs stabilization in D-(–)-fructose
The result of the successful stabilization of the O-complexing atoms was confirmed by apparent stability of the obtained Re-based NMs. While the raw-ReOxNPs tended to sediment in several min after their synthesis, the monosaccharide-stabilized ReOxNPs and polysaccharide biopolymer-stabilized ReOxNPs were stable for 2 months and more, showing no signs of agglomeration.
The Role of Saccharides in the Production and Stabilization of ReOxNPs
It was found that the monosaccharide and the polysaccharide biopolymer played the primary role in the effective stabilization of the resultant Re-based NMs. Nevertheless, there was yet another issue that needed to be considered. A relatively large difference between the TEM-defined size of the raw- and saccharide-stabilized ReOxNPs and the respective ones determined by DLS suggested that fructose and Arabic gum also contributed to some extent to the production of the ReOxNPs.
The operation of dc-APGD in contact with liquids, acting as the FLC of the system, in the open-to-air atmosphere is responsible for a number of RONS, H radical as well as eaq in these liquids that are involved in the NPs formation [29]. In this way, the FLC solution, containing NH4ReO4, is irradiated by H radicals and e−aq, leading to the reduction of Re(VII) ions [27]. On the other hand, some of RONS that remain dissolved in the Re(VII) solution are responsible for the prolonged and shifted in time oxidation process [29]. As the result, the synthesis products are raw-ReOxNPs that can be stabilized latter with fructose of Arabic gum.
Therefore, in the proposed method, at first, the operation of dc-APGD in contact with the Re(VII) solution in the continuous flow CAPP-based reaction-discharge system led to the production of the certain RONS along with the H radical and e−aq in this solution. These reactive individuals were responsible for the formation of the crystalline centers of the ReOxNPs. With the lack of saccharides, the dissolved reactive individuals further promoted the reduction of the Re(VII) ions, and the formation of the octahedral raw-ReOxNPs (Fig. 5A). In this case, as evidenced by the XPS analysis the reduction was limited, as a larger quantities of Re7+ were detected. Thought, in the case of the stabilization of the ReOxNPs by saccharides, the solution containing the Re(VII) ions and the crystalline centers of the ReOxNPs was immediately mixed with the solutions of fructose or Arabic gum. This resulted in the stabilization of the progenitors of the ReOxNPs due to the O–complexation as displayed in Fig. 7. Then, the remaining reactive individuals promoted the further reduction of the Re(VII)ions, but in this case, the size and shape of the ReOxNPs were already limited by fructose or Arabic gum that capped the resultant ReOxNPs. This could be the reason why the monosaccharide-stabilized ReOxNPs or the polysaccharide biopolymer-stabilized ReOxNPs revealed the different morphology as compared to the raw-ReOxNPs (Fig. 5A vs. Fig. 5C, D).
To sum up, the saccharide stabilization of ReOxNPs played a triple role on the properties of the synthesized NMs. First, the saccharides prevented Re-based NPs from sedimentation. While the raw-ReOxNPs agglomerated and sedimented in the form of black powder within just few minutes after their synthesis, the saccharide-stabilized ones remain stable for months with no signs of agglomeration. Second, the presence of excessive O-based moieties caused the reduction of ReO4− was apparently enhanced. Third, the O–complexation prevents the ReOxNPs crystalline centres from overgrowth. As the result, the saccharide stabilized ReOxNPs are much smaller than non-stabilized ones.
Catalytic Deactivation of Antibiotics with Biopolymer-Stabilized ReOxNPs
The synthesized Re-based NMs were used for deactivation of FRz and ChRP through conversion of their –NO2 groups to –NH2 groups (see Fig. 2) [47]. It was hypothesized that the saccharide-stabilized ReOxNPs would lead to catalytic deactivation of FRz and ChRP under mild conditions. If so, the developed Re-based NMs could be a very tempting”stabile&green” alternative that could address the major challenges related to the occurrence of residual antibiotics and the multidrug resistance among pathogenic microorganisms towards which they are widely applied [3].
The catalytic reaction was carried out in the presence of NaBH4 under the supervision of UV/Vis spectrophotometry (see “Catalytic Reduction of Antibiotics Triggered by the ReOxNPs Application” section for details). First, the reduction reaction of FRz using the raw-ReOxNPs and the saccharide-stabilized ReOxNPs was carried out. The collected UV/Vis absorption spectra are displayed in Fig. 9.

