
Abstract
Some hereditary ataxias are treatable and the insight required for this has come from an in depth knowledge of the phenotypes and clinical biochemistry of the conditions. This has required both fundamental and translational clinical research. Prof John Blass was fortunate to begin his career at what we can now recognise as a golden era for such studies and he worked upon two important conditions; Refsum’s disease and Friedreich’s ataxia. More recently the mitochondrial encephalomyopathies have been described and similar investigative work has been undertaken upon them. Ubiquinone, CoQ10, deficiency is the most recently recognised encephalomyopathy and is itself treatable. Though rare, it is becoming increasingly recognised and patients are benefiting from the same scholarly approach to its’ investigation as was afforded Refsums’ disease and Friedreich’s ataxia.



Similar content being viewed by others
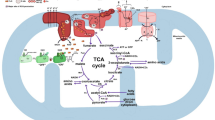


References
Refsum’s Heredopathia actica polyneuritiformis (1946) Acta Psychiatr Scand Suppl 38:9
Klenk E, Kahike W (1963) On the presence of 3,7,11,15-tetramethylhexadecanoic acid (phytanic acid) in the cholesterol esters and other fractions of the organs in a case of a disease of unknown origin (possibly heredopathia atactica polyneuritiformis, Refsum’s syndrome). Hoppe Seylers Z Physiol Chem 333:133–139
Steinberg D, Avigan J, Mize C, Eldjarn L, Try K, Refsum S (1965) Conversion of U-C14-phytol to phytanic acid and its oxidation in heredopathia atactica polyneuritiformis. Biochem Biophys Res Commun 19:783–789
Blass JP, Avign J, Steinberg D (1969) Alpha-hydroxy fatty acids in hereditary ataxic polyneuritis (Refsum’s disease). Biochim Biophys Acta 29(187):36–41
Tsai SC, Avigan J, Steinberg D (1969) Studies on the alpha oxidation of phytanic acid by rat liver mitochondria. J Biol Chem 244:2682–2692
Tsai SC, Herndon JH Jr, Uhlendorf BW, Fales HM, Mize CE (1967) The formation of alpha-hydroxy phytanic acid from phytanic acid in mammalian tissues. Biochem Biophys Res Commun 28:571–577
Mize CE, Herndon JH Jr, Blass JP, Milne GW, Follansbee C, Laudat P, Steinberg D (1969) Localization of the oxidative defect in phytanic acid degradation in patients with Refsum’s disease. J Clin Invest 48:1033–1040
Jansen GA, Ofman R, Ferdinandusse S, Ijlst L, Muijsers AO, Skjeldal OH, Stokke O, Jakobs C, Besley GTN, Wraith JE, Wanders RJA (1997) Refsum disease is caused by mutations in the phytanoyl-CoA hydroxylase gene. Nat Genet 17:190–193
Garrod AE (1909) Inborn errors of metabolism Oxford University Press. England
Eldjarn L, Try K, Stokke O, et al (1966) Dietary effects on serum-phytanic-acid levels and on clinical manifestations in heredopathia atactica polyneuritiformis. Lancet 1:691–693
Kark RA, Engel WK, Blass JP, et al (1971) Heredopathia actica polyneuritiformis (Refsum’s Disease): a second trial of dietary therapy in two patients. Birth Defects Orig Artic Ser 1:53–55
Djupesland G, Flottorp G, Refsum S (1983) Phytanic acid storage disease: hearing maintained after 15 years of dietary treatment. Neurology 33:237–240
Blass JP, Avigan J, Uhlendorf BW (1970) A defect in pyruvate decarboxylase in a child with an intermittent movement disorder. J Clin Invest 49:423–432
Blass JP, Lonsdale D, Uhlendorf BW, Hom E (1971) Intermittent ataxia with pyruvate-decarboxylase deficiency. Lancet 7712:1302
Blass JP, Kark AP, Engel WK (1971) Clinical studies of a patient with pyruvate decarboxylase deficiency. Arch Neurol 25:449–460
Reynolds SF, Blass J (1976) A possible mechanism for selective cerebellar damage in partial pyruvate dehydrogenase deficiency. Neurology 26:625–628
