
Abstract
Manganese (Mn) is an essential trace element required for ubiquitous enzymatic reactions. Chronic overexposure to this metal may promote potent neurotoxic effects. The mechanism of Mn toxicity is not well established, but several studies indicate that oxidative stress play major roles in the Mn-induced neurodegenerative processes. Silymarin (SIL) has antioxidant properties and stabilizes intracellular antioxidant defense systems. The aim of this study was to evaluate the toxic effects of MnCl2 on the mouse neuroblastoma cell lines (Neuro-2A), to characterize the toxic mechanism associated with Mn exposure and to investigate whether SIL could efficiently protect against neurotoxicity induced by Mn. A significant increase in LDH release activity was observed in Neuro-2A cells associated with a significant decrease in cellular viability upon 24 h exposure to MnCl2 at concentrations of 200 and 800 μM (P < 0.05) when compared with control unexposed cells. In addition, exposure cells to MnCl2 (200 and 800 μM), increases oxidant biomarkers and alters enzymatic and non enzymatic antioxidant systems. SIL treatment significantly reduced the levels of LDH, nitric oxide, reactive oxygen species and the oxidants/antioxidants balance in Neuro-2A cells as compared to Mn-exposed cells. These results suggested that silymarin is a powerful antioxidant through a mechanism related to its antioxidant activity, able to interfere with radical-mediated cell death. SIL may be useful in diseases known to be aggravated by reactive oxygen species and in the development of novel treatments for neurodegenerative disorders such as Alzheimer or Parkinson diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chronic environmental or occupational exposure to manganese (Mn) in mammals is identified as a dystonic neurodegenerative movement disorders that results from excessive accumulation of Mn in multiple brain regions and presents with neurologic symptoms resembling feature of Parkinson diseases [1]. Mn exposure can cause motor deficits in mammals that are characterized by extrapyramidal motor abnormalities, such as rigidity, hypokinesia, tremor, and behavioural disturbances [2]. Although the pathological changes and symptoms of Mn exposure are well known, the primary mechanisms of neurotoxicity have yet to be established. Several mechanisms of Mn neurotoxicity have been proposed, including disruption of mitochondrial metabolism [3], disturbance of neurotransmitter metabolism [4], alteration of iron homeostasis [5] and induction of oxidative stress via direct or indirect formation of free radicals in both neurons and astrocytes [6–8]. Upon entering cells via facilitated diffusion and receptor-gated cation channels [9], Mn avidly accumulates in mitochondria [10] and inhibits the complexes of the electron transport chain [11], which impairs oxidative phosphorylation and ATP production [12]. The resulting decreased energy production and alters mitochondrial permeability transition, leading to excessive Ca2+ influx and massive reactive oxygen species (ROS) production [13]. Moreover, superoxide, produced in the mitochondrial electron transport chain (ETC.) [14], may catalyze the transition shift of Mn2+–Mn3+ through a set of reactions similar to those mediated by superoxide dismutase and thus lead to the increased oxidant capacity of this metal [15].
Knowing that oxidative stress plays a central role in Mn-induced neurotoxicity, several authors have investigated the potential neuroprotective effect of antioxidants against Mn-induced toxicity. In mitochondrial preparations, N-acetylcysteine (NAC), glutathione, and vitamin C prevent ROS production caused by high concentrations of Mn [16]. In cultured astrocytes, Chen and Liao [17] observe that NAC attenuates the pro-oxidant effects of Mn. Ex vivo, Hazell et al. [18] report that NAC blocks the astrocytosis caused by acute exposure to Manganese.
Though several synthetic antioxidants are available, a growing trend has been targeted toward the use of natural products (polyphenols, flavonoids, vitamins, carotenes and lycopenes) as antioxidants in view of toxicity condemn [19]. Bioflavonoids are the ubiquitous group of polyphenolic substances that are present in most plants [20]. Silymarin is a flavonoid isolated from the fruits and seeds of the milk thistle (S. marianum). It is classified as a benzopyranone [21] consisting of a mixture of polyphenolic flavonoid like silibin, isosilybin, silydianin and silychristin. It has been reported to possess multiple pharmacological activities and considered as an anti-hepatotoxic agent for the treatment of various diseases [22]. In addition several recent studies have shown the potential hepatopreventive and therapeutic efficacy of silymarin in different animal models and cell culture systems [23, 24]. Its positive effects have been ascribed to the putative anti-oxidant, anti-inflammatory and anti-proliferative properties based on the modulation of specific signalling pathway, transcription factor and gene expression [25]. But its potential protection for central nervous system (CNS) and the mechanisms of action are still unclear.
In the current study, we established the mechanism of SIL-mediated attenuation of reactive oxygen species (ROS) generation and the possible involvement of its neuroprotection against Mn toxicity. The preventive effect of silymarin against oxidative stress promoted by Mn was studied by assessing biochemical parameters in Neuro-2A cell, in order to evaluate enzymatic antioxidant defenses, hydrogen peroxide content, glutathione redox, lipid peroxidation and protein carbonyl content. We further addressed whether the neuroprotective effects of SIL against Mn toxicity were mediated via their antioxidant properties.
Materials and Methods
Materials
Lactate deshydrogenase (LDH) kit was purchased from Biomaghreb ref. 20012 (Ariana, Tunis, Tunisia). Minimum Essential Medium (MEM) was purchased from Invitrogen (Glasgow, UK), foetal bovine serum (FBS) was provided by Gibco (Invitrigen, USA). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), 5,50-dithi-obis-(2-nitrobenzoic acid) (DTNB), Silymarin and all other chemical products used in this study were obtained from Sigma Chemical Co. (St. Louis, France). Culture plates and flasks were purchased from IWAKI (Japan).