UV/Vis absorption spectra displaying catalytic reduction of FRz using A the raw-ReOxNPs, B the fructose-stabilized ReOxNPs, and C the Arabic gum-stabilized ReOxNPs
The rate and the efficiency of –NO2 groups reduction were estimated by monitoring the changes of the absorbance reading at λmax of 364 nm (a characteristic band of FRz [48]). First, the spectrum of the FRz solution was recorded (black lines with the highest absorbance, Fig. 9). After the addition of the reducing agent, the absorbance reading slightly decreased (blue lines with the second-greatest absorbance) as the result of the sample dilution. Afterwards, the raw-ReOxNPs-based catalyst was added. This resulted in the immediate blue-shift (Fig. 9A) of λmax from 364 to 358 nm. Then, as the reaction proceeded, the band was further shifted, manifesting its final location at 340 nm that could be recognized as a fingerprint of the FRz amino-derivative. The described observation was done for the process carried out with the use of raw-, and fructose-stabilized ReOxNPs. When Arabic gum-stabilized ReOxNPs were applied, the shift of the band at 364 nm was observed at smaller frequencies and finally was located at 378 nm. This observation does not exclude the formation of the furanamine derivative, as this may be observed in different environments up to the region of ~ 411 nm [49]. Such shifts could be attributed to the various factors, including, for example, the influence of functional groups [49]. This suggests that the Arabic gum could also interact with the reaction product besides the ReOxNPs.
The catalytic activity was determined by recording the corresponding spectra at different time intervals (Fig. 9) until the reaction significantly decreased its rate. The reduction of the –NO2 group in FRz was evidenced by a gradual decrease of the absorbance at λmax of 358 nm (Fig. 9A). Simultaneously with the fading of the band at λmax of 358 nm a series of new maxima in the range of λmax of 213–238 nm appeared (see Figure S5, Supplementary Materials, for details). These were previously recognized as a fingerprint of different amino-moieties [50,51,52,53,54], suggesting that the catalytic reaction led to the formation of amino-derivatives of FRz, as the product of its deactivation.
Similar observations were made in the case of the saccharides-stabilized ReOxNPs. Both the fructose- and the Arabic gum-stabilized ReOxNPs were responsible for a gradual decrease of the absorption maxima at λmax of ~ 360 nm with the simultaneous appearance of bands at λmax of 213–238 nm. This was linked with the simultaneous reduction of FRz and the production of its amino-derivative. At this point it should be also noticed, that the applied raw-ReOxNPs were characterized by the greater concentration of Re as compared to the saccharide-stabilized catalysts (1000 vs. 100 mg L−1). This difference arises from the fact, that the raw-ReOxNPs diluted in the same way as those introduced into fructose, and Arabic gum solutions (see “Synthesis of Raw- and Saccharide-Stabilized ReOxNPs” section for details), revealed neglible catalytic activity. This phenomenon could be caused by the rapid sedimentation of raw-ReOxNPs that has been discussed above. In this context, the undiluted raw-ReOxNPs must had offered an excessive concentration of Re that suppressed the effect made by the sedimentation. For this reason, the undiluted raw-ReOxNPs were applied in this study. Because of this, to enable the direct comparison between all the used samples, Re concentration was also considered. It was done further in the manuscript.
Because the absorption maxima of –NH2 groups tend to shift (due to the ability of N atoms to be protonated) and thus they are not specific, the color reaction with ninhydrin [28] was carried out to verify whether the FRz reduction reaction indeed resulted in the production of its amino-derivative. Based on the test, the catalytic process indeed resulted in the production of excessive –NH2 groups.
Because the absorbance readings given in Fig. 9 were proportional to the concentration of FRz, it was possible to assess the reduction yield (%) as well as the rate of the process, as determined using the pseudo-first order kinetics. Generally, the raw-ReOxNPs led to the 73.4% conversion of FRz within 19 min; the fructose-stabilized ReOxNPs led to the 80.4% conversion of FRz within 15 min, and the Arabic gum-stabilized ReOxNPs led to the 74.8% conversion of FRz within 15 min. At the same time, the pseudo-first order rate constants (k1) of –NO2 groups reduction were 0.067, 0.11, and 0.081 min−1, respectively. However, it must be remembered, that the factual mass of Re introduced together with 0.2 mg of raw-ReOxNPs was 0.2 mg, while the mas of Re used with saccharide-stabilized samples was 0.02 mg. Despite this tenfold difference both of the stabilized catalysts outperformed the raw-ReOxNPs.