Letellier T, Heinrich R, Malgat M, Mazat JP (1994) The kinetic basis of threshold effects observed in mitochondrial diseases: a systemic approach. Biochem J 302:171–174
Davey GP, Peuchen S, Clark JB (1998) Energy thresholds in brain mitochondria. Potential involvement in neurodegeneration. J Biol Chem 273:12753–12757
Cedarbaum JM, Blass JP (1986) Mitochondrial dysfunction and spinocerebellar degenerations. Neurochem Pathol 4:43–63
Sheu KF, Blass JP, Cedarbaum JM, et al (1988) Mitochondrial enzymes in hereditary ataxias. Metab Brain Dis 3:151–160
Campuzano V, Montermini L, Molto MD, et al (1996) Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271:1423–1427
Durr A, Cossee M, Agid Y, et al (1996) Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N Engl J Med 335:1169–1175
Beal MF (1998) Mitochondrial dysfunction in neurodegenerative diseases. Biochim Biophys Acta 1366:211–223
Calabrese V, Lodi R, Tonon C, et al (2005) Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich’s ataxia. J Neurol Sci 233:145–162
Pandolfo M (2001) Molecular basis of Friedreich ataxia. Mov Disord 16:815–821
Emond M, Lepage G, Vanasse M, Pandolfo M (2000) Increased levels of plasma malondialdehyde in Friedreich ataxia. Neurology 55:1752–1753
Rustin P, von Kleist-Retzow JC, Chantrel-Groussard K, et al (1999) Effect of idebenone on cardiomyopathy in Friedreich’s ataxia: a preliminary study. Lancet 354:477–479
Aure K, Benoist JF, Ogier de Baulny H, Romero NB, Rigal O, Lombes A (2004) Progression despite replacement of a myopathic form of coenzyme Q10 defect. Neurology 63:727–729
Artuch R, Aracil A, Mas A, et al (2004) Cerebrospinal fluid concentrations of idebenone in Friedreich ataxia patients. Neuropediatrics 35:95–98
Artuch R, Brea-Calvo G, Briones P, et al (2006) Cerebellar ataxia with coenzyme Q10 deficiency: diagnosis and follow-up after coenzyme. J Neurol Sci 246:153–158
Turunen M, Olsson J, Dallner G (2004) Metabolism and function of coenzyme Q. Biochim Biophys Acta 1660:171–199
Shults CW (2003) Coenzyme Q10 in neurodegenerative diseases. Curr Med Chem 10:1917–1921
Linnane AW, Eastwood H (2004) Cellular redox poise modulation; the role of coenzyme Q10, gene and metabolic regulation. Mitochondrion 4:779–789
Di Giovanni S, Mirabella M, Spinazzola A, et al (2001) Coenzyme Q10 reverses pathological phenotype and reduces apoptosis in familial CoQ10 deficiency. Neurology 57:515–518
Baker SK, Tarnopolsky MA (2003) Targeting cellular energy production in neurological disorders. Expert Opin Investig Drugs 12:1655–1679
Kalen A, Appelkvist EL, Chojnacki T, Dallner G (1990) Nonaprenyl-4-hydroxybenzoate transferase, an enzyme involved in ubiquinone biosynthesis, in the endoplasmic reticulum-Golgi system of rat liver. J Biol Chem 265:1158–1164
Duncan AJ, Heales SJ, Mills K, Eaton S, Land JM, Hargreaves IP (2005) Determination of coenzyme Q10 status in blood mononuclear cells, skeletal muscle, and plasma by HPLC with di-propoxy-coenzyme Q10 as an internal standard. Clin Chem 51:2380–2382
Ernster L, Dallner G (1995) Biochemical, physiological and medical aspects of ubiquinone function. Biochim Biophys Acta 1271:195–204
Hirano M, Quinzii CM, Dimauro S (2006) Restoring balance to ataxia with coenzyme Q10 deficiency. J Neurol Sci 246:11–12
Fischer JC, Ruitenbeek W, Gabreels FJ, et al (1986) A mitochondrial encephalomyopathy: the first case with an established defect at the level of coenzyme Q. Eur J Pediatr 144:441–444
Ogasahara S, Engel AG, Frens D, Mack D (1989) Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proc Natl Acad Sci USA 86:2379–2382
Lamperti C, Naini A, Hirano M, et al (2003) Cerebellar ataxia and coenzyme Q10 deficiency. Neurology 60:1206–1208