Cell Culture
Neuro-2A mouse neuroblastoma cell line, obtained from the ATCC (CCL-131), was routinely grown in 75 cm2 flasks and maintained in MEM supplemented with 10% FBS, 3,500 units/ml polymyxin B sulfate, 1,400 units/ml neomycine sulfate and 7.5 mg/ml streptomycin base. Cultures were maintained in a humidified atmosphere with 5% CO2 at 37°C. Cell dissociation was achieved with 0.05% trypsin-0.02% verséne (triplex III). Briefly, cells were seeded on 96, 24 and 6-well culture plates in medium at an approximate density of 1 × 105 cells/ml. After 24 h stabilization, neuronal cells were co-cultured with medium containing various concentrations of MnCl2 (200 and 800 μM) and/or silymarin (10, 50, and 100 μM) for 24 h. The concentration of MnCl2 was selected based on previously reported cytotoxic levels in cultured cells [26]. For stock solution, MnCl2 was dissolved in MilliQ Plus sterilized water at the concentration of 800 mM. Silymarin was dissolved in dimethylsulfoxide (DMSO) to obtain 100 mM. The experimental concentrations were freshly prepared in the basal medium with a final DMSO concentration 0.1%.
Cell Viability Assay and Morphological Assessment in Neuro-2A Cells
Cell viability was determined colorimetrically using the MTT reduction assay. Neuro-2A cells were seeded in 96-well plate (1 × 105 cells/well) for 24 h and were exposed following the above cell treatment protocol. Briefly, at the end of treatment, 10 μl of MTT [5 mg/ml in phosphate buffered saline (PBS)] solution were added to each well and the plates were incubated at 37°C for additional 4 h. Then, the medium containing MTT was removed and 200 μl of DMSO was added to each well to achieve solubilization of the formazan crystal formed in viable cells. The absorbance was read at 570 nm with DMSO used as the blank. Cell viability was expressed as a percentage of the control culture. Changes on the cell morphology were observed with an Olympus model (CK2, Japan) inverted phase-contrast microscope fitted with a digital camera to capture images.
LDH Assay
Membrane integrity was evaluated by measuring the LDH activity in the culture medium of control and treated cells. After 24 h treatment, the culture medium was removed and the LDH activity was measured following the manufacturer’s protocol (Biomagreb, ref. 20012 Ariana, Tunis, Tunisia). The percent of LDH activity released was determined as percent of enzyme activity in the incubation medium to the total LDH activity. The absence of the viable cells corresponds to 100% of LDH activity in the incubation medium.
Measurement of Intracellular ROS
The production of intracellular ROS was assessed by 2′,7′- dichlorofluorescin diacetate (DCFDA) oxidation. Twenty-four hours after treatment with SIL and/or Mn, cells were incubated with 20 μM DCFH-DA at 37°C for 1 h and washed twice with PBS. The fluorescence intensity of DCF was measured in a microplate-reader at an excitation wavelength of 485 nm and an emission wavelength of 530 nm. Results were expressed as percentage of controls.
Measurement of Cellular H2O2
The H2O2 concentration was measured in Neuro-2A by a modification of the ferrous oxidation-xylenol orange (FOX1 reagent) assay adapted to microtiter plates [27, 28]. 50 μl of Neuro-2A cells (1.0 × 106 cells/ml) were added to 950 μl of FOX 1 reagent and incubated for 30 min at room temperature. This assay is based on the ability of H2O2 to oxidize the ferrous Fe2+-ions to the ferric Fe3+-ions which react with xylenol orange to a coloured complex. The absorbance of the samples was read at 570 nm and the concentration of H2O2 was determined using standard peroxide solutions in the same microtiter plate.
Determination of Stress Markers and Antioxidant Enzymes
Preparation of the Cellular Extracts
After the different treatments, Neuro-2A cells were trypsinized and harvested. The pellets obtained after centrifugation (2 min, 1.500 rpm, 4°C) were resuspended and washed twice in isotonic Tris-HCl buffer (20 mM, pH 7.4). Cells suspension was lysed by 5 freeze-thaw cycles (liquid nitrogen −37°C). After centrifugation of the lysates at 8.000×g for 10 min at 4°C they were stored at −80°C, until analysis.
Assay of Antioxidant Enzymes
Catalase (CAT) activity was assayed by the decomposition of hydrogen peroxide according to the method of Aebi [29]. A decrease in absorbance due to H2O2 degradation was monitored at 240 nm for 1 min and the enzyme activity was expressed as μmol H2O2 consumed/min/mg protein.
Superoxide dismutase SOD (MnSOD and Cu/ZnSOD) activities were evaluated by measuring the inhibition of pyrogallol activity as described by Marklund and Marklund [30]. Based on the competition between pyrogallol oxidation by superoxide radicals and superoxide dismutation by SOD. The specific Cu/Zn-SOD inhibition by potassium cyanide allowed the Mn-SOD determination in the same conditions. Assays were monitored by spectrophotometry at 420 nm. One unit (U) corresponded to the enzyme activity required to inhibit the half of pyrogallol oxidation. SOD activity was expressed as U/mg protein.
Glutathione peroxidase (GPx) activity was measured according to Flohe and Gunzler [31]. The enzyme activity was expressed as nmoles of GSH oxidized/min/mg protein.
Assays of Oxidant Markers
Thiol, Disulfide (SH/SS Redox State) and GSH Assays
Thiols (SH) and disulfides (SS) were determined according to Zahler and Cleland [32]. The method is based on the reaction of DTNB with the samples producing a yellow product, thionitrobenzoic acid (TNB). The disulfide content was calculated through the difference between the two determinations. The SH/S-S ratio was also calculated. Results were expressed as nmol TNB/mg protein.
Total Cellular Glutathione (GSH) levels were determined as described previously by Fairbanks and Klee [33]. Distilled water was used as blank and standards were prepared from GSH. The slope of the reaction rate was used to calculate the GSH content.
Lipid Peroxidation End Products and Protein Carbonyl Assays
The extend of lipid peroxidation in Neuro-2A cell homogenates, was determined by measuring the release of thiobarbituric acid reactive substance (TBARS) in terms of malondialdehyde (MDA) formation according to Draper and Hadley method [34]. Results were expressed as nmol of MDA/mg of cellular protein.
The protein carbonyl PCO content of Neuro-2A homogenates was determined according to the method of Evans et al. [35]. Results were expressed as nmol DNPH conjugated/mg protein.
Assay of Na+-K+-ATPase and Ca2+-ATPase Activities
Na+-K+-ATPase and Ca2+-ATPase were measured as previously described [16, 36]. The inorganic phosphate–molybdate complex was quantitated by spectrophotometer at 690 nm. The Na+-K+-ATPase activity was calculated as the difference between the total ATPase activity and the ATPase activity inhibited by ouabain, the specific-inhibitor of Na+-K+-ATPase. In the presence of Ca2+, Ca2+- ATPase activity appeared and was measured in the presence of Ca2+, Na+, K+, Mg2+ ions and ouabain. The ATPase activity was expressed as μmol Pi/hour/mg of protein.