Based on these results it could be stated that the saccharide-stabilized ReOxNPs revealed the greater catalytic activity than this for the raw-ReOxNPs despite a significant difference in the concentration of Re between raw and stabilized samples. This implies that the saccharides played the primary role not only in the synthesis and the stabilization of the Re-based NMs, but also they enhanced their catalytic activity. This phenomenon could be caused by two factors. Firstly, the saccharide-stabilization prevented agglomeration and sedimentation of the ReOxNPs (see “Stabilization of ReOxNPs” section for details). Secondly, the mechanism of the stabilization led to the production of the ReOxNPs that were much smaller as compared to the raw-ReOxNPs (see “Characterization of the ReOxNPs Obtained Usig a CAPP-Based Approach” and “Stabilization of ReOxNPs” sections for details). At this point, it should be also noted that the ReOxNPs stabilized by fructose revealed the approx. 25% greater k1 value as compared to this assessed for the Arabic gum-stabilized ReOxNPs (0.11 vs. 0.081 min−1).
The raw-ReOxNPs and the saccharide-stabilized ReOxNPs were used for catalytic deactivation of ChRP. The experiment conditions remained the same, and the recorded UV/Vis spectra are displayed in Fig. 10.

UV/Vis absorption spectra of catalytic reduction of ChRP using A the raw-ReOxNPs, B the fructose-stabilized ReOxNPs, and C the Arabic gum-stabilized ReOxNPs
The ChRP characteristic absorption maximum at λmax of 278 nm, attributed to the N–O electron transition in –NO2 groups [55], gradually faded after the raw-ReOxNPs catalyst was applied. The presence of –NH2 groups was also verified in product of processed ChRP by using Kaiser test. The saccharide-stabilized ReOxNPs were not effective. The fructose-stabilized and the Arabic gum-stabilized ReOxNPs led to only 8 and 5% conversion (within 10 min) of –NO2 groups, respectively. In turn, the application of the raw-ReOxNPs led to 88% conversion of –NO2 groups within 10 min with the rate constant k1 = 0.21 min−1. This result was even greater as compared to the same catalyst used for the reduction reaction of FRz (k1 = 0.067 min−1, 73.4% conversion within 19 min). At the same time, the application of raw-ReOxNPs with Re concentration equal to this obtained in the saccharide-stabilized catalysts (100 mg L−1) was unsuccessful. Based on these observations, it was concluded that the lack of the activity towards catalytic reduction of ChRP could be linked with both, not enough amount of Re as well as with saccharides themselves. It was previously recognized that ChRP interacts with oligosaccharides, leading to the promotion of glucokinase and the activation of fructose 1-phosphate (as a part of the process of the saccharides metabolism) in the absence of insulin [56], also ChRP conjugates facilitate the uptake of glucose and fructose by Acetobacter xylinum [56]. Hence, it was concluded that although saccharides successfully stabilized the ReOxNPs, they could also prevent ChRP from reduction.
As evidenced in Figs. 9, 10, and S5, the catalytic reaction revealed a complex nature. The absorption maxima at λmax 364, and 278 nm assigned to the –NO2 groups tend to be shifted, while the maxima attributed to the –NH2 groups are usually unspecific. This is caused by the ability of –NH2 groups to be protonated, and therefore, they tend to shift UV/Vis spectrum on the regular basis. In the present studies, the FRz and ChRP amino-derivatives were assigned to maxima at λmax 205–235 nm. For these reasons, to confirm the reduction of native antibiotics and changes in their concentration (and suspected conversion to amino-derivatives), the HPLC–DAD analysis was employed. The chromatogram shown in Fig. 11 (extracted for 365 nm) displays the samples of FRz before and after the reduction process with the main signal derived from FRz (retention time 5.5 min). In turn, the chromatogram shown in Fig. 12 (extracted for 280 nm) displays the samples of ChRP before and after the reduction process with the main signal derived from ChRP (retention time 5.9 min). No additional signals were found that suggested that in these chromatographic conditions any other conversion products could not be detected. Few relatively low signals present in the chromatograms after the catalytic reduction were likely derived from the saccharides stabilizers used (stated after comparison with chromatogram of blank samples).

