
Abstract
Ovarian cancer represents the most common gynaecological malignancy and has the highest mortality of all female reproductive cancers. It has a rare predilection to develop brain metastases (BM). In this study, we evaluated the mutational profile of ovarian cancer metastases through Next-Generation Sequencing (NGS) with the aim of identifying potential clinically actionable genetic alterations with options for small molecule targeted therapy. Library preparation was conducted using Illumina TruSight Rapid Capture Kit in combination with a cancer specific enrichment kit covering 94 genes. BRCA-mutations were confirmed by using TruSeq Custom Amplicon Low Input Kit in combination with a custom-designed BRCA gene panel. In our cohort all eight sequenced BM samples exhibited a multitude of variant alterations, each with unique molecular profiles. The 37 identified variants were distributed over 22 cancer-related genes (23.4%). The number of mutated genes per sample ranged from 3 to 7 with a median of 4.5. The most commonly altered genes were BRCA1/2, TP53, and ATM. In total, 7 out of 8 samples revealed either a BRCA1 or a BRCA2 pathogenic mutation. Furthermore, all eight BM samples showed mutations in at least one DNA repair gene. Our NGS study of BM of ovarian carcinoma revealed a significant number of BRCA-mutations beside TP53, ATM and CHEK2 mutations. These findings strongly suggest the implication of BRCA and DNA repair malfunction in ovarian cancer metastasizing to the brain. Based on these findings, pharmacological PARP inhibition could be one potential targeted therapeutic for brain metastatic ovarian cancer patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ovarian cancer (OC) represents the most common gynaecological malignancy and has the highest mortality in the developed world [1]. The vast majority of OCs are of epithelial origin [2] and the most frequent subtype of epithelial OCs is the serous carcinoma [3]. More than 70% of OCs are diagnosed in advanced stages with detectable transperitoneal spread [4]. The 5-year survival rate is approximately 30% [5]. Despite progress in the diagnosis and treatment of these tumors, the rates of relapse and distant metastasis remain high [1]. Given the high recurrence rate and poor long-term survival of women with advanced stage disease, there is a strong need to document the unique metastatic patterns of epithelial OC by comparing the differences in genetic profiles between primary and metastatic tissue specimens [6].
Clinical observation and retrospective clinical studies suggest that serous OCs grow very efficiently within the peritoneal cavity, but rarely metastasize beyond it [7]. Accordingly, brain metastases (BM) from OC is a very rare phenomenon and a late-stage disease manifestation, usually occurring in the context of widely disseminated disease [1]. The incidence of BM from epithelial OC ranges from 0.29 to 5% [8, 9]. However, the incidence of BM seems to be increasing (11.6%) since the introduction of platinum-based chemotherapy [10] and the more frequent use of sensitive detection methods [11]. BM are associated with a large burden of neurological symptoms and a poor survival prognosis [12]. Treatments vary widely, including chemotherapy, steroids, whole brain radiation therapy, surgical resection, and stereotactic radiosurgery [13].
To date, most research efforts have focused on defining the molecular characteristics of primary OC [6, 14, 15]. Only very limited data are available on molecular aberrations in BM of OC [16,17,18]. In this study, we evaluated the mutational profile of OC metastases through Next-Generation Sequencing (NGS) with the aim of identifying potential clinically actionable genetic alterations with options for small molecule targeted therapy.
Methods
Patients
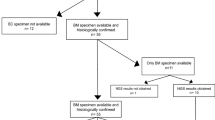
Only BM of ovarian carcinomas were included into this retrospective observational study. In total, ten ovarian cancer BM of the same number of patients were identified and subsequently investigated by NGS. All patients were female with a median age of 51 years (range 36–65 years) at diagnosis of ovarian cancer. The median interval between primary diagnosis of ovarian cancer and diagnosis of BM was 30.5 months (range 0–61 months). Nine of the ten patients were treated with a platinum-based chemotherapy before the diagnosis of BM. Supplementary Table S1 shows further patient´s baseline characteristics. For histological subtypes, nine were serous carcinoma and one small cell carcinoma of the ovary.
Publicly available datasets
Genomic mutational profiles of ovarian serous cystadenocarcinoma were obtained from the cBioPortal for Cancer Genomics (http://www.cbioportal.org) [19, 20]. This TCGA provisional dataset is directly from the TCGA data center and consists of 316 sequenced tumors of clinically annotated stage-II–IV high-grade serous ovarian cancer. TCGA performed exome capture and massively parallel sequencing on the Illumina GAIIx platform (236 samples) or ABI SOLiD 3 platform (80 samples) [14].