Measurement of Nitrite, Tumor Necrosis Factor α (TNFα) and Interleukin 6 (IL-6)
Neuro-2A cells were seeded on 24-well plates. After being treated with SIL and/or Mn, the supernatants were collected and stored at −80°C until assays. As an indicator of nitric oxide production, the amount of nitrite accumulated in culture supernatant was determined with a colorimetric assay using Griess reagent as previously described [37]. The concentration of nitrite in samples was determined using a sodium nitrite standard curve.
TNFα and IL-6 levels were detected by mouse TNFα and IL-6 enzyme-linked immunosorbent assay sandwich method (ELISA) kits according to the procedures provided by the manufacturers. Microtiter (96-well) plates were coated with anti-mouse IL-6 and TNF-α (BD Pharmingen) in coating buffer at 4°C overnight. After the plates were blocked and washed, 100 μl supernatant was added, and plates were incubated for 1 h at room temperature. The plates were then washed and treated with biotinylated antibodies against IL-6 and TNF-α, then Extravidin—peroxidase conjugate (Sigma). A standard curve generated by using purified recombinant IL-6, and TNF-α was used to determine cytokine concentration.
Protein Assay
The protein concentration was determined using the BCA™ Bicinchoninic acid protein assay kit (Pierce Biotechnology, Rockford, IL, USA) with bovine serum albumin as a standard.
Statistical Analysis
Results were expressed as mean ± standard deviation (mean ± SD). All analysis was carried out with GraphPad Prism 4.02 for Windows (GraphPad Software, San Diego, CA). Significant differences between treatment effects were determined by 1-way ANOVA, followed by Tukey’s post-hoc test for multiple comparisons with statistical significance of P < 0.05.
Results
Protective Effect of SIL on Mn-Induced Toxicity in Neuro-2A Cells
To determine the toxic effect of Mn, the cell viability and cell membrane damage were analysed by MTT and LDH release assay, respectively. Cytotoxicity was proportionally enhanced with increased Mn concentrations (200 and 800 μM). As illustrated in Fig. 1a, the viability of cells treated with 200 and 800 μM MnCl2 for 24 h showed a significant dose-dependent decrease (P < 0.05) compared to control group. As shown in Fig. 1b, cells treated with the same doses induced significant increase (P < 0.05) of the LDH level in the culture medium indicative of over cytotoxicity.

The effect of silymarin on Mn-induced toxicity in Neuro-2A cells. (a) Dose-dependent effects on cell viability was assessed using MTT assay. (b) Cell death was quantified by the release of LDH, from Neuro-2A cells into the medium treated with Mn and/or various concentrations of Silymarin exposure for 24 h. (c) The morphology of the treated cells was examined by phase contrast photomicrography using olympus inverted microscope. Data represent the group mean ± SD of 6 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 versus vehicle control group. # P < 0.05, ## P < 0.01, ### P < 0.001 versus Mn-treated alone group
The effect of SIL on Mn-induced toxicity was investigated by testing three concentrations of the flavonoid (10, 50 and 100 μM). The treatment of Neuro-2A cells with SIL alone (10 and 50 μM) showed no toxicity after 24 h of exposure. However, the highest concentration of SIL (100 μM) did not provide significantly greater antioxidant activity than 50 μM, and the culture began to exhibit signs of toxicity with significant reduction (P < 0.05) in cell viability compared to that of control. These data showed a dose-dependent effect of SIL with 50 μM evoking the greatest protection against Mn toxicity. Based on those results, this concentration was used in subsequent studies to determine the neuroprotective effects against Mn toxicity.
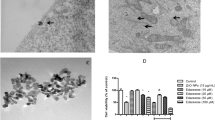
In addition, we observed the effect of SIL on morphology in Neuro-2A cells treated with Mn. As shown in Fig. 1c, under phase contrast microscopy, untreated Neuro-2A cells were long fusiform in shape with slender processes. After treatment of Mn, cells lost their normal spindle-like morphology, shrank or became round. They were detached and some even floated into the medium. Co-treatment with SIL (50 μM) markedly ameliorated Mn-induced morphological alterations. Cells treated with SIL alone displayed normal morphology.
Inhibitory Effect of SIL on ROS Production in Mn-Induced Neuro-2A Cells
Figure 2 presented the inhibitory effect of SIL on ROS production in Mn-induced Neuro-2A cells. Compared to control, treatment of Neuro-2A cells with Mn at 200 and 800 μM for 24 h increased ROS by 30 and 40% (P < 0.05) and cellular H2O2 by 140 and 200%, respectively. However, co- treatment with 50 μM of SIL significantly reduced the levels of Mn-induced ROS and H2O2 production (P < 0.05) compared to Mn treated cells. There were no changes in ROS and H2O2 levels after treatment with 50 μM of SIL alone (Fig. 2).

The effects of silymarin on ROS generation were measured by increased fluorescence with the DCFH-DA assay (a) as well as on cellular H2O2 accumulation (b) induced by manganese in Neuro-2A cells. Cells were co-cultured with Mn (200 and 800 μM) and SIL (50 μM) for 24 h. The values are mean ± SD of 6 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 versus vehicle control group. # P < 0.05, ## P < 0.01, ### P < 0.001 versus Mn-treated alone group
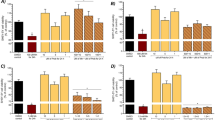
Effect of SIL on Antioxidant Enzyme Activities in Mn-Induced Neuro-2A Cells
Intracellular levels of antioxidant enzymes such as Mn-SOD, Cu-Zn SOD, CAT and GPx in Neuro-2A cells following exposure to MnCl2 and SIL for 24 h were measured by spectrophotometric analysis (Table 1). Neuro-2A cells exposed to MnCl2 showed a significant decrease in the specific activities of Mn-SOD, Cu-Zn SOD, CAT and GPx when compared to cells treated with a vehicle control (P < 0.05). Whereas, their activity was increased significantly (P < 0.05) when Mn-treated cells were co-cultured with SIL as compared to Mn-treated cells.