HPLC–DAD (365 nm) chromatograms of furazolidone (FRz) of the samples: not subjected to the reduction process (A), and after the catalytic reduction of FRz using B the raw-ReOxNPs, C the Arabic gum-stabilized ReOxNPs, and D the fructose-stabilized ReOxNPs

HPLC–DAD (280 nm) chromatograms of chloramphenicol (ChRP) of the samples: not subjected to the reduction process (A), and after the catalytic reduction of ChRP using B the raw-ReOxNPs, C the Arabic gum-stabilized ReOxNPs, and D the fructose-stabilized ReOxNPs
The HPLC–DAD analysis pointed out that conversion of FRz and ChRP was successful. Table 2 displays the % values of the FRz and ChRP maximum possible reductions, determined based on the chromatographic signals. The results of both the –NO2 groups conversion as well as the decrease of the antibiotics concentrations resulting from the process were rather consistent. The differences in the yields of conversion were probably caused by a delay in performing the HPLC–DAD analysis, carried out approximately 48 h after the start of the catalytic reaction. Hence, the value defined as the antibiotic loss (Table 2) might be recognized as the maximum possible conversions of FRz and ChRP, while the value assigned as –NO2 conversion gives a perspective on a catalyst activity within fixed reaction time. Among all of the considerations, that include the above-mentioned rate constants and the Antibiotics loss, a difference in the concentration of Re applied in the catalytic systems must had been considered as well. For this reason, Table 2 displays the set of data related to the catalytic reduction processes.
To sum up, it could be generally concluded that the applied methodology (based on NMs with ReOxNPs) efficiently led to the conversion of FRz and ChRP. The pseudo-first order kinetics modelling revealed, that within the given time of the catalytic reaction (15 min), the saccharide-stabilization of the ReOxNPs significantly enhanced the mass-normalized rate constants (km, Table 2) of FRz reduction. As the result, the fructose-stabilized ReOxNPs revealed 80.4% of –NO2 groups conversion with km = 5.5 min−1 mg−1, while the corresponding values for the raw-ReOxNPs were 54% of –NO2 groups conversion, and km = 0.34 min−1 mg−1 (Table 2). However, as evidenced by HPLC–DAD analysis, the maximum possible yields of FRz reduction were 91% and 97% for fructose-ReOxNPs and raw-ReOxNPs, respectively. This may suggest, that both, raw- as well as fructose-ReOxNPs can be recognized as successful. The selection of the best catalyst depends on the preference the process could be carried out, i.e. whether it is necessary to convert maximum concentration of antibiotic within fixed amount of time or it is required to achieved the highest possible antibiotic loss. It must be remembered tough, that the raw-ReOxNPs are unstable as they tend to quickly sediment, while the fructose-stabilized ReOxNPs do not. Furthermore, both of the saccharide-stabilized ReOxNPs outperformed the raw-ReOxNPs in the terms of reaction kinetics despite the fact, the actual amount of stabilized Re was significantly smaller (tenfold) difference. This means that the advantages of using saccharide-stabilized catalysts can be considered on to different levels, i.e. by increasing the reaction kinetics, and by the decrease of amount of Re to be applied. On the other, hand, when considering the mass-normalized rate constants in the context of the XPS analysis, it seems that there is indeed a link between the ratio of Re7+/Re4+,Re6+ (see Table 1) and the catalytic activity achieved by the catalysts (see Table 2). A significant difference between these values can be a further support for the conclusion, that the saccharides enhanced the reduction of ReO4− and thus led to the fabrication of more efficient catalysts.
The kinetics modeling suggests, that fructose-ReOxNPs led to the efficient, almost entire conversion of FRz. Moreover, fructose not only provided the stabilization of the ReOxNPs but also boosted rate constants the ReOxNPs-based catalyst can reveal (Table 2). In turn, the stabilization of ReOxNPs with Arabic gum led to the formation of a catalyst revealing the similar boost of mass-normalized kinetics. Despite lower antibiotic loss the polysaccharide biopolymer still provided proper stability of the ReOxNPs and enabled catalytic reduction of FRz under the mild conditions (ambient temperature, pressure and water-based environment).
Simultaneously, a different tendency is observed in the case of the reaction carried out on ChRP. In this case, although the raw-ReOxNPs led to the 100% conversion of the antibiotic to its amino-derivative, the saccharide-stabilized ReOxNPs showed the negligible catalytic activity towards the ChRP reduction. The mass-normalized rate constants (km, Table 2) achieved by the saccharide-stabilized ReOxNPs were over twice lower as compared to the values revealed by the raw-ReOxNPs. This may suggest the primal role of Re concentration in the catalytic reduction of ChRP.
Conclusions
Here a series of new NMs, based on the ReOxNPs, were obtained. The premise of this work was to verify whether the stabilization of ReOxNPs by sacharides (fructose, Arabic gum) enables the catalytic reduction of nitro-based antibiotics to their amino-analogues under the mild conditions. For this purpose, a series of nanocatalysts were synthesized to enable the assessment of the saccharide-stabilization. This included the raw-ReOxNPs, the monosaccharide (fructose)-stabilized ReOxNPs, and the polysaccharide (Arabic gum)-stabilized ReOxNPs.