Illumina sequencing panel
Illumina TruSight Rapid Capture Kit
Genomic DNA was isolated from formalin fixed, paraffin embedded (FFPE) tissue sections using QIAamp DNA FFPE Tissue Kit (Qiagen) according to the user manual. Quantification was performed with Qubit® dsDNA HS Assay Kit (Invitrogen) according to the instructions provided by the manufacturer. The quality of DNA samples was controlled prior to sample selection for NGS using a qPCR-based assay. Library preparation was conducted using TruSight Rapid Capture Kit in combination with a cancer-specific enrichment kit covering 94 genes provided by Illumina Inc. (Supplementary Table S2). Subsequently 150 bp paired-end sequencing was performed on a MiSeq System (Illumina, USA) with reagent kit v2. The read alignment and variant calling were performed with BaseSpace BWA Enrichment v1.0 App. The sequence is aligned with BWA Genome Alignment Software and variant calling is performed with GATK using the human reference sequence hg19/GRCh37. These variants were then annotated using the Illumina VariantStudio data analysis software.
A number of steps were used to filter nucleotide variants identified in the screening: First, intronic variants and synonymous SNVs were excluded. Subsequently, variants annotated in dbSNP without pathogenic relevance for the clinical phenotype were removed. The remaining variants were further filtered by excluding all variants that are not annotated as “pathogenic” or “likely pathogenic” by in silico prediction tools. This set was further filtered by excluding all variants showing a poor quality (read depth <30; alternative variant frequency <5). Manual and thorough observation of the variants was performed to exclude false variants. The resulting filtered sets of all eight samples were then combined to a total of 37 variants.
TruSeq Custom Amplicon Low Input Kit
Based on the presented results we aimed to focus on the analysis of both BRCA genes of interest and therefore used the TruSeq Custom Amplicon Low Input Kit (Illumina) in combination with a custom-designed BRCA gene panel. Selected FFPE samples were subjected to dual-pool amplicon-based library preparation. Subsequent sequencing of pooled libraries was performed on the MiniSeq sequencing platform using MiniSeq High Output Reagent Kit for 300-cycles.
Paired-end sequencing resulted in average 6115644.86 (6.1 Mio) paired-end passed filter reads per sample and mean amplicon coverage of 6774. Data analysis was conducted using on board Amplicon DS pipeline. Sequencing data was aligned to the reference genome UCSC hg19 using banded Smith–Waterman algorithm and variant calling was performed with Illumina Somatic variant caller. Filtering of all datasets was conducted manually according to predefined (custom) criteria.
Statistical analysis
The statistical analysis was performed with SPSS (19.0). Mainly descriptive statistic was used. For association analyses with clinical data, Spearmen Correlation and Mann Whitney U test was applied. Due to the observational and hypothesis generating character of this study, we did not adjust for multiple testing [21]. The threshold for statistical significance was set at a p-value of less than 0.05.
Results
NGS mutational profile
Somatic mutations in OC BM
In two of the total ten cases, data quality was low, probably due to the fact that DNA was isolated from FFPE material and may have been degraded. These were excluded from further analysis. The remaining eight samples were successfully sequenced. The mean count of total aligned reads obtained per sample was 2.88 million (range 1.88–6.39 million) and the minimum sequencing depth was 543.5X.
Overall, 37 variants were detected with known or likely pathogenic effect. A total of 3231 variants were excluded because they were known polymorphisms, likely benign variants or did not pass the quality criteria specified in the Supplementary Methods section. The number of gene mutations per sample ranged from 3 to 7 with a median of 4.5. Three samples (37.5%) showed 3 mutations, one sample (12.5%) showed 4 mutations, two samples (25%) showed 5 mutations, and two samples (25%) showed 7 mutations (Table 1). The 37 variants consisted of 6 frameshift variants, 2 splice variants, another 3 were stop mutations, and the remaining 26 were missense variants (Table 1). In addition, base transitions (purine–purine and pyrimidine–pyrimidine) were considerably more frequent (61.29%) than transversions (purine–pyrimidine, vice versa) (Supplementary Figure S3). In our cohort all eight sequenced BM samples exhibited a multitude of variant alterations, each with unique molecular profiles. A few samples showed common mutations, however, each sample exhibited different overall molecular profiles. In summary, 20 of 37 variants are known COSMICs (catalogue of somatic mutations in cancer) and the remaining 17 were considered potentially biologically significant.
