
Abstract
Cupriavidus sp. are model organisms for heavy metal(loid) resistance and aromatic compound’s degradation studies and these characteristics make them a perfect candidate for biotechnological purposes. Bacterial strain S14E4C (identified as Cupriavidus campinensis) was isolated from a playground by enrichment method in a 0.25 mM containing medium. The analysis revealed that this bacterium is able to tolerate high concentrations of heavy metal(loid)s: Cd up to 19.5 mM, Pb to 9 mM, Hg to 5.5 mM and As to 2 mM in heavy metal(loid) salt containing nutrient medium. The whole genome data and analysis of the type strain of C. campinensis CCUG:44526T have not been available so far, thus here we present the genome sequencing results of strain S14E4C of the same species. Analysis was carried out to identify possible mechanisms for the heavy metal resistance and to map the genetic data of C. campinensis. The annotation pipelines revealed that the total genome of strain S14E4C is 6,375,175 bp length with a GC content of 66.3% and contains 2 plasmids with 295,460 bp (GC content 59.9%) and 50,483 bp (GC content 63%). In total 4460 coding sequences were assigned to known functions and 1508 to hypothetical proteins. Analysis proved that strain S14E4C is having gene clusters such as czc, mer, cus, chr, ars to encode various heavy metal resistance mechanisms that play an important role to survive in extreme environments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Extensive use of metals and chemicals in the industrial processes have resulted in accumulation of large quantities of effluents containing toxic heavy metals in the environment, and these effluents pose environmental disposal problems due to their non-degradable and persistent characters [1]. In biological systems, heavy metals have been reported to interact with cell components such as DNA and different proteins, causing DNA damage and block functional groups of important molecules or transport channels. Hence, microorganisms have evolved unique characteristics to tolerate/resist heavy metals by various mechanisms, such as detoxification, bioprecipitation, bioaccumulation, etc., which proved to be an ideal tool in bioremediation of heavy metal contaminated environments. Therefore, understanding the impact of heavy metal(loid)s on microorganisms and mechanisms of metal resistance is crucially important and imperative in order to remove and recover heavy metals from polluted environments.
Cupriavidus is a genus of the family Burkholderiaceae that is well known for its heavy-metal resistance and diverse metabolic capabilities in different niches, especially from heavy metal and organic-chemical contaminated soils [2]. The β-proteobacterium Cupriavidus (formerly Wautersia, Ralstonia) campinensis was isolated first time from Campine, the geographical region of northeast Belgium [3]. This bacterium was found to be highly resistant to heavy metals [3] and following analysis confirmed the ability of degradation 2,4-dichlorophenoxyacetic acid by other strain [4] as well, as the extrachromosomal genetic determinants were transferable to related bacteria. In the present study the whole genome of Cupriavidus campinensis strain S14E4C was sequenced and analysed in detail, as well as heavy metal resistance genes and genomic potentials were characterised.
Materials and methods
Isolation of the bacterial strain
C. campinensis S14E4C was isolated from the heavy metal contaminated playground (GPS coordinates: 48°05′42.0″N 19°47′32.8″E) of Salgótarján (Hungary), a former industrial city. Sample was obtained by scraping away the upper (grassy) surface of the soil with a sterile knife and the upper 10–15 cm thick soil layer was placed into sterile plastic tubes, then transported to the laboratory at 4 °C temperature condition. The sample was enriched with 0.25 mM of Cd2+ salt containing broth (DSM medium 1, without agar) by constantly shaking (270 rpm) for two weeks at room temperature. Isolation of strains was performed in a random manner from the growing colonies by standard dilution plate technique. Bacterial strain S14E4C was maintained at 28–30 °C on nutrient agar medium (DSM medium 1) supplemented with 0.25 mM cadmium (Cd).
DNA isolation and identification
Identification of strain S14E4C was done based on 16S rRNA gene sequencing: the genomic DNA of strain S14E4C was extracted using a DNA extraction kit (DNeasy Power Lyzer Microbial Kit, Qiagen, Germany). To confirm the identities of the isolates the 16S rRNA gene was amplified (PCR) from the extracted genomic DNA using the universal primers 27f (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492r (5′-GGCTACCTTGTT ACGACTT-3′) [5] (LGC Genomics, Berlin, Germany). The 16S rRNA gene sequence of strain S14E4C was compared with references in the EzTaxon database [6] and the NCBI Nucleotide database using BLAST [7] to identify closely related bacteria.