Van Maldergem L, Trijbels F, DiMauro S, et al (2002) Coenzyme Q-responsive Leigh’s encephalopathy in two sisters. Ann Neurol 52:750–754
Rotig A, Appelkvist EL, Geromel V, et al (2000) Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet 356:391–395
Rahman S, Hargreaves I, Clayton P, Heales S (2001) Neonatal presentation of coenzyme Q10 deficiency. J Pediatr 139:456–458
Horvath R, Schneiderat P, Schoser BG, et al (2006) Coenzyme Q10 deficiency and isolated myopathy. Neurology 66:253–255
Lalani SR, Vladutiu GD, Plunkett K, et al (2005) Isolated mitochondrial myopathy associated with muscle coenzyme Q10 deficiency. Arch Neurol 62:317–320
Quinzii CM, Kattah AG, Naini a, et al (2005) Coenzyme Q deficiency and cerebellar ataxia associated with an aprataxin mutation. Neurology 64:539–541
Clark JB, Bates TE, Boakye P, Kuimov A, Land JM (1996) Investigation mitochondrial defects in brain and skeletal muscle. In: Turner AJ, Bachelor (eds) In a practical approach to the investigation of metabolic disease. IRL Press at Oxford University Press, Oxford. pp 151–174
Davey GP, Peuchen S, Clark JB (1998) Energy thresholds in brain mitochondria. Potential involvement in neurodegeneration. J Biol Chem 273:12753–12757
Steele PE, Tang PH, DeGrauw AJ, Miles MV (2004) Clinical laboratory monitoring of coenzyme Q10 use in neurologic and muscular diseases. Am J Clin Pathol 121(Suppl):S113–S120
Ullmann U, Metzner J, Schulz C, et al (2005) A new Coenzyme Q10 tablet-grade formulation (all-Q) is bioequivalent to Q-Gel and both have better bioavailability properties than Q-SorB. J Med Food 8:397–399
Bhagavan HN, Chopra RK (2006) Coenzyme Q10: absorption, tissue uptake, metabolism and pharmacokinetics. Free Radic Res 40:445–453
Author information
Authors and Affiliations
Corresponding author
Additional information
A dedication to Professor John P. Blass.
Appendix
Appendix
Vignette 1
SA was a lady of 47 years. She had shown psychomotor retardation since early childhood. She was markedly ataxic, unable to walk and requiring a wheelchair or having to bottom shuffle. She also had poor vision and had hypertrophic cardiomyopathy. She was investigated for mitochondrial disease and underwent a muscle biopsy. The results of skeletal muscle respiratory chain enzyme analysis are shown below:
SA
-
Complex I: 0.137 ref interval 0.104–0.268
-
Complex II-III: 0.015 ref interval 0.040–0.204
-
Complex IV: 0.012 ref interval 0.014–0.034
Activities are expressed as a ratio of specific activity to citrate synthase, except for Complex IV, which is expressed as a pseudo first order rate constant relative to citrate synthase. This corrects for mitochondrial enrichment.
The low complex II+III suggested a possible CoQ10 deficiency. This was measured in skeletal muscle as described [37] and confirmed the diagnosis.
Skeletal muscle CoQ10: 33 pmol/mg protein [140-580 pmol/mg of protein] and in isolated mononuclear cells: 20 pmol/mg protein [37–133 pmol/mg protein].
High dose CoQ10 was commenced [300 mg per day]. Over the next six months the patient improved markedly and became able to walk albeit with assistance. A repeat assessment of mononuclear cell CoQ10 was within the reference interval.
111 pmol/mg protein [37–133 pmol/mg protein].
Rights and permissions
About this article
Cite this article
Land, J.M., Heales, S.J.R., Duncan, A.J. et al. Some Observations upon Biochemical Causes of Ataxia and a New Disease Entity Ubiquinone, CoQ10 Deficiency. Neurochem Res 32, 837–843 (2007). https://doi.org/10.1007/s11064-006-9222-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-006-9222-8