Effect of SIL on Lipid Peroxidation Product and Protein Carbonylation in Mn-Treated Neuro-2A Cells
We tested next the effect of SIL on the lipid peroxidation product (MDA) and protein carbonyls (PCO) in Mn-treated cells as presented in Fig. 3. A significant increase (P < 0.05) in MDA and PCO levels was observed in Mn-treated cells compared to a vehicle control. Co-administration of Silymarin at 50 μM significantly decreased (P < 0.01) the levels of these parameters. Further, there were no changes in the MDA and protein carbonyl levels after silymarin treatment as compared to control cells.

The effects of the Mn exposure (200 and 800 μM) and/or SIL (50 μM) on MDA and protein carbonyl levels in Neuro-2A lysates. MDA are expressed as nmol formed/mg protein and protein carbonyl (nmol DNPH conjugated/mg protein). Results represent mean ± SD (n = 6). *P < 0.05, ** P < 0.01, *** P < 0.001 versus vehicle control group. # P < 0.05, ## P < 0.01, ### P < 0.001 versus Mn-treated alone group
Effect of SIL on ATPase Activity in Mn-Induced Neuro-2A Cells
Table 2 illustrated Na+-K+-ATPase and Ca2+ activities in Neuro-2A cells. Mn exposure caused a significant inhibition (P < 0.05) in the Na+-K+-ATPase and Ca2+ activities in Neuro-2A cells compared to those of control group. Co-incubation cells with SIL at 50 μM significantly (P < 0.05) ameliorated the decreasing activities of these enzymes obtained in the Mn-treated cells.
Effect of SIL on Thiol (SH), Disulfide (SS) Contents, as Well as on SH/SS Ratio and GSH in Mn-Treated Neuro-2A Cells
The in vitro effect of SIL on SH/SS redox status in Mn-treated cells was studied by measuring thiol and disulfide contents. Table 3 showed that Mn significantly decreased the total thiol content and increased disulfide levels in a dose-dependent manner (P < 0.01). The SH/SS ratio was significantly reduced. These effects were significantly mitigated by 50 μM of silymarin compared to those of Mn-treated cells. These biochemical parameters did not differ between control and silymarin treated cells.
The effects of varying concentrations of MnCl2 on intracellular GSH were also evaluated and presented in Table 3. Mn treatment led to a significant decrease in GSH level (P < 0.05) compared to that of control group. Co-treatment with SIL significantly improved GSH levels in the cell lysates when Neuro-2A cells were treated with Mn only.
Effect of SIL on MnCl2 Generated of Proinflammatory Cytokines and Nitrite in Neuro-2A Cells Culture
As shown in Fig. 4, and compared to control cultures, no significant release of either TNFα or IL-6 was detected in the medium of Neuro-2A cells exposed to Mn. Since, the elevated accumulation of nitrite (P < 0.05), in a concentration-dependent manner, was observed compared to a vehicle control. Co-incubation of Mn treated cells with SIL significantly attenuated NO levels without reaching control values.

The effects of the Mn exposure (200 and 800 μM) and/or SIL (50 μM) on the production of nitrite (a), TNFα (b), and IL-6 (c). The data are expressed as mean ± SD from triplicate assays. ***P < 0.001 versus vehicle control group. # P < 0.05, ### P < 0.001 versus Mn-treated alone group
Discussion
Manganese, 1 of the essential elements for the living body, is required for lipid, protein and carbohydrate metabolism and also in the function and the development of the brain [38]. Its excessive amounts cause parkinsonian symptoms and which were implicated in the pathogenesis of Parkinson’s disease (PD) via several pathways. In vitro models represent an effective system for determining the mechanisms involved in neurodegenerative effects of Mn. The present study focused on characterising the mode of cell death in Neuro-2A cells treated with Mn and the possible mechanisms in neuroprotective effects of silymarin. To our knowledge, this is the first work which showed that silymarin protected Neuro-2A cells against Mn-induced ROS generation and glutathione depletion.
Neuronal injury was assessed by measuring MTT and LDH released activity. The MTT assay has been used to study neuronal injury, which quantifies mitochondrial activity by measuring the formation of a dark blue formazan product formed by the reduction of the tetrazolium ring of MTT [39] LDH is an indicator of the integrity of cell membrane, normally existing in the cell was released into the medium from damaged neurons. In our study, Mn used at 200 and 800 μM for 24 h induced toxicity of Neuro-2A cells as evidenced by an increase of Lactate deshydrogenase (LDH) release and a decrease of cellular viability (MTT assay) which was supported by morphological examinations. These results are consistent with earlier observations in other cell types [40–42] corroborating that impairment of mitochondria function is one of the key mechanisms in Mn cytotoxicity. However, Gunter et al. [43] demonstrate that Mn2+, preferentially accumulated in mitochondria, can disrupt oxidative phosphorylation and enhances the generation of reactive oxygen species (ROS). The increase of ROS, as demonstrated by both DCFH-DA and FOX detection of cellular H2O2, support these findings. In our present study, the co-incubation of Neuro-2A cells with silymarin protects them against cytotoxicity induced by Mn. These observations indicate that the neuroprotective effect of silymarin, at least partially, is attributed to its antioxidant and free radical scavenging properties. Similar results report that silymarin decreases lipid peroxidation, a sensitive marker of oxidative stress, in liver microsome and isolated hepatocytes [44]. Furthermore, it has been demonstrated that the antioxidative activity of silymarin is related to the scavenging of free radicals [45, 46].