The ReOxNPs were successfully synthesized and formed as a blend of different Re oxides and Re0. Based on the obtained results it was concluded that the mono- and polysaccharide indeed led to the efficient stabilization of the resultant ReOxNPs via the chelation mechanism, and furthermore played a significant role in the synthesis itself, leading to the production of the much smaller (2–20 nm) ReOxNPs as compared to the raw-ReOxNPs (~ 100 nm). The raw- as well as mono- and polysaccharide-stabilized ReOxNPs led to the efficient reduction of FRz to its amino analogue. Simultaneously, it was observed that the stabilization of the ReOxNPs with fructose enhanced the rate constant of the catalytic reaction, despite applying smaller mass of Re-active sites. This resulted resulting in the greater yield of –NO2 groups conversions within fixed time, as compared to other unstabilized nanocatalyst. Both of the stabilizing saccharides provided the catalytic activity under mild conditions, and significantly boosted the mass-normalized rate constants of FRz and ChRP reduction. This allowed, on the one hand, to make the catalytic reduction quicker, and on the other hand, opens a possibility of significant decrease of Re that needs to be applied in the catalytic system.
Although the raw-ReOxNPs led to the 100% conversion of ChRP, both the mono- and polysaccharide-stabilized ReOxNPs revealed the negligible catalytic activity towards ChRP reduction. This phenomenon was likely linked to the concentration of Re-based catalyst that needs to be applied, and saccharide-ChRP interactions, disabling the reduction of the antibiotic.
Based on the obtained results it can be stated that the ReOxNPs is indeed a prospective NM that could be used for the catalytic reduction of nitro-based antibiotics. Also, it was found that the use of the monosaccharide or the polysaccharide biopolymer may provide stability of the resultant NMs and in the case of FRz also preserves or even enhances their catalytic activity.
Patents
The CAPP-based synthesis method of NPs is protected by Polish Patent No. Pat.231602, dedicated to A.D, P.J, and P.P.
Abbreviations
- AOP:
-
Advanced oxidation processes
- ATR FT-IR:
-
Attenuated total reflectance fourier transform infrared spectroscopy
- CAPP:
-
Cold atmospheric pressure plasma
- ChRP:
-
Chloramphenicol
- dc-APGD:
-
Direct current atmospheric pressure glow discharge
- DLS:
-
Dynamic light scattering
- EDAX:
-
Energy dispersive X-ray scattering
- FLC:
-
Flowing liquid cathode
- FRz:
-
Furazolidone;
- HER:
-
Hydrogen evolution reaction
- HPLC–DAD:
-
High-performance liquid chromatography with a diode-array detector
- NMs:
-
Nanomaterials
- NPs:
-
Nanoparticles
- ReOxNPs:
-
Rhenium oxide nanoparticles
- RONS:
-
Reactive oxygen and nitrogen species
- SAED:
-
Selected area electron diffraction
- SEM:
-
Scanning electron microscopy
- TEM:
-
Transmission electron microscopy
- UV/Vis:
-
UV/Vis absorption spectrophotometry
- XPS:
-
X-Ray photoelectron spectroscopy
References
Xu W, Zhu X, Wang X, Deng L, Zhang G (2006) Residues of enrofloxacin, furazolidone and their metabolites in Nile tilapia (Oreochromis niloticus). Aquaculture 254(1–4):1–8
Kalita S, Devi B, Kandimalla R, Sharma KK, Sharma A, Kalita K et al (2015) Chloramphenicol encapsulated in poly-ε-caprolactone–pluronic composite: nanoparticles for treatment of MRSA-infected burn wounds. Int J Nanomed 10:2971