Mutated genes in OC BM
In a next step we searched for genes that were recurrently mutated in our cases. We tested 94 genes, and 72 genes displayed no alterations (Supplementary Table S2). The 37 identified variants were distributed over 22 genes (23.4%). The number of mutated genes per sample ranged from 3 to 7 with a median of 4.5, identical with the number of mutations. Just two of the 37 revealed mutations were known cancer hotspots: TP53 p.G266V and TP53 p.R248Q, the latter actually in OC [22], and TP53 p.R248Q mutation has been reported to aggregate and to be associated with metastasis [23]. The most commonly altered genes were TP53 and BRCA1 with 6/8 and 5/8 events, respectively. Aberrations in ATM were found in 3/8 samples. BRCA2, CHEK2, NBN and RB1 showed aberrations in 2/8 cases, each. Furthermore, 15 other genes showed aberrations, however less frequently (n = 1) (Table 1).
The most common combination was TP53 and BRCA1 mutations (n = 4), followed by TP53 and ATM (n = 3), and TP53 and BRCA2 (n = 2). The number of mutations was not associated with age (p > 0.05, Spearmen Correlation). There was a weak association between the tumor stage and the number of genomic abnormalities in BM, which did not reach statistical significance (p > 0.05, Mann Whitney U test). A trend in longer overall survival was seen in BRCA1, BRCA2 and TP53 mutation carriers compared with non-carriers, whereas just 1/8 patients revealed no mutations in these 3 genes and in addition showed a short overall survival. In summary, 7 out of 8 BM samples revealed either a BRCA1 or a BRCA2 pathogenic mutation. Furthermore, all eight BM samples showed mutations in at least one DNA repair gene (BRCA1/2, ATM, CHEK2) (Table 1).
Genomic profile of ovarian serous cystadenocarcinoma (TCGA dataset)
Due to the lack of paired samples, i.e. corresponding primary tumor biopsies (PT) to each of our BM samples, we analyzed a publicly available dataset encompassing 316 ovarian serous cystadenocarcinoma (TCGA, Provisional) to be able to obtain at least a comparison between PT and BM. We focused mainly on the 94 genes in this TCGA dataset, which we analyzed in our BM samples (Supplementary Table S2). Overall, 7/22 genes, which were mutated in the BM samples, showed no alterations in the PM, whereas the remaining 15 genes were mutated as well. Notably, in agreement with the BM samples, TP53 (86.71%), BRCA1 (3.48%), and BRCA2 (3.48%) were the most frequently altered genes. Additionally, NF1 (4.43%), RB1 (2.85%), and KIT (2.22%) were frequently mutated in the PT in contrast to the BM samples. Furthermore, in the PT samples BRCA1 and BRCA2 mutations were mutually exclusive. This is in agreement with our BM samples and with the findings of Mafficini et al. [24]. In addition, the ratio of BRCA1 (11/316) to BRCA2 (11/316) was balanced in the PT samples, whereas BRCA2 (2/8) was less commonly mutated in the BM samples than BRCA1 (5/8). The most common combination of gene alterations in the PT samples was TP53 and NF1 (n = 11), followed by TP53 and BRCA2 (n = 9), and TP53 and BRCA1 (n = 8). Analysis of the remaining 72/94 genes, which were not mutated in the BM samples, revealed that 43/72 were altered in the PT dataset, amongst which EGFR (2.22%), APC (2.2%), and SMARCB1 (1.58%) have to be highlighted. When focusing on the 94 genes analyzed in the BM samples, we found the median number of gene mutations per PT sample and median number of mutated genes to be 1.0 (range 0–5).