Oxidative stress occurs when the antioxidant defence system is overwhelmed by the production of reactive oxygen species. The latter can induce lipid peroxidation and protein oxidation, hence potentially causing cell death [47, 48]. Therefore, increasing the antioxidant capacity of neurons will provide a potential strategy to protect neurons from Mn-induced oxidative damage. In this study, cells treated with Mn increased significantly MDA and protein carbonyl levels with a significant depressed level in the activity of the antioxidant enzymes, including isoforms of SOD, CAT and GPx in Neuro-2A cells. These results indicated that the cytotoxic actions of Mn in Neuro-2A cells were mediated, at least in part, by an oxyradical mechanism involving overproduction of ROS and down regulation of certain key antioxidant enzymes. Both events would lead to oxidative stress in Mn-exposed cells. Treatment with silymarin improved MDA, protein carbonyl levels and antioxidant enzyme activities. Based on these findings, it is possible that silymarin might be able to down regulate Mn-induced oxidative damages through the inhibition of ROS generation and indirectly by activation of antioxidative system including GSH biosynthesis. One of the defense mechanisms that protect the cell from oxidative damage is the dismutation of superoxide anion radical to hydrogen peroxide by SOD which is further converted to water by GPx, using GSH as a substrate. The generated hydrogen peroxide can be converted in spontaneous reaction catalyzed by metals such as iron (Fenton reaction) to the highly reactive hydroxyl radical, reacting with various biological macromolecules including polyunsaturated fatty acids by which the process of lipid peroxidation is initiated [49]. In our study, a clear-cut enhancement of GPx activity caused by SIL might conceal an expected increase in GSH levels. As a result, this facilitated defense mechanism shifts the conversion of hydrogen peroxide to water thereby reducing the formation of highly reactive hydroxyl radical, and eventually leading to a lipid peroxidation decrease and hydrogen peroxide accumulation.
Previous studies demonstrate that hydrogen peroxide (H2O2) can provoke cellular injuries, including the oxidation of redox sensitive cysteinyl thiols in proteins [50, 51]. In oxidative conditions, S-nitrosation is a reversible modification able to protect protein cysteinyl thiols against unwanted and possibly irreversible oxidation into sulfenic, sulfinic and sulfonic acids and intra/intermolecular disulfide bonds [52, 53]. As an abundant cellular antioxidant, GSH, like its precursors, has a protective action in oxidative stress conditions. In cells, GSH is oxidized to the GSSG disulfide by the action of GSH-peroxidase through a mechanism that consumes H2O2 [54]. In its S-nitrosated form, S-nitroso-glutathione (GSNO), GSH could act as a transporter and a stable reservoir of NO [55], as does Cystein (Cys) [56]. Redox sensitive Cystein residues of cellular proteins (–SH) become S-oxidized (–SOx) providing an important non-enzymatic cellular means to eliminate H2O2 [50]. Excessive oxidation can however, severely damage proteins by the formation of irreversible S-S bonds and the conversion of SH groups into sulfinic and sulfonic acids. These attacks on sulfhydryls have been detected in Mn-treated cells. GSH was reported to provide cell protection possibly by consuming H2O2, generating extracellular GSSG and protein S-glutathionylated adducts (–SSG) [57] and thus decreasing the amount of oxidant that can attack sulfhydryls. Considering that the total thiol content was significantly reduced by Mn while disulfide levels were enhanced, a significant reduction in SH/SS ratio was observed, suggesting that Mn was able to disturb the thiol redox status in Neuro-2A cells lysates. Given the numerous processes that are dependent upon cellular redox state, alteration of SH/SS ratio is considered as an important parameter indicative of oxidative stress. GSH is used to detoxify ROS and itself is converted to the oxidized form, GSSG. Our results are in accordance with previous observation which demonstrates the reduction of total GSH in the SK-N-SH (neuron-like) cells induced by manganese [58]. Several explanations by which silymarin regulates Mn-induced oxidative stress remain possible: (1) As a polyphenolic flavonoid, silymarin has a strong free radical-scavenging activity [46]. It reacts with a damaging free radical and forms a flavonoid radical, with a great stability and breaks the free radical chain reaction [59]. SIL, having potential antioxidative properties is known for its anti-lipid peroxidative capability against a variety of oxidative stress and thus can suppress oxidative damage [60–62]. (2) The neurotoxicity of Mn is mediated by forming H2O2, which generates lipid peroxidation adducts and protein carbonyl modifications in various cellular compartments. Incubation with the Fe3+ chelator desferrioxamine has been shown to decrease the toxicity of Mn. It is possible that silymarin attenuates the Mn-induced oxidative stress by chelating metal ions [63, 64]. (3) Oxidative stress occurs when the antioxidative defenses are broken down by the overproduction of reactive oxygen species. Expanding the antioxidant capacity of neuronal cells will provide a potential strategy to protect neurones from oxidative damage [65, 66]. Silymarin is known to induce the expression of antioxidative enzymes [67, 68]. Therefore, it is possible that silymarin alleviates oxidative damage indirectly by activation of antioxidative system including GSH biosynthesis.
Free radicals can attack polyunsaturated fatty acids (PUFA) in the biomembrane and initiate lipid peroxide chain reactions, leading to membrane dysfunction thereby destroying his special arrangement and impairing local enzyme activities. Na+-K+-ATPase, a membrane-bound enzyme responsible for maintaining Na+ and K+ gradients across the plasma cell membranes [69], was reported to be inhibited by ROS in primary cultured neurons [70]. ATP-driven plasma membrane calcium-pump (Ca2+-ATPase), of crucial importance in maintaining a low resting intra-neuronal Ca2+ particularly in excitable cells such as neurons, is involved in the final common pathway leading to cell death [71]. The decrease in the activities of Na+-K+-ATPase and Ca2+-ATPase observed in Neuro-2A cells supports these findings. Previous studies have found that Mn2+ could alter the balance between the simultaneous uptake and release of Ca2+ across the mitochondrial inner membrane [72]. In fact, it has been shown that oxidative stress increases intracellular Ca2+ concentrations leading to activation of endonucleases which degrade DNA and, ultimately, contribute to cell death. Furthermore, it has been demonstrated that ROS overproduction may inhibit Ca2+-ATPases and lead to alter regulation of Ca2+ levels and cell death. In the current study, we found that the co-incubation of Neuro-2A cells with silymarin effectively alleviated the decline of Na+-K+-ATPase and Ca2+-ATPase activities caused by Mn and subsequently stabilized transmembrane protein.