Magureanu M, Bilea F, Bradu C, Hong D (2021) A review on non-thermal plasma treatment of water contaminated with antibiotics. J Hazard Mater 417:125481
de Souza Santos LV, Meireles AM, Lange LC (2015) Degradation of antibiotics norfloxacin by Fenton, UV and UV/H2O2. J Environ Manag 154:8–12
Liu X, Zhou Y, Zhang J, Luo L, Yang Y, Huang H et al (2018) Insight into electro-fenton and photo-fenton for the degradation of antibiotics: mechanism study and research gaps. Chem Eng J 347:379–397
Naddeo V, Ricco D, Scannapieco D, Belgiorno V (2012) Degradation of antibiotics in wastewater during sonolysis, ozonation, and their simultaneous application: operating conditions effects and processes evaluation. Int J Photoenergy 2012:1
Dzimitrowicz A, Jamroz P, Cyganowski P, Bielawska-Pohl A, Klimczak A, Pohl P (2021) Application of cold atmospheric pressure plasmas for high-throughput production of safe-to-consume beetroot juice with improved nutritional quality. Food Chem 336:127635
Krolow M, Hartwig C, Link G, Raubach C, Pereira J, Picoloto R et al (2013) Synthesis and characterisation of carbon nanocomposites. NanoCarbon 2011. Springer, Berlin, pp 33–47
Buzea C, Pacheco II, Robbie K (2007) Nanomaterials and nanoparticles: sources and toxicity. Biointerphases 2(4):MR17–MR71
Elnashaie SS, Danafar F, Rafsanjani HH (2015) Nanotechnology for chemical engineers. Springer
Dzimitrowicz A, Cyganowski P, Pohl P, Jermakowicz-Bartkowiak D, Terefinko D, Jamroz P (2018) Atmospheric pressure plasma-mediated synthesis of platinum nanoparticles stabilized by poly(vinylpyrrolidone) with application in heat management systems for internal combustion chambers. Nanomaterials 8(8):619
Cheong S, Watt JD, Tilley RD (2010) Shape control of platinum and palladium nanoparticles for catalysis. Nanoscale 2(10):2045–2053
Cyganowski P, Dzimitrowicz A, Jamroz P, Jermakowicz-Bartkowiak D, Pohl P (2021) Rhenium nanostructures loaded into amino-functionalized Resin as a nanocomposite catalyst for hydrogenation of 4-nitrophenol and 4-nitroaniline. Polymers 13(21):3796
Cyganowski P, Terefinko D, Jamroz P, Pohl P, Dzimitrowicz A (2021) Non-thermal atmospheric pressure plasma as a powerful tool for the synthesis of rhenium-based nanostructures for the catalytic hydrogenation of 4-nitrophenol. RSC Adv 11(61):38596–38604
Zhu M, Shao HP, Zhai HL, Meng Y, Liu R, Ren C (2021) Rhenium nanoparticles for the delivery of HSP 90 inhibitors: a new drug delivery platform designed by molecular dynamics simulation. J Mol Liq 347:117995
Sakkas PM, Argirusi M, Sourkouni G, Argirusis C (2020) Rhenium oxide nanoparticles–Sonochemical synthesis and integration on anode powders for solid oxide fuel cells. Ultrason Sonochem 69:105250
Karthick K, Subhashini S, Teepikha M, Kumar R, Markandaraj SS, Kundu S (2020) Employing DNA scaffold with rhenium electrocatalyst for enhanced HER activities. Appl Surf Sci 528:147049
Kessler VG, Seisenbaeva GA (2012) Rhenium nanochemistry for catalyst preparation. Minerals 2(3):244–257
Kirilin AV, Tokarev AV, Manyar H, Hardacre C, Salmi T, Mikkola J-P et al (2014) Aqueous phase reforming of xylitol over Pt-Re bimetallic catalyst: effect of the re addition. Catal Today 223:97–107
Hurley KD, Zhang Y, Shapley JR (2009) Ligand-enhanced reduction of perchlorate in water with heterogeneous Re−Pd/C catalysts. J Am Chem Soc 131(40):14172–14173
Veerakumar P, Thanasekaran P, Lin K-C, Liu S-B (2017) Well-dispersed rhenium nanoparticles on three-dimensional carbon nanostructures: efficient catalysts for the reduction of aromatic nitro compounds. J Colloid Interface Sci 506:271–282