Confirmation of BRCA-mutations in four patients by using the TruSeq Custom Amplicon Low Input Panel
For conformational analysis in addition to BM samples, primary tumor and/or normal FFPE tissue samples were fortunately available in some cases. We were able to confirm BRCA-mutations in four patients using the TruSeq Custom Amplicon Low Input Kit. We validated the BRCA-mutations in patients #5 and #7, but no additional patient-matched tissues were available for testing of the mutational origin. We tested available patient-matched normal tissue additionally to BM in patient #6. In this case, the BRCA1 mutation was found only in the tumor, thus confirming that this mutation was of somatic origin. Furthermore, the BRCA1 mutation p.Gln1777ProfsTer74 was already described in the context of OC by Mafficini and colleagues [24]. We tested PT and normal tissue additionally to BM in patient #4. We detected c.4039A>G (p.Arg1347Gly) in all tissues including normal tissue and concluded that this missense mutation is a germline mutation. The c.4039A>G is a variant of uncertain significance according to ACMG guidelines (RCV000034747.1). Interestingly the c.4039A>G was detected in all tissues at different frequencies: normal tissue showed 50%, PT revealed 75% and BM showed 100%. Thus, we conclude that the loss of heterozygosity/BRCA1-deletion is a somatic mutation and occurred in part of the PT sample and in the entire BM sample. In summary, all BRCA-mutations were confirmed by an independent analysis, and in two cases we could furthermore prove-due to additional normal tissue-that they were of somatic origin.
Discussion
To the best of our knowledge, we present here for the first time results of NGS in BM of OC. Too little is currently known about the genomic makeup of OC BM to offer a potential pathway for targeted therapeutics in this disease. Because of the rarity of BM of OC, the number of studied patients in general as well as in our cohort has been small. With this NGS-based study, we aimed to get some insight for a better understanding of this rare phenomenon. We successfully sequenced eight BM samples of primary OC and detected 37 variants in total, distributed over 22 cancer-related genes (23.4%). Mutations per analyzed BM sample ranged from 3 to 7 with a median of 4.5. The most commonly altered genes were BRCA1/2, TP53, ATM, and among others, CHEK2. Consistently, TP53, BRCA1, and BRCA2 were the most frequently altered genes in the TCGA ovarian serous cystadenocarcinoma data. Moreover, in line with other studies on the pattern of somatic mutations in human cancer genomes [15, 25], we observed a higher rate of transitions than transversions. Even if metastatic tumor spread is a very complex process consisting of many different events, the mutational spectrum of our BM specimens was surprisingly simple, which is consistent with the findings of Beltrame et al., who reported that the genomic architecture of relapsed disease was less heterogenous than that of the primary disease [26]. Nevertheless, no two BM shared an identical genetic profile, which is similar to published data about metastatic breast cancer [27, 28].
One of our major findings was the unexpectedly high number of BRCA1/2 mutations in BM of OC (7 out of 8). The National Genome Atlas study identified somatic BRCA1 and BRCA2 mutations as a significant feature of high grade serous OC [14], but the characterized mutational frequencies were much lower [2, 20]. The BRCA1 gene is involved in DNA repair, cell-cycle checkpoint control, chromatin remodeling, transcriptional regulation and mitogenesis, while the BRCA2 has an important role in homologous recombination [29]. In our BM samples, 2/8 revealed a BRCA2 mutation. Koul and colleagues hypothesized that BRCA1 might have a possible role in ovarian tumors metastasizing to the brain [30], which is mostly in line with our mutational results. In our cohort, 5/8 patients showed a BRCA1 mutation and 2/8 a BRCA2 mutation. Patient #4 was an interesting case, because we had PT and normal tissue available in addition to BM. A BRCA1-deletion occurred in part of the PT sample and in the entire BM sample, suggesting that there was a positive selection of BRCA1 mutation in BM compared to PT. In conclusion, mutated BRCA1 and BRCA2 seem to be important in the development of BM.
It was not surprising that TP53 and ATM were mutated in our BM samples of primary OC. Previous studies have highlighted that TP53, that encodes the tumor suppressor protein p53, is the most frequently altered gene in serous OC [2, 17]. In addition, the majority of our genetic alterations identified in TP53 were predicted to be deleterious, and 2 of the BM mutations were known hotspot mutations (p.G266V and p.R248Q) (http://cancerhotspots.org/). Based on these findings one can hypothesize that TP53 plays a key role in BM specimens of primary OC. In our samples mutations in TP53 and ATM (n = 3), and BRCA1/2 and ATM (n = 3) are common combinations. ATM is a major regulator of DNA damage detection and repair [31]. In response to DNA damage, ATM controls the initial phosphorylation of a wide variety of downstream proteins such as TP53 and BRCA1 [32]. Moreover, ATM mutations can lead to deficiencies in DNA repair, which in return may give rise to cancer [33]. Therefore, it can be hypothesized that ATM does not promote OC metastasizing to the brain isolatedly but through the DNA damage-recognition and repair pathway together with TP53 and BRCA1.