Upon activation, Neuro-2A cells secrete numerous bioactive factors such as interleukins, TNF-α and nitric oxide (NO). The nature of the released factors appears to be dependent on the nature of the causative agents. For example, lipopolysaccharide (LPS) was reported to increase the release of IL-6, TNF-α and NO [73]. In this study, NO was released but no significant release of cytokines TNFα and IL-6 was detected in Neuro-2A cells cultures treated with MnCl2 (200, 800 μM). This result is consistent with findings reported by others who demonstrate that in the mouse N9 microglial cells where no release of TNF-α, IL-1β and IL-6 was detected after treatment with MnCl2 at doses lower than 500 μM [73]. Moreover, a significant accumulation of nitrite was observed in the Neuro-2A cells cultures following treatment with MnCl2 at two doses used (200, 800 μM). Similar results were observed in primary mouse astroglia treated for 8 h with 10 μM MnCl2 [74] or C6 glioma cells treated for 24 h with 100 μM MnCl2 [75]. Although the NO synthesized by neuronal nitric oxide synthase (NOS) facilitates the formation of memories under physiological conditions [76], NO production from inducible nitric oxide synthase (iNOS) is deleterious under pathological conditions. Because of its independence of elevated intracellular Ca2+ levels [77], iNOS catalyzes a high-output pathway of NO production [78] that is able to cause neuronal peroxynitrite-mediated damage and dysfunction [79]. Silymarin significantly inhibited the increase of extra-cellular nitrite (NO) as reported by us and suppressed the expression or activation of iNOS in several tissues including a glial cell line as it has been reported by others [49, 80].
The results from the present study confirm, for the first time, that silymarin could alleviate the toxicity induced by Mn in Neuro-2A cells. The beneficial effect may be attributed to the prevention of oxidative damage, measured in terms of the amount of peroxidized lipid and the level of GSH. Currently, the exact mechanism of SIL-induced neuroprotection and its potential therapeutic applications in neurodegenerative disorders are still unclear and warrant further investigations.
References
Perl DP, Olanow CW (2007) The neuropathology of manganese-induced Parkinsonism. J Neuropathol Exp Neurol 66:675–682
Newland MC (1999) Animal models of manganese’s neurotoxicity. Neurotoxicology 20:15–432
Tamm C, Sabri F, Ceccatelli S (2008) Mitochondrial-mediated apoptosis in neural stem cells exposed to manganese. Toxicol Sci 101(2):310–320
Fitsanakis VA, Au C, Erikson KM, Aschner M (2006) The effects of manganese on glutamate, dopamine and gamma-aminobutyric acid regulation. Neurochem Int 48(6–7):426–433
Kwik-Uribe C, Smith DR (2006) Temporal responses in the disruption of iron regulation by manganese. J Neurosci Res 83(8):1601–1610
Gunter KK, Aschner M, Miller LM, Eliseev R, Salter J, Anderson K, Hammond S, Gunter TE (2005) Determining the oxidation states of manganese in PC12 and nerve growth factor-induced PC12 cells. Free Radic Biol Med 39:164–181
Milatovic D, Yin Z, Gupta RC, Sydoryk M, Albrecht J, Aschner JL, Aschner M (2007) Manganese induces oxidative impairment in cultured rat astrocytes. Toxicol Sci 98:198–205
Zhang F, Xu Z, Gao J, Xu B, Deng Y (2008) In vitro effect of manganese chloride exposure on energy metabolism and oxidative damage of mitochondria isolated from rat brain. Environ Toxicol Pharmacol 26(2):232–236
Gavin CE, Gunter KK, Gunter TE (1999) Manganese and calcium transport in mitochondria: implications for manganese toxicity. Neurotoxicology 20:445–454
Lai JCK, Chan AWK, Minski MJ, Lim L (1985) Roles of metal ions in brain development and aging. In: Gabay S, Harris J, Ho BT (eds) Metal ions in neurology and psychiatry. Alan Liss, NY, pp 49–67
Zhang S, Fu J, Zhou Z (2004) In vitro effect of manganese chloride exposure on reactive oxygen species generation and respiratory chain complexes activities of mitochondria isolated from rat brain. Toxicol In Vitro 18:71–77
Brouillet EP, Shinobu L, Mc Garvey U, Hochberg F, Beal MF (1993) Manganese injection into the rat striatum produces excitotoxic lesions by impairing energy metabolism. Exp Neurol 120:89–94
Gavin CE, Gunter KK, Gunter TE (1992) Mn2+ sequestration by mitochondria and inhibition of oxidative phosphorylation. Toxicol Appl Pharmacol 115:1–5
Turrens JF, Boveris A (1980) Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J 191:421–427
Gunter TE, Gavin CE, Aschner M, Gunter KK (2006) Speciation of manganese in cells and mitochondria: a search for the proximal cause of manganese neurotoxicity. Neurotoxicology 27(5):765–776
Meng N, Zhao J, Zhao B, Cheng Y, WangW ZhangY et al (2008) A novel butyrolactone derivative inhibited smooth muscle cell migration and proliferation and maintained endothelial cell functions through selectively affecting Na, K-ATPase activity and mitochondria membrane potential during in vitro angiogenesis. J Cell Biochem 104(6):2123–2130
Chen CJ, Liao SL (2002) Oxidative stress involves in astrocytic alterations induced by manganese. Exp Neurol 175:216–225
Hazell AS, Normandin L, Norenberg MD, Kennedy G, Yi JH (2006) Alzheimer type II astrocytic changes following sub-acute exposure to manganese and its prevention by antioxidant treatment. Neurosci Lett 396:167–171
Zeisel SH (2004) Antioxidant suppress apoptosis. J Nutr 134:3179–3180
Nijveldt RJ, Nood EV, Hoorn DEV, Boelens PG, Norren KV, Van Leeuwen PA (2001) Flavonoids: a review of probable mechanisms of action and potential applications 1–3. Am J Clin Nutr 74:418–425
