
Abstract
In this work, we present the theoretical quantum chemical calculation of UV–Vis spectroscopy and electronic effect delocalization of π-conjugated molecular system variation based of benzodifuran (BDF) derivatives with 6-substituted by furan, thiophene or pentafluorophenyl cycles by azomethine bonds. For the geometrical optimization, dipole moments and frontier molecular orbital HOMO and LUMO energies we use the Becke’s functional B3LYP with a polarized and diffused 6-31+G(d,p) basis set. For the first (β) and second (γ) hyperpolarizabilities calculation we use the time-dependent Hartree–Fock (TDHF) method with 6-31+G(d,p) basis set. In particular, we discuss correlation between the chemical structures of benzodifuran based derivatives and their NLO properties.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Conjugated and functionalized organic materials have attract scientific consideration in recent years thanks to their potential applications as organic semiconducting materials in electronic and optoelectronics devices such as field effect transistors, photovoltaic cells, electroluminescent diodes (Sahraoui et al. 2009; Pron et al. 2010; Klauk 2010; Cheng et al. 2009; Mishra and Bauerle 2012). The π-conjugation trough the tetrathiafulvalene (TTF) core with highly delocalized pi-electron and their theoretical calculations establish an important contribution and ply crucial role for NLO applications (Terkia-Derdra et al. 2000; Sahraoui et al. 1995). The investigation of the benzodifuran derivatives building block in the π-conjugated side chains has been continued with the study of benzodifuran in the polymer backbone. It is a promising strategy to adjust the HOMO/LUMO levels and control the nonlinear optical properties by putting furan, thiophene or pentafluorophenyl cycles by azomethine bonds in different positions. Zhang et al. (2014) and coworkers has been prepared benzodifuran-dione as an newly electron accepting material in D-A copolymers. Moussalem et al. (2014) published a molecules based on benzodifuran substituted by bithiophene electron donating group and an electron deficient pentafluorobenzene group attached on the same furan fused ring with a deep HOMO level as an attractive for organic photovoltaics (OPV) application. Kulyk et al. (2016) propose benzodifuran-based derivatives incorporated into PMMA for nonlinear study where the NLO response of polymer guest–host systems come from the hyperpolarizability of chromophores incorporated into them.
Chromophores with delocalized π-electron systems capable of exhibiting a charge transfer usually show large molecular hyperpolarizability. When chromophores in a polymer are aligned in a noncentrosymmetric system after poling, this increases the NLO properties of the polymer system (Lee 2003; Yesodha et al. 2004; El et al. 2011). This study is devoted to the understanding of the relationship between chemical structure of benzodifuran-based derivatives and their nonlinear optical properties in molecular scale for application in photonics. To expose the structural and electronic features as well as nonlinear optical properties the theoretical calculations have been performed using B3LYP density functional theory and the time-dependent Hartree–Fock method. The second important purpose of this work was to compare the obtained theoretical results (evaluated for gas-phase) with experimental measurements and finding the explanation of origin of NLO phenomena in the investigate systems. The theoretical calculations of electronic structure and second and third-order NLO properties of S1–S6 (Fig. 1) provide useful information for the design of novel efficient systems. In this work the effect of charge transfer caused by attaching the pentafluorobenzene group in different way to the benzodifuan core on the NLO properties will be discussed.

Optimized chemical structure of S1–S6 (DFT/B3LYP/6-31+G(d,p))
2 Theoretical simulations
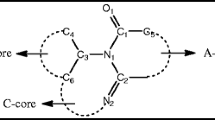
In this work, GAUSSIAN 09 program package (Frisch et al. 2009) was used to perform the energy levels HOMO and LUMO calculations for S1–S6 by density functional theory DFT/B3LYP, which is called the hybrid method based on three Becke parameters (Becke 1993) using the base assembly 6-31+G(d,p) level of theory. A lot of research work indicates the effectiveness of the DFT method in geometry optimization and electric property calculations. Full optimization in the gas phase the molecular geometry of all the six molecules have been made starting from the structural parameters obtained from the XRD analysis deposited with the Cambridge Crystallographic Data under reference CCDC: 858095 (Moussallem et al. 2012). Then the structure was expanded with subsequent elements and optimized using the method given above. The optimized structures of these three compounds are shown in Fig. 1. Our calculation results are presented in Table 1.