The mutation frequencies for NF1, RB1, and KIT in our BM samples were not in the range described for ovarian cancer specimens, they were less frequent. EGFR (2.22%), APC (2.2%), and SMARCB1 (1.58%) have to be stressed here as well, because they were mutated in the TCGA dataset, but not in our BM samples. Beltrame and colleagues reported that somatic mutations showed a low rate of concordance between primary and recurrent disease in stage III–IV epithelial ovarian cancer, which may explain both these phenomena, or may be due to an underrepresentation of the mutational burden in the BM samples because of the small sample size [26].
In this study, we focused on identifying potentially actionable somatic mutations in metastatic OC. In total, 7/8 BM samples analyzed showed BRCA1/2 mutations and, moreover, all 8 samples revealed mutations in at least one DNA repair gene (BRCA1/2, ATM, CHEK2). It is known that cells which are BRCA deficient and then undergo Poly (adenosine diphosphate-ribose) polymerase (PARP) inhibition (PARPi), suffer cell death [34]. Accordingly, Mateo and colleagues concluded that, if a cell was lacking homologous repair capacity because of BRCA1, BRCA2 or ATM dysfunction or loss, then PARPi would lead to synthetic lethality due to cell cycle arrest and subsequent apoptosis [35]. Additionally, McCabe et al. showed via in vitro studies that, among others, ATM and CHEK2 abnormalities resulted in sensitivity to PARPi, suggesting that PARPi would be beneficial for a variety of genes involved in the DNA damage response [36]. The PARPi Olaparib has shown significant clinical activity in BRCA-mutated platinum-sensitive recurrent serous OC and has been approved for clinical use [37,38,39,40]. Based on these facts and on our findings, pharmacological PARPi could be one potential targeted therapeutic for brain metastatic OC patients, especially because PARPi Olaparib has shown evidence of crossing the blood–brain barrier [41]. Multiple PARP inhibitors are already at different stages of clinical development for the management of OC [42, 43].
Metastatic OC remains largely incurable, and thus a larger population based study and molecular genetic analyses of OCs metastatic to the brain are needed for a better understanding of the role of key genes like BRCA1/2, TP53, ATM, and CHEK2 in this rare phenomenon. Because of the rarity of OC BM our sample size is relatively small. Therefore, the acquisition of sufficiently large cohorts consisting of matched samples of this rare disease will require international collaborations [2].
Taken together, our NGS study of OC BM revealed a prominent number of BRCA-mutations beside TP53, ATM and CHEK2 mutations. These findings strongly suggest the implication of BRCA and DNA repair malfunction in OC metastasizing to the brain. For any conclusive statement as to whether the DNA damage-recognition and repair pathway plays a key role in this phenotype, the implementation of a similar study with a larger cohort and functional analyses is needed. PARPi represents one of the most promising treatment options for OC patients in general and for OC patients with metastatic BM in particular.