Fraschini F, Demartini G, Esposti D (2002) Pharmacology of silymarin. Clin Drug Invest 22:51–65
Gazák R, Walterová D, Kren V (2007) Silybin and silymarin: new and emerging applications in medicine. Curr Med Chem 14:315–338
Wagoner J, Negash A, Kane OJ et al (2010) Multiple effects of silymarin on the hepatitis C virus life cycle. Hepatology 51(6):1912–1921
Polyak SJ, Morishima C, Lohmann V, Pal S, Lee DY, Liu Y, Graf TN, Oberlies NH (2010) Identification of hepatoprotective flavonolignans from silymarin. Proc Nat Acad Sci USA 107(13):5995–5999
Katiyar SK, Roy AM, Baliga MS (2005) Silymarin induces apoptosis primarily through a p53-dependent pathway involving Bcl-2/Bax, cytochrome c release, and caspase activation. Mol Cancer Ther 4:207–216
Dos Santos AP, Milatovic D, Au C, Yin Z, Batoreu MC, Aschner M (2010) Rat brain endothelial cells are a target of manganese toxicity. Brain Res 1326:152–161
Ou P, Wolff SP (1996) A discontinuous method for catalase determination at near physiological concentrations of H2O2 and its application to the study of H2O2 fluxes within cells. J Biochem Biophys Methods 31:59–67
Dringen R, Kussmaul L, Hamprecht B (1998) Detoxification of exogenous hydrogen peroxide and organic hydroperoxides by cultured astroglial cells assessed by microtiter plate assay. Brain Res Protoc 2:223–228
Aebi H (1984) Catalase in vitro. Methods Enzymol 105:121–126
Marklund S, Marklund G (1974) Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur J Biochem 47:469–474
Flohe L, Gunzler WA (1984) Assays of glutathione peroxidase. Methods Enzymol 105:114–121
Zahler WL, Cleland WW (1968) A specific and sensitive assay for disulfides. J Biol Chem 243:716–719
Fairbanks VF, Klee GG (1999) Biochemical aspects of hematology. In: Burtis CA, Ashwood ER (eds) Tietz textbook of clinical chemistry, 3rd edn. WB Saunders Company, Philadelphia, pp 1642–1644
Draper HH, Hadley M (1990) Malondialdehyde determination as index of lipid peroxidation. Methods Ezymol 86:421–431
Evans P, Lyras L, Halliwell B (1999) Measurement of protein carbonyls in human brain tissue. Methods Enzymol 300:145–156
Carfagna MA, Ponsler GD, Muhoberac BB (1996) Inhibition of ATPase activity in rat synaptic plasma membranes by simultaneous exposure to metals. Chem Biol Interact 100(1):53–65
Archer S (1993) Measurement of nitric oxide in biological models. FASEB J 7:349–359
Prohaska JR (1987) Function of trace elements in brain metabolism. Physiol Rev 67:858–901
Lobner D (2000) Comparison of the LDH and MTT assays for quantifying cell death: validity for neuronal apoptosis? J Neurosci Methods 96(2):147–152
Yin Z, Aschner JL, Dos Santos AP, Aschner M (2008) Mitochondrial-dependent manganese neurotoxicity in rat primary astrocyte cultures. Brain Res 1203:1–11
Marreilha dos Santos AP, Santos D, Au C, Milatovic D, Aschner M, Batoreu MC (2008) Antioxidants prevent the cytotoxicity of manganese in RBE4 cells. Brain Res 1236:200–205
Lee ES, Yin Z, Milatovic D, Jiang H, Aschner M (2009) Estrogen and tamoxifen protect against Mn-induced toxicity in rat cortical primary cultures of neurons and astrocytes. Toxicol Sci 110:156–167
Gunter TE, Gavin CE, Aschner M, Gunter KK (2006) Speciation of manganese in cells and mitochondria: A search for the proximal cause of manganese neurotoxicity. Neurotoxicology 27:765–776
Bosisio E, Benelli C, Pirola O (1992) Effect of the flavanolignans of Silybum marianum L on lipid peroxidation in rat liver microsomes and freshly isolated hepatocytes. Pharmacol Res 25:147–154
De Groot H, Rauen U (1998) Tissue injury by reactive oxygen species and the protective effects of flavonoids. Fundam Clin Pharmacol 12:249–255
Trouillas P, Marsal P, Svobodová A, Vostálová J, Gazák R, Hrbác J et al (2008) Mechanism of the antioxidant action of silybin and 2,3-dehydrosilybin flavonolignans: a joint experimental and theoretical study. J Phys Chem A 112:1054–1063
Kirkland JB (1991) Lipid peroxidation, protein thiol oxidation and DNA damage in hydrogen peroxide-induced injury to endothelial cells: role of activation of poly (ADP-ribose) polymerase. Biochim Biophys Acta 1092:319–325
Facchinetti F, Dawson VL, Dawson TM (1998) Free radicals as mediators of neuronal injury. Cell Mol Neurobiol 18:667–682
Reiter RJ (1995) Oxidative processes and antioxidative defense mechanisms in the aging brain. FASEB J 9:526–533
Adimora NJ, Jones DP, Kemp ML (2010) A model of redox kinetics implicates the thiol proteome in cellular hydrogen peroxide responses. Antioxid Redox Signal 13:731–743
Wang G, Liem DA, Vondriska TM, Honda HM, Korge P, Pantaleon DM, Qiao X, Wang Y, Weiss JN, Ping P (2005) Nitric oxide donors protect murine myocardium against infarction via modulation of mitochondrial permeability transition. Am J Physiol Heart Circ Physiol 288:1290–1295
Dalle-Donne I, Rossi R, Giustarini D, Colombo R, Milzani A (2007) S-glutathionylation in protein redox regulation. Free Radic Biol Med 43:883–898
Sun J, Steenbergen C, Murphy E (2006) S-nitrosylation: NO-related redox signaling to protect against oxidative stress. Antioxid Redox Signal 8:1693–1705
Lim CC, Bryan NS, Jain M, Garcia-Saura MF, Fernandez BO, Sawyer DB, Handy DE, Loscalzo J, Feelisch M, Liao R (2009) Glutathione peroxidase deficiency exacerbates ischemia-reperfusion injury in male but not female myocardium: insights into antioxidant compensatory mechanisms. Am J Physiol Heart Circ Physiol 297:2144–2153
Romero JM, Bizzozero OA (2006) Extracellular S-nitrosoglutathione, but not S-nitrosocysteine or N2O3, mediates protein S-nitrosation in rat spinal cord slices. J Neurochem 99:1299–1310