The frequency-dependent first and second hyperpolarizabilities were calculated using ab initio method implemented at the self-consistent field level of theory known as TDHF/6-31+G(d,p) methodology. Both the addition of d polarization functions on the carbon and nitrogen atoms and the addition of p functions on hydrogen atoms and diffuse functions are critical in order to have a precise estimation of the hyperpolarizabilities. The frequency-dependent β(− 2ω,ω,ω) and γ(− 3ω,ω,ω,ω) computations at considered ω = 0.04282 a.u. (λ = 1064 nm wavelength) were calculated using GAMESS (Gordon and Schmidt 2005) program within the previously mentioned computational method for isolated molecules placed in vacuum environment. The first and second order hyperpolarizabilities calculated for S1–S6 are presented in Tables 2 and 3.
3 Results and discussion
The knowledge of the linear optical properties is very important for the characterization of materials in many optical and nonlinear optical application. The UV–visible absorption spectra of S1–S6 molecules were calculated using DFT/B3LYP functional and 6-31+G(d,p) basis set. Positions of the experimental and theoretical first absorption peaks (λmax) are collected in Table 1. The spectra calculated for isolated molecules are presented in Fig. 2. The theoretical spectrum was made in the gas phase. As we can see experimental peak position (λmax) are shifted toward longer wavelengths in respect to theoretical spectral peak position about 5–20 nm. This is connected to the solvatochromic solvent effect. The investigated molecular structure exhibit electronic transitions in the UV and UV–Vis regions. The UV–Vis absorption bands are attributed to the charge transfer from the electron-donor and acceptor groups. The computed position of absorption peaks are in good agreement with the experimental results noted for S1–S6 and therefore the DFT/B3LYP functional is convenient for molecules with separated electron-donor and acceptor groups.

Theoretical UV–Vis absorption spectra of S1–S6 molecules calculated by DFT//B3LYP/6-31+G(d,p) methodology in vacuum
Interpretations of orbital energies give useful predictions. In general the HOMO and LUMO energy level depend on electron donating strength of donor and electron—withdrawing strength of acceptor, accordingly (Leclerc et al. 2006). The HOMO and LUMO are most likely place where reaction will occur. The reaction is likely to occur there because the electrons in the HOMO have the highest energy and therefore the electrons are most willing to react. LUMO is probably the site of binding because any invasive electrons for other molecules will fill into the LUMO. Comparing the energies of these orbitals create an idea of how reactive a molecule is what is important parametric properties of the molecules. The HOMO, LUMO energies and HOMO–LUMO energy gaps for S1–S6 has been given in Table 1.
At the DFT/B3LYP level the HOMO energy of S1 (− 7.40619 eV) is more negative than the HOMO for the remaining molecules see Table 1 and the LUMO energy of S1 is − 4.19277 eV. Therefore The HOMO–LUMO gap S1 is relatively higher than energy gap value for the remaining molecules. High energy gap and the data here suggested that S1 is relatively less reactive and more stable than molecule S2–S6. The smaller value of the energy gap explains eventually charge transfer interaction takes place inside molecules. Hard particles have large energy gaps and the soft particles have small HOMO–LUMO energy gaps.
Figure 3 shows the visualized structures of studied complexes and show the population of electrons on their orbitals. The HOMO–LUMO map of these molecule indicate the presence of inter molecular charge transfer in molecules. The HOMO for S1 is mainly located on the benzodifuran and carboxyl polar functional grup. LUMO is localized on the benzodifuran acceptor group. The introduction of one pentafluorophenyl attached to the benzodifuran core in S2 making the benzodifuran stem as an electron donating material as the charge is shifted in the LUMO on the pentafluorophenyl attached by azomethine bond.

Frontiers molecular orbitals HOMO and LUMO of S1–S6 compounds
For the remaining four investigated molecules (S3–S6) the HOMO is also located on the benzodifuran and pentafluorophenyl and other ligads. LUMO for S3–S6 is located mainly on benzodifuran and two ligands attached by azometine bond.
The introduction of two pentafluorophenyl attached to the benzodifuran core in S3–S6 changes the character of the charge transfer in the molecule, the center of the molecule—benzodifuran becomes an accepting group which can be clearly seen in the Fig. 3. This electron delocalization (see Fig. 3) can be attributed to the strong electron withdrawing nature of these groups having a major impact on the NLO effect and is defined as intramolecular charge transfer (Abbas 2012).