References
Nafisi H, Cesari M, Karamchandani J, Balasubramaniam G, Keith JL (2015) Metastatic ovarian carcinoma to the brain: an approach to identification and classification for neuropathologists. Neuropathology 35(2):122–129. doi:10.1111/neup.12172
Hollis RL, Gourley C (2016) Genetic and molecular changes in ovarian cancer. Cancer Biol Med 13(2):236–247. doi:10.20892/j.issn.2095-3941.2016.0024
Bookman MA (2016) Optimal primary therapy of ovarian cancer. Ann Oncol 27(Suppl 1):i58–i62. doi:10.1093/annonc/mdw088
Goodman MT, Howe HL, Tung KH, Hotes J, Miller BA, Coughlin SS, Chen VW (2003) Incidence of ovarian cancer by race and ethnicity in the United States, 1992–1997. Cancer 97(10 Suppl):2676–2685. doi:10.1002/cncr.11349
Siegel R, Ma J, Zou Z, Jemal A (2014) Cancer statistics, 2014. Cancer J Clin 64(1):9–29. doi:10.3322/caac.21208
Lee JY, Yoon JK, Kim B, Kim S, Kim MA, Lim H, Bang D, Song YS (2015) Tumor evolution and intratumor heterogeneity of an epithelial ovarian cancer investigated using next-generation sequencing. BMC Cancer 15:85. doi:10.1186/s12885-015-1077-4
Lengyel E (2010) Ovarian cancer development and metastasis. Am J Pathol 177(3):1053–1064. doi:10.2353/ajpath.2010.100105
Larson DM, Copeland LJ, Moser RP, Malone JM Jr, Gershenson DM, Wharton JT (1986) Central nervous system metastases in epithelial ovarian carcinoma. Obstet Gynecol 68(6):746–750
Barker GH, Orledge J, Wiltshaw E (1981) Involvement of the central nervous system in patients with ovarian carcinoma. Br J Obstet Gynaecol 88(7):690–694
Hardy JR, Harvey VJ (1989) Cerebral metastases in patients with ovarian cancer treated with chemotherapy. Gynecol Oncol 33(3):296–300
Marchetti C, Ferrandina G, Cormio G, Gambino A, Cecere S, Lorusso D, De Giorgi U, Bogliolo S, Fagotti A, Mammoliti S, Narducci F, Bergamini A, Scollo P, Biglia N, Breda E, Tamberi S, Marinaccio M, Angioli R, Salerno L, Eusebi MC, Loizzi V, Scambia G, Panici PB (2016) Brain metastases in patients with EOC: Clinico-pathological and prognostic factors. A multicentric retrospective analysis from the MITO group (MITO 19). Gynecol Oncol 143(3):532–538. doi:10.1016/j.ygyno.2016.09.025
Preusser M, Berghoff AS, Koller R, Zielinski CC, Hainfellner JA, Liebmann-Reindl S, Popitsch N, Geier CB, Streubel B, Birner P (2015) Spectrum of gene mutations detected by next generation exome sequencing in brain metastases of lung adenocarcinoma. Eur J Cancer 51(13):1803–1811. doi:10.1016/j.ejca.2015.06.107
Teckie S, Makker V, Tabar V, Alektiar K, Aghajanian C, Hensley M, Beal K (2013) Radiation therapy for epithelial ovarian cancer brain metastases: clinical outcomes and predictors of survival. Radiat Oncol 8:36. doi:10.1186/1748-717x-8-36
Cancer Genome Atlas Research Network (2011) Integrated genomic analyses of ovarian carcinoma. Nature 474(7353):609–615. doi:10.1038/nature10166
Ab Mutalib NS, Syafruddin SE, Md Zain RR, Mohd Dali AZ, Mohd Yunos RI, Saidin S, Jamal R, Mokhtar NM (2014) Molecular characterization of serous ovarian carcinoma using a multigene next generation sequencing cancer panel approach. BMC Res Notes 7:805. doi:10.1186/1756-0500-7-805
Brastianos PK, Carter SL, Santagata S, Cahill DP, Taylor-Weiner A, Jones RT, Van Allen EM, Lawrence MS, Horowitz PM, Cibulskis K, Ligon KL, Tabernero J, Seoane J, Martinez-Saez E, Curry WT, Dunn IF, Paek SH, Park SH, McKenna A, Chevalier A, Rosenberg M, Barker FG 2nd, Gill CM, Van Hummelen P, Thorner AR, Johnson BE, Hoang MP, Choueiri TK, Signoretti S, Sougnez C, Rabin MS, Lin NU, Winer EP, Stemmer-Rachamimov A, Meyerson M, Garraway L, Gabriel S, Lander ES, Beroukhim R, Batchelor TT, Baselga J, Louis DN, Getz G, Hahn WC (2015) Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discov 5(11):1164–1177. doi:10.1158/2159-8290.cd-15-0369