Zhang Y, Hogg N (2005) S-Nitrosothiols: cellular formation and transport. Free Radic Biol Med 38:831–838
Klatt P, Lamas S (2000) Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur J Biochem 267:4928–4944
Dukhande VV, Malthankar-Phatak GH, Hugus JJ et al (2006) Manganese induced neurotoxicity is differentially enhanced by glutathione depletion in astrocytoma and neuroblastoma cells. Neurochem Res 31:1349–1357
Weber KC, Honório KM, Bruni AT, da Silva AB (2006) The use of classification methods for modeling the antioxidant activity of flavonoid compounds. J Mol Model 12:915–920
Tripathi M, Singh BK, Kakkar P (2009) Glycyrrhizic acid modulates t-BHP induced apoptosis in primary rat hepatocytes. Food Chem Toxicol 47:339–347
Lu P, Mamiya T, Lu LL, Mouri A, Niwa M, Hiramatsu M, Zou LB, Nagai T, Ikejima T, Nabeshima T (2009) Silibinin attenuates amyloid β25 – 35 peptide-induced memory impairments: implication of inducible nitric-oxide synthase and tumor necrosis factor-α in mice. J Pharmacol Exp Ther 331(1):319–326
Lu P, Mamiya T, Lu L, Mouri A, Niwa M, Kim HC, Zou LB, Nagai T, Yamada K, Ikejima T, Nabeshima T (2010) Silibinin attenuates cognitive deficits and decreases of dopamine and serotonin induced by repeated methamphetamine treatment. Behav Brain Res 207(2):387–393
Gharagozloo M, Khoshdel Z, Amirghofran Z (2008) The effect of an iron (III) chelator, silybin, on the proliferation and cell cycle of jurkat cells: a comparison with desferrioxamine. Eur J Pharmacol 589(1–3):1–7
Roth JA, Feng L, Dolan KG, Lis A, Garrick MD (2002) Effect of the iron chelator desferrioxamine on manganese-induced toxicity of rat pheochromocytoma (PC12) cells. J Neurosci Res 68(1):76–83
Ahlemeyer B, Bauerbach E, Plath M, Steuber M, Heers C, Tegtmeier F et al (2001) Retinoic acid reduces apoptosis and oxidative stress by preservation of SOD protein level. Free Radic Biol Med 30:1067–1077
Alcaraz-Zubeldia M, Montes S, Ríos C (2001) Participation of manganese-superoxide dismutase in the neuroprotection exerted by copper sulfate against 1-methyl 4-phenylpyridinium neurotoxicity. Brain Res Bull 55:277–279
Valenzuela A, Aspillaga M, Vial S, Guerra R (1989) Selectivity of silymarin on the increase of the glutathione content in different tissues of the rat. Planta Med 5:420
Müzes G, Deák G, Láng I, Nékám K, Gergely P, Fehér J (1991) Effect of the bioflavonoid silymarin on the in vitro activity and expression of superoxide dismutase enzyme. Acta Physiol Hung 78:3–9
Magistretti PJ, Pellerin L (1999) Astrocytes couple synaptic activity to glucose utilization in the brain. News Physiol Sci 14:177–182
Rodrigo R, Bachler JP, Araya J, Prat H, Passalacqua W (2007) Relationship between (Na+-K+)-ATPase activity, lipid peroxidation and fatty acid profile in erythrocytes of hypertensive and normotensive subjects. Mol Cell Biochem 303:73–81
Baggaley EM, Elliott AC, Bruce JI (2008) Oxidant-induced inhibition of the plasma membrane Ca2+-ATPase in pancreatic acinar cells: role of the mitochondria. Am J Physiol Cell Physiol 295(5):1247–1260
Allshire A, Bernardi P, Saris NE (1985) Manganese stimulates calcium flux through the mitochondrial uniporter. Biochim Biophys Acta 807(2):202
Filipov NM, Seegal RF, Lawrence DA (2005) Manganese potentiates in vitro production of proinflammatory cytokines and nitric oxide by microglia through a nuclear factor kappa B-dependent mechanism. Toxicol Sci 84:139–148
Barhoumi R, Faske J, Liu X, Tjalkens RB (2004) Manganese potentiates lipopolysaccharide-induced expression of NOS2 in C6 glioma cells through mitochondrial-dependent activation of nuclear factor kappaB. Brain Res Mol Brain Res 122:167–179
Moreno JA, Sullivan KA, Carbone DL, Hanneman WH, Tjalkens RB (2008) Manganese potentiates nuclear factor-kappaB-dependent expression of nitric oxide synthase 2 in astrocytes by activating soluble guanylate cyclase and extracellular responsive kinase signaling pathways. J Neurosci Res 86:2028–2038
Yamada K, Noda Y, Nakayama S, Komori Y, Sugihara H, Hasegawa T, Nabeshima T (1995) Role of nitric oxide in learning and memory and in monoamine metabolism in the rat brain. Br J Pharmacol 115:852–858
Cho HJ, Xie QW, Calaycay J, Mumford RA, Swiderek KM, Lee TD, Nathan C (1992) Calmodulin is a subunit of nitric oxide synthase from macrophages. J Exp Med 176:599–604
Xie QW, Cho HJ, Calaycay J, Mumford RA, Swiderek KM, Lee TD, Ding A, Troso T, Nathan C (1999) Cloning and characterization of inducible nitric oxide synthase from mouse macrophages. Science 256:225–228
Tran MH, Yamada K, Nakajima A, Mizuno M, He J, Kamei H, Nabeshima T (2003) Tyrosine nitration of a synaptic protein synaptophysin contributes to amyloid beta-peptide-induced cholinergic dysfunction. Mol Psychiatr 8:407–412
Wang MJ, Lin WW, Chen HL, Chang YH, Ou HC, Kuo JS, Hong JS, Jeng KC (2002) Silymarin protects dopaminergic neurons against lipopolysaccharide induced neurotoxicity by inhibiting microglia activation. Eur J Neurosci 16:2103–2112
Acknowledgments
The present work was supported by the grants of DGRST (Appui à la Recherche Universitaire de base, ARUB 99/UR/08-73) Tunisia.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Khaled Trabelsi and Hamadi Fetoui contributed equally to this work.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Chtourou, Y., Trabelsi, K., Fetoui, H. et al. Manganese Induces Oxidative Stress, Redox State Unbalance and Disrupts Membrane Bound ATPases on Murine Neuroblastoma Cells In Vitro: Protective Role of Silymarin. Neurochem Res 36, 1546–1557 (2011). https://doi.org/10.1007/s11064-011-0483-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-011-0483-5