Non linear optics deals with the interaction of applied electromagnetic fields in various materials to generate a new electromagnetic field, altered in frequency, phase, or other physical properties. When a molecule is subjected to an external electric field Eloc, it causes the charges of the atoms to oscillate. This effect can distort the molecular electron density distribution, an effect that can be described in terms of change in dipole moment μ, which can be expressed by a power series in the field strength as in Eq. (1):
The tensors α, β and γ defined by the above equation are linear polarizability, the second-order or quadratic hyperpolarizability (the first hyperpolarizability) and the third order or cubic polarizability (the second hyperpolarizability) respectively. However, when a molecule is subjected to an intense electric field the second and third terms in Eq. (1) become important, such that nonlinear optical behaviour can be observed. For a medium to exhibit second-order nonlinear susceptibility it should not possess a center of symmetry, i.e. it should be noncentrosymmetric. All media and molecules exhibit third-order response. Basically all forms of matter exhibit nonlinear optical phenomena. But to be useful in a device, the material must exhibit a high degree of nonlinearity at a reasonable power level. Second-order NLO studies focussed on unsymmetrical derivatives; the basic requirements for large second-order hyperpolarizability values being an asymmetric charge distribution, with high donor and acceptor substituents at positions para to each other and an optimized π-electron conjugation pathway. To find a correlation between structure property and NLO phenomena, we have extended our study for the computing the first and second order hyperpolarizabilities.
The βtot value has been calculated using the following expression (Thanthiriwatte and Nalin de Silva 2002).
The obtained results are collected in Table 2. As described above the β values should not be observed in centrosymmetric materials. Only in S2 as an uncentrosymmetric molecule the value of the odd hyperpolarizability tensor should be non-zero. However, these molecules have been synthesized for the study of third-order non-linear optical properties, despite the centrosymmetric structure, they also exhibit second-order non-linear properties evaluated experimentally (see Table 4).
So we decided to calculate the first order hyperpolarizabilities for these molecules that also turned out to be non-zero values of β (Table 2). Generally, an increase in the β value occurs together with bathochromic effect (see Tables 1 and 2) due to a larger π-conjugated length and/or stronger donor and acceptor ability what is clearly seen for compounds S1, S3–S6.
Received theoretical results suggest that better second order nonlinear optical properties possess the noncentrosymmetric compound S2 (βtot = 424.804 × 10−30 esu) with added strong electron-acceptor pentafluophenyl group. The calculated first hyperpolarizability of this molecule was compared with first order hyperpolarizabilities of organometallic compounds which are known with very high βtotal values within limits 10–1300 × 10−30 esu (Migalska-Zalas 2016; Hurst et al. 2001) and with first-order hyperpolarizability (βtotal) of Urea Sulphamic acid which is a reference material in SHG amounting to 1.023 × 10−30 esu (Senthilvelan et al. 2017). The calculated hyperpolarizability of S2 is many times greater than that of urea Sulphamic acid and also urea (0.1947 × 10−30 esu) and comparable with the beta value obtained for organometallic materials. The calculated first hyperpolarizabilities for the remaining molecules are very small but not equal zero.
The molecule S5 containing the tiophene ring occurring in the radical substituted by azomethine bond is characterized by a fairly large beta value (βtot = 14.8805 × 10−30 esu) which is bigger than value obtained for urea. This study reveals that the investigated compounds have β values what is connected with the noncetrosymmetry in the charge density distribution in the ground and excited states presented in Fig. 2.
The tortuosity of the phentafluorophenyl ring in the molecules S3–S6 also brought about inferior planarity in whole molecule, then these molecules can organize through intermolecular interactions along different dimensions and resulted in spherical nanostructures which may affect the increase of NLO property of macroscopic material as seen in Table 4.
The average third-order hyperpolarizability γtot values have been computed using the Eq. (3) (Bogaard and Orr 1975):
The theoretical second order hyperpolarizability tensor components are presented in Table 3. The calculated hyperpolarizabilities of S3–S6 (γtot = 1054.114 − 1559.255 × 10−36 esu) are about three times larger than for the molecule with the formula S2 (γtot = 403.807 × 10−36 esu) and two orders of magnitude bigger than for compound S1. Leupacher and Penzkofer (1985) reported the second hyperpolarizabilities of methylene blue (γ = 32.00 × 10−36 esu) and the value of γtot obtained for S3–S6 are two order higher (see Table 3.). Received theoretical results are in good agreement with the experimental data which show that better third order nonlinear optical properties have been obtained for compounds with long π-conjugated chains. The only deviation was obtained for the S3 molecule for which the third order hyperpolarizability is higher than for the non-centrosymmetric S2 molecule. The carried out studies also indicate that the third-order nonlinear response increases thanks to the multiple conjugate bonds present in S4 and S5 molecule. The further adding of n-butyl groups causes saturation of second order hyperpolarizability values in S6 molecule. In these materials as in other compounds recently published the big optical nonlinearity is caused by strong intramolecular charge transfer between donors and acceptors though a conjugated transmitter.