Zhang K, Suh JM, Choi J-W, Jang HW, Shokouhimehr M, Varma RS (2019) Recent advances in the nanocatalyst-assisted NaBH4 reduction of nitroaromatics in water. ACS Omega 4(1):483–495
Wei H, Liu X, Wang A, Zhang L, Qiao B, Yang X et al (2014) FeO x-supported platinum single-atom and pseudo-single-atom catalysts for chemoselective hydrogenation of functionalized nitroarenes. Nat Commun 5:5634
Corma A, Serna P (2006) Chemoselective hydrogenation of nitro compounds with supported gold catalysts. Science 313(5785):332–334
Zhao P, Feng X, Huang D, Yang G, Astruc D (2015) Basic concepts and recent advances in nitrophenol reduction by gold-and other transition metal nanoparticles. Coord Chem Rev 287:114–136
Zhao H, Wang Y, Wang R (2014) In situ formation of well-dispersed palladium nanoparticles immobilized in imidazolium-based organic ionic polymers. Chem Commun 50(74):10871–10874
Dzimitrowicz A, Motyka-Pomagruk A, Cyganowski P, Jamroz P, Terefinko D, Pohl P et al (2021) Cold atmospheric pressure plasmas as versatile tools for effective degradation of a mixture of hazardous and endocrine disturbing compounds from liquid wastes. J Environ Chem Eng 9:106718
Muting D, Kaiser E (1963) Quantitative estimation of α-amino N in biological material by the ninhydrin reaction. Hoppe-Seyler’s Z Physiol Chem 332:276–281
Chen Q, Li J, Li Y (2015) A review of plasma–liquid interactions for nanomaterial synthesis. J Phys D: Appl Phys 48:424005
Kundu S, Ma L, Dai W, Chen Y, Sinyukov AM, Liang H (2017) Polymer encapsulated self-assemblies of ultrasmall rhenium nanoparticles: catalysis and SERS applications. ACS Sustain Chem Eng 5(11):10186–10198
Dhenadhayalan N, Mythily R, Kumaran R (2014) Fluorescence spectral studies of gum Arabic: multi-emission of gum Arabic in aqueous solution. J Lumin 155:322–329
Chen Z, Ye H, Chen Y, Xu J (2021) Tuning the dispersity and sphericity of silver particles by wrapping mechanism using Arabic gum. Mater Lett 293:129665
Meyer R, Arnold W (1991) The electrode potential of the Tc (IV)-Tc (VII) couple. Radiochim Acta 55(1):19–22
Lukens WW, McKeown DA, Buechele AC, Muller IS, Shuh DK, Pegg IL (2007) Dissimilar behavior of technetium and rhenium in borosilicate waste glass as determined by X-ray absorption spectroscopy. Chem Mater 19(3):559–566
Vanysek P (2000) Electrochemical series. In: CRC Handbook of Chemistry and Physics, 84th (Ed). Published online: CRC Press
Yi J, Miller JT, Zemlyanov DY, Zhang R, Dietrich PJ, Ribeiro FH et al (2014) A reusable unsupported rhenium nanocrystalline catalyst for acceptorless dehydrogenation of alcohols through γ-C–H activation. Angew Chem 126(3):852–855
Jeong Y-K, Lee YM, Yun J, Mazur T, Kim M, Kim YJ et al (2017) Tunable photoluminescence across the visible spectrum and photocatalytic activity of mixed-valence rhenium oxide nanoparticles. J Am Chem Soc 139(42):15088–15093
Ratter K (2017) Epitaxial rhenium, a clean limit superconductor for superconducting qbits. Université Grenoble Alpes
Kaasalainen M, Aseyev V, von Haartman E, Karaman DŞ, Mäkilä E, Tenhu H et al (2017) Size, stability, and porosity of mesoporous nanoparticles characterized with light scattering. Nanoscale Res Lett 12(1):74. https://doi.org/10.1186/s11671-017-1853-y
NIST Standard Reference Database 20 tabase 20. http:/srdata.nist.gov/xps/, 2003. http:/srdata.nist.gov/xps/.