Ross JS, Ali SM, Wang K, Palmer G, Yelensky R, Lipson D, Miller VA, Zajchowski D, Shawver LK, Stephens PJ (2013) Comprehensive genomic profiling of epithelial ovarian cancer by next generation sequencing-based diagnostic assay reveals new routes to targeted therapies. Gynecol Oncol 130(3):554–559. doi:10.1016/j.ygyno.2013.06.019
Pentsova EI, Shah RH, Tang J, Boire A, You D, Briggs S, Omuro A, Lin X, Fleisher M, Grommes C, Panageas KS, Meng F, Selcuklu SD, Ogilvie S, Distefano N, Shagabayeva L, Rosenblum M, DeAngelis LM, Viale A, Mellinghoff IK, Berger MF (2016) Evaluating cancer of the central nervous system through next-generation sequencing of cerebrospinal fluid. J Clin Oncol 34(20):2404–2415. doi:10.1200/jco.2016.66.6487
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2(5):401–404. doi:10.1158/2159-8290.cd-12-0095
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6(269):pl1. doi:10.1126/scisignal.2004088
Bender R, Lange S (2001) Adjusting for multiple testing–when and how? J Clin Epidemiol 54(4):343–349
Seagle BL, Yang CP, Eng KH, Dandapani M, Odunsi-Akanji O, Goldberg GL, Odunsi K, Horwitz SB, Shahabi S (2015) TP53 hot spot mutations in ovarian cancer: selective resistance to microtubule stabilizers in vitro and differential survival outcomes from the Cancer Genome Atlas. Gynecol Oncol 138(1):159–164. doi:10.1016/j.ygyno.2015.04.039
Mullany LK, Wong KK, Marciano DC, Katsonis P, King-Crane ER, Ren YA, Lichtarge O, Richards JS (2015) Specific TP53 mutants overrepresented in ovarian cancer impact CNV, TP53 activity, responses to nutlin-3a, and cell survival. Neoplasia 17(10):789–803. doi:10.1016/j.neo.2015.10.003
Mafficini A, Simbolo M, Parisi A, Rusev B, Luchini C, Cataldo I, Piazzola E, Sperandio N, Turri G, Franchi M, Tortora G, Bovo C, Lawlor RT, Scarpa A (2016) BRCA somatic and germline mutation detection in paraffin embedded ovarian cancers by next-generation sequencing. Oncotarget 7(2):1076–1083. doi:10.18632/oncotarget.6834
Rubin AF, Green P (2009) Mutation patterns in cancer genomes. Proc Natl Acad Sci USA 106(51):21766–21770. doi:10.1073/pnas.0912499106
Beltrame L, Di Marino M, Fruscio R, Calura E, Chapman B, Clivio L, Sina F, Mele C, Iatropoulos P, Grassi T, Fotia V, Romualdi C, Martini P, Noris M, Paracchini L, Craparotta I, Petrillo M, Milani R, Perego P, Ravaggi A, Zambelli A, Ronchetti E, D’Incalci M, Marchini S (2015) Profiling cancer gene mutations in longitudinal epithelial ovarian cancer biopsies by targeted next-generation sequencing: a retrospective study. Ann Oncol 26(7):1363–1371. doi:10.1093/annonc/mdv164
Wheler JJ, Parker BA, Lee JJ, Atkins JT, Janku F, Tsimberidou AM, Zinner R, Subbiah V, Fu S, Schwab R, Moulder S, Valero V, Schwaederle M, Yelensky R, Miller VA, Stephens MP, Meric-Bernstam F, Kurzrock R (2014) Unique molecular signatures as a hallmark of patients with metastatic breast cancer: implications for current treatment paradigms. Oncotarget 5(9):2349–2354. doi:10.18632/oncotarget.1946
Muller KE, Marotti JD, de Abreu FB, Peterson JD, Miller TW, Chamberlin MD, Tsongalis GJ, Tafe LJ (2016) Targeted next-generation sequencing detects a high frequency of potentially actionable mutations in metastatic breast cancers. Exp Mol Pathol 100(3):421–425. doi:10.1016/j.yexmp.2016.04.002
O’Donovan PJ, Livingston DM (2010) BRCA1 and BRCA2: breast/ovarian cancer susceptibility gene products and participants in DNA double-strand break repair. Carcinogenesis 31(6):961–967. doi:10.1093/carcin/bgq069
Koul A, Loman N, Malander S, Borg A, Ridderheim M (2001) Two BRCA1-positive epithelial ovarian tumors with metastases to the central nervous system: a case report. Gynecol Oncol 80(3):399–402. doi:10.1006/gyno.2000.6085
Anacker DC, Moody CA (2016) Modulation of the DNA damage response during the life cycle of human papillomaviruses. Virus Res. doi:10.1016/j.virusres.2016.11.006
Zhou BB, Elledge SJ (2000) The DNA damage response: putting checkpoints in perspective. Nature 408(6811):433–439. doi:10.1038/35044005
Harper JW, Elledge SJ (2007) The DNA damage response: ten years after. Mol Cell 28(5):739–745. doi:10.1016/j.molcel.2007.11.015