The molecules with long π-conjugated chains are characterized with comparatively higher values of χ(3) due to delocalized π-electrons, which traveling freely along the conjugated structure of molecules and are the key factor to high nonlinearities in organic materials.
4 Conclusions
Theoretical studies on these molecules showed that the strong second order hyperpolarizability characterizing the nonlinear optical efficiency of the molecule depends on the length of the π-conjugated way and the force on the donor and acceptor substituents. A source of second order non-linear-optical properties of these materials is connected with the noncetrosymmetry in the charge density distribution in the ground and excited states. The tortuosity of the phentafluorophenyl ring can affect the intermolecular interactions along different dimensions resulted in spherical nanostructures and increase the second order NLO property of the material. Within this framework it was observed that the nonlinear response of the third-order increases thanks to the multiple conjugate bonds present in S4 and S5 molecule further addition of n-butyl groups causes saturation in S6 molecule. Good compatibility has been achieved between the theoretical and experimental results. Studied benzodifuran derivatives showed the contrast in NLO response for various substituents making them interesting for using in photonics devices.
References
Abbas, H.: First principle calculation of the photophysical properties of silylated Coumarins 120 and 151. Comput. Theor. Chem. 992, 55–58 (2012)
Becke, A.D.: Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993)
Bogaard, M.P., Orr, B.J.: Electric dipole polarizabilities of atoms and molecules. In: Buckingham, A.D. (ed.) MTP International Review of Science: Physical Chemistry, Series 2, vol. 2, pp. 149–194. Butterworths, London (1975)
Cheng, Y.J., Yang, S.H., Hsu, C.-S.: Synthesis of conjugated polymers for organic solar cell applications. Chem. Rev. 109, 5868–5923 (2009)
El Ouazzani, H., Iliopoulos, K., Pranaitis, M., Krupka, O., Smokal, V., Kolendo, A., Sahraoui, B.: Second- and third- order nonlinearities of novel push − pull azobenzene polymers. J. Phys. Chem. B 115, 1944–1949 (2011)
Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A., Nakatsuji H., Caricato M., Li X., Hratchian H.P., Izmaylov A.F., Bloino J., Zheng G., Sonnenberg J.L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J.A. Jr, Peralta J.E., Ogliaro F., Bearpark M., Heyd J.J., Brothers E., Kudin K.N., Staroverov V.N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J.C., Iyengar S.S., Tomasi J., Cossi M., Rega N., Millam J.M., Klene M., Knox J.E., Cross J.B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R.E., Yazyev O., Austin A.J., Cammi R., Pomelli C., Ochterski J.W., Martin R.L., Morokuma K., Zakrzewski V.G., Voth G.A., Salvador P., Dannenberg J.J., Dapprich S., Daniels A.D., Farkas Ö., Foresman J.B., Ortiz J.V., Cioslowski J., Fox D.J.: Gaussian 09, Revision D.01. Gaussian, Inc., Wallingford CT (2009)
Gordon, M.S., Schmidt, M.W.: Advances in electronic structure theory: GAMESS a decade later. In: Dykstra, C.E., Frenking, G., Kim, K.S., Scuseria, G.E. (eds.) Theory and Applications of Computational Chemistry: The First Forty Years, pp. 1167–1189. Elsevier, Amsterdam (2005)
Hurst, S.K., Cifuentes, M.P., Morrall, J.P.L., Lucas, N.T., Whittall, I.R., Humphrey, M.G., Asselberghs, I., Persoons, A., Samoc, M., Luther-Davies, B., Willis, A.C.: Organometallic complexes for nonlinear optics. 22.1Quadratic and cubic hyperpolarizabilities of trans-bis(bidentate phosphine)ruthenium σ-arylvinylidene and σ-arylalkynyl complexes. Organometallics 20, 4664–4675 (2001)
Klauk, H.: Organic thin-film transistors. Chem. Soc. Rev. 39, 2643–2666 (2010)