Okal J, Tylus W, Kȩpiński L (2004) XPS study of oxidation of rhenium metal on γ-Al2O3 support. J Catal 225(2):498–509
High Resolution XPS of organic polymers: the scienta ESCA300 database (Beamson, G.; Briggs, D.) (1993) Journal of Chemical Education 70(1):A25
Guo J, Zhang X (2004) Metal–ion interactions with sugars. The crystal structure and FTIR study of an SrCl2–fructose complex. Carbohydr Res 339(8):1421–1426
Long DA (2004) Infrared and Raman characteristic group frequencies. tables and charts Journal of Raman spectroscopy. John Wiley & Sons Ltd, Chichester
Ibekwe CA, Oyatogun GM, Esan TA, Oluwasegun KM (2017) Synthesis and characterization of chitosan/gum arabic nanoparticles for bone regeneration. Am J Mater Sci Eng 5(1):28–36
Polowczyk I, Cyganowski P, Urbano BF, Rivas BL, Bryjak M, Kabay N (2017) Amberlite IRA-400 and IRA-743 chelating resins for the sorption and recovery of molybdenum(VI) and vanadium(V): equilibrium and kinetic studies. Hydrometallurgy 169:496–507. https://doi.org/10.1016/j.hydromet.2017.02.017
Corbett MD, Chipko BR (1978) Synthesis and antibiotic properties of chloramphenicol reduction products. Antimicrob Agents Chemother 13(2):193–198
Jain R, Jain N, Jain DK, Patel VK, Rajak H, Jain SK (2017) Novel UV spectrophotometer methods for quantitative estimation of metronidazole and furazolidone using mixed hydrotropy solubilization. Arab J Chem 10(2):151–156
Fu W-Q, Zhu G-N, Shi J-B, Tong B, Cai Z-X, Dong Y-P (2019) Synthesis and properties of photodegradable poly (furan-amine) s by a catalyst-free multicomponent cyclopolymerization. Chin J Polym Sci 37(10):981–989
Das S, Palani Natarajan P, König B (2017) Teaching old compounds new tricks: DDQ-photocatalyzed C−H amination of arenes with carbamates, urea, and N-heterocycles. Chemistry 23(72):18161
Wu Z, Huang F, Chen Y, Xu H, Meti MD, Fan Y et al (2018) Conformation change of trypsin induced by acteoside as studied using multiple spectroscopic and molecular docking methods. Int J Food Prop 21(1):301–312
Mumtaz M, Rasool N, Ahmad G, Kosar N, Rashid U (2021) N-Arylation of protected and unprotected 5-bromo 2-aminobenzimidazole as organic material: non-linear optical (NLO) properties and structural feature determination through computational approach. Molecules 26(22):6920
Ferro EI, Perrin J, Dawson OGJ, Vuorinen T (2021) Tertiary amine-catalyzed generation of chlorine dioxide from hypochlorous acid and chlorite ions. Wood Sci Technol 55(1):67–81
website: http://tera.chem.ut.ee/~manna/spe/base.htm; UV-VIS spectra of neutral bases and their protonated conjugate cationic acids in acetonitrile. Accessed 02 Dec 2021
Shi J-H, Zhou Y-f (2011) Inclusion interaction of chloramphenicol and heptakis (2, 6-di-O-methyl)-β-cyclodextrin: phase solubility and spectroscopic methods. Spectrochim Acta Part A Mol Biomol Spectrosc 83(1):570–574
Benziman M, Eizen N (1971) Pyruvate-phosphate dikinase and the control of gluconeogenesis in acetobacter xylinum. J Biol Chem 246(1):57–61
Acknowledgements
The Authors would like to acknowledge M.Sc. Ewelina Cholewa for participating in the developing the procedure of rhenium nanostructures stabilization within the polymeric matrix as well as M.Sc. Dominik Terefinko for technical support.
Funding
A. D. and P.C. are thankful to the National Science Centre (NCN), Poland, for supporting this study according to the research projects nos. UMO-2019/35/D/ST8/04107 (A.D.) and UMO-2020/39/D/ST8/01352 (P.C.). Furthermore, A. D. and P.C. are also supported by Polish Ministry of Education and Science within the programme for outstanding young scientists.
Author information
Authors and Affiliations
Contributions
PC and AD have conceptualized this work. MAK, PJ, and AD performed the synthesis of raw-ReOxNPs using the patented plasma-based synthesis method of NPs. MAK, and AD incorporated the raw-ReOxNPs within the biopolymeric matrix. PC, MAK, and AD prepared the antibiotics solutions. MAK and AD analyzed the optical (UV/Vis absorption spectrophotometry) and selected granulometric properties (DLS) of obtained nanomaterials. PC conducted the ATR FT-IR measurements. AZ performed the TEM, SAED, SEM, and EDAX analyses. PC and MAK tested the catalytic activity of obtained biopolymer-stabilized ReOxNPs in addition to raw-ReOxNPs towards degradations of studied antibiotics. MC revealed the degradation of antibiotics after their treatment by proper Re nanostructures, analyzed, and described the obtained results. MMM and AB performed the XPS analysis and wrote the part of discussion related to this. P.C. and A.D. analyzed the obtained results and wrote the first draft of this manuscript. PP and PJ took a part in the discussion and corrected the first draft of manuscript. Additionally, PP provided founding for publication of this article in open access mode. All Authors have revised the manuscript and agreed to its final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cyganowski, P., Caban, M., Khan, M.A. et al. The Use of Cold Atmospheric Pressure Plasma for the Synthesis of Saccharide-Stabilized Re Nanostructures Enabling Effective Deactivation of Nitro-Based Antimicrobial Agents. Plasma Chem Plasma Process 43, 199–224 (2023). https://doi.org/10.1007/s11090-022-10285-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11090-022-10285-9