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434(7035):917–921. doi:10.1038/nature03445
Mateo J, Boysen G, Barbieri CE, Bryant HE, Castro E, Nelson PS, Olmos D, Pritchard CC, Rubin MA, de Bono JS (2016) DNA repair in prostate cancer: biology and clinical implications. Eur Urol. doi:10.1016/j.eururo.2016.08.037
McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O’Connor MJ, Tutt AN, Zdzienicka MZ, Smith GC, Ashworth A (2006) Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res 66(16):8109–8115. doi:10.1158/0008-5472.can-06-0140
Ledermann JA, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott C, Meier W, Shapira-Frommer R, Safra T, Matei D, Fielding A, Spencer S, Rowe P, Lowe E, Hodgson D, Sovak MA, Matulonis U (2016) Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: an updated analysis from a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Oncol 17(11):1579–1589. doi:10.1016/s1470-2045(16)30376-x
Moschetta M, George A, Kaye SB, Banerjee S (2016) BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann Oncol 27(8):1449–1455. doi:10.1093/annonc/mdw142
Matulonis UA, Penson RT, Domchek SM, Kaufman B, Shapira-Frommer R, Audeh MW, Kaye S, Molife LR, Gelmon KA, Robertson JD, Mann H, Ho TW, Coleman RL (2016) Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: a multistudy analysis of response rates and safety. Ann Oncol 27(6):1013–1019. doi:10.1093/annonc/mdw133
Oza AM, Cibula D, Benzaquen AO, Poole C, Mathijssen RH, Sonke GS, Colombo N, Spacek J, Vuylsteke P, Hirte H, Mahner S, Plante M, Schmalfeldt B, Mackay H, Rowbottom J, Lowe ES, Dougherty B, Barrett JC, Friedlander M (2015) Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trial. Lancet Oncol 16(1):87–97. doi:10.1016/s1470-2045(14)71135-0
Forster MD, Dedes KJ, Sandhu S, Frentzas S, Kristeleit R, Ashworth A, Poole CJ, Weigelt B, Kaye SB, Molife LR (2011) Treatment with olaparib in a patient with PTEN-deficient endometrioid endometrial cancer. Nat Rev Clin Oncol 8(5):302–306. doi:10.1038/nrclinonc.2011.42
Meehan RS, Chen AP (2016) New treatment option for ovarian cancer: PARP inhibitors. Gynecol Oncol Res Pract 3:3. doi:10.1186/s40661-016-0024-7
Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, Fabbro M, Ledermann JA, Lorusso D, Vergote I, Ben-Baruch NE, Marth C, Madry R, Christensen RD, Berek JS, Dorum A, Tinker AV, du Bois A, Gonzalez-Martin A, Follana P, Benigno B, Rosenberg P, Gilbert L, Rimel BJ, Buscema J, Balser JP, Agarwal S, Matulonis UA (2016) Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. doi:10.1056/NEJMoa1611310
Acknowledgements
Open access funding provided by Austrian Science Fund (FWF). Special thanks to Mag. Gertrude Kainz for improving the use of English in our final manuscript and to Dr. Gerwin Heller for his scientific support. All sequencing was performed in cooperation with the Core Facility Genomics of the Medical University Vienna.
Funding
This work was supported by the Austrian Science Fund (FWF) P26011 (L.K.) and FWF-P 29251-B28 (L.K.) and the European Training Network MSCA-ITN-2015-ETN ALKATRAS No 675712 (L.K.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have declared no conflicts of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.

11060_2017_2459_MOESM3_ESM.jpg
Supplementary Figure S3. Pie chart of the substitutions revealed in the 8 BM samples sequenced in this study. The majority of the 54 substitutions were transitions (purine–purine and pyrimidine–pyrimidine) (JPG 2430 KB)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

{kind=link}
Cite this article
Balendran, S., Liebmann-Reindl, S., Berghoff, A.S. et al. Next-Generation Sequencing-based genomic profiling of brain metastases of primary ovarian cancer identifies high number of BRCA-mutations. J Neurooncol 133, 469–476 (2017). https://doi.org/10.1007/s11060-017-2459-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-017-2459-z