Kulyk, B., Kerasidou, A.P., Soumahoro, L., Moussallem, C., Gohier, F., Frére, P., Sahraoui, B.: Optimization and diagnostic of nonlinear optical features of π-conjugated benzodifuran-based derivatives. RSC Adv. 6, 14439–14447 (2016)
Leclerc, N., Michaud, A., Sirois, K., Morin, J.F., Leclerc, M.: Synthesis of 2,7-carbazolenevinylene-based copolymers and characterization of their photovoltaic properties. Adv. Funct. Mater. 16(13), 1694–1704 (2006)
Lee, K.S.: Nonlinear optical, photorefractive and two-photon absorption polymers polymers for photonics applications: II. Springer, Berlin (2003)
Leupacher, W., Penzkofer, A.: Third-order nonlinear susceptibilities of dye solutions determined by third-harmonic generation. Appl. Phys. B Lasers Opt. 36, 25–31 (1985)
Migalska-Zalas, A.: Theoretical study of the effect of π-conjugated transmitter of D–π: a ruthenium systems on the quadratic NLO properties. Opt. Quant. Electron. 48, 183–191 (2016)
Mishra, A., Bauerle, P.: Small molecule organic semiconductors on the move: promises for future solar energy technology. Angew. Chem. Int. Ed. 51, 2020–2067 (2012)
Moussalem, C., Segut, O., Gohier, F., Allain, M., Frére, P.: Facile access via green procedures to a material with the benzodifuran moiety for organic photovoltaics. ACS Sustain. Chem. Eng. 2, 1043–1048 (2014)
Moussallem, C., Gohier, F., Mallet, Ch., Allain, M., Frere, P.: Extended benzodifuran–furan derivatives as example of π-conjugated materials obtained from sustainable approach. Tetrahedron 68, 8617–8621 (2012)
Pron, A., Gawrys, P., Zagorska, M., Djurado, D., Demadrille, R.: Electroactive materials for organic electronics: preparation strategies, structural aspects and characterization techniques. Chem. Soc. Rev. 39, 2577–2632 (2010)
Sahraoui, B., Sylla, M., Bourdin, J.P., Rivoire, G., Zaremba, J., Nguyen, T.T., Sallé, M.: Third-order nonlinear optical properties of ethylenic tetrathiafulvalene derivatives. J. Mod. Opt. 42(10), 2095–2107 (1995)
Sahraoui, B., Luc, J., Meghea, A., Czaplicki, R., Fillaut, J.L., Migalska-Zalas, A.: Nonlinear optics and surface relief gratings in alkynyl–ruthenium complexes. J. Opt. A: Pure Appl. Opt. 11, 024005–024031 (2009)
Senthilvelan, N., Rajarajan, G., Jegatheesan, A., Sivakumar, S.: Growth and spectroscopic characterization of urea sulfamic acid crystal: a second-order nonlinear material. Rasayan J. Chem. 10(1), 218–225 (2017)
Terkia-Derdra, N., Andreu, R., Sallé, M., Levillai, E., Orduna, J., Garín, J., Ortí, E., Viruela, R., Pou-Amérigo, R., Sahraoui, B., Gorgues, A., Favard, J.-F., Riou, A.: π conjugation across the tetrathiafulvalene core: synthesis of extended tetrathiafulvalene derivatives and theoretical analysis of their unusual electrochemical properties. Chem. Eur. J. 6(7), 1199–1213 (2000)
Thanthiriwatte, K.S., Nalin de Silva, K.M.: Non-linear optical properties of novel fluorenyl derivatives-ab initio quantum chemical calculations. J. Mol. Struct. Theochem. 617, 169–175 (2002)
Yesodha, S.K., Pillai, C.K.S., Tsutsumi, N.: Stable polymeric materials for nonlinear optics: a review based on azobenzene systems. Prog. Polym. Sci. 29, 45–74 (2004)
Zhang, G., Li, P., Tang, L., Ma, J., Wang, X., Lu, H., Kang, B., Cho, K., Qiu, L.: A bis(2-oxoindolin-3-ylidene)-benzodifuran-dione containing copolymer for high-mobility ambipolar transistors. Chem. Commun. 50, 3180–3183 (2014)
Acknowledgements
Calculations have been carried out in Wroclaw Centre for Networking and Supercomputing (http://www.wcss.pl), Grant No. 282. The authors would like to acknowledge Prof. Bouchta SAHRAOUI for helpful discussion (MOLTECH-Anjou UMR CNRS 6200, Angers Cedex, France).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Migalska-Zalas, A., EL Korchi, K. & Chtouki, T. Enhanced nonlinear optical properties due to electronic delocalization in conjugated benzodifuran derivatives. Opt Quant Electron 50, 389 (2018). https://doi.org/10.1007/s11082-018-1659-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11082-018-1659-x