Genome sequencing and assembly
The whole genome shotgun and paired-end sequencing of the strain S14E4C was performed by the Genomics Facility RTSF, Michigan State University (USA), on an Illumina MiSeq platform using the MiSeq standard v2 chemistry. Low quality reads, with excess “N” and low quality score, duplication reads, and adaptor contamination were filtered out from the sequence set. Subsequently, high quality reads were assembled using the SPAdes v3.10.0 assembler in careful mode [8] and the existence of plasmids in the genome was identified by plasmid SPAdes (v3.5.0) tool [9]. The assembly quality was checked by QUAST v2.3 [10] and coverage was calculated by coverage calculator master v0.0.1 (https://github.com/GenomicaMicrob/coverage_calculator).
Genome annotation and analysis
Genome annotation, prediction of genome features and functions were analysed by various tools, such as RAST (Rapid Annotation using Subsystem Technology) [11], PATRIC 3.5.38 [12] and DDBJ Fast Annotation and Submission Tool (DFAST) [13] web interfaced pipelines. The annotation results of tools were combined in order to cover throughout the genome. Additionally, after submission the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) (https://www.ncbi.nlm.nih.gov/genome/annotation_prok/) annotated the genome. Functional genes that were investigated as having possible roles in metabolic pathways were checked by KEGG database [14] on PATRIC 3.5.38. Phylogenetic classification of proteins encoded in the S14E4C genome were based on clusters of orthologues group (COG) functions [15].
Determination of minimum inhibitory concentration (MIC) and antibiotic resistance
MIC values of the 4 heavy metal(loid)s (Cd, Hg, Pb and As) for C. campinensis S14E4C were determined using nutrient medium (DSM medium 1) supplemented with the following heavy metal(loid) salts (CdSO4, HgCl2, Pb(NO3)2 or As2O3, respectively). Analysis has started with 0.25 mM of relevant salt concentrations and after one week the adapted cultures transferred to elevated concentrations (1.5–9 mM for Pb, 0.65–19.5 mM for Cd, 1.46–5.5 mM for Hg, 1–2 mM for As) media. Additionally, heavy metal tolerance of the strain S14E4C was checked by low phosphate Tris-salt mineral medium with heavy metal salt additives (CdSO4, HgCl2, Pb(NO3)2 or As2O3,) in various concentrations (1.5–2.4 mM for Pb, 0.65–2 mM for Cd, 1.5–1.85 mM for Hg, 0.5–1 mM for As).
Antibiotic resistance of strain S14E4C was tested on nutrient agar medium using disk diffusion method of EUCAST regulation (www.eucast.org) Version 8.0 (January 2020).
Degradation of aromatic compounds
The strain was tested for its ability to degrade phenanthrene and naphthalene as aromatic compounds. Cultivation was performed in a 48 ml sterile Bushnell Haas Broth (BHB) containing 2 ml bacterial suspension (50 μl trace element solution, and 1 mg/l phenanthrene and 5 mg/l naphthalene) in sterile 100 ml sealed serum bottle at 24 °C on a magnetic stirrer. Three replicates were tested and the variations were measured after 3 and 7 days by using SPME GC-FID [16,17,18].
Phylogenetic analysis
To determine the phylogenetic relationships among Cupriavidus species, the public and completed 16S rRNA gene sequences of the corresponding Cupriavidus type strains were gathered from the Arb-Silva database [19] and aligned by SINA 1.2.11 aligner in SILVA ACT (Alignment Classification and Tree Service) service before creating Maximum Likelihood (ML) tree. A rooted phylogenetic tree based on 16S rRNA gene sequence similarity of the genera Cupriavidus was created using CIPRES Science Gateway’s MrBayes tool [20] and the closely related bacterium Polynucleobacter cosmopolitanus CIP 109840T (AJ550672) was used as an outgroup. Phylogenetic tree was visualized by FigTree v1.4.4 [21]. In addition, PATRIC [12] presents the reference and representative genomes and uses them as part of the Comprehensive Genome Analysis in phylogenomic analysis. The closest reference and representative genomes have been identified by Mash/MinHash [22]. In order to determine the phylogenetic position of this genome, PATRIC chose global protein families (PGFams) [23] from these genomes. Then these protein sequences were aligned with MUSCLE [24] and the nucleotides were plotted to the protein alignment of each these sequences. The amino acid and nucleotide alignments were linked into a data matrix, and RaxML tool [25] was used to analyse this matrix using quick bootstrapping [26] and produce support values in the tree.
Results and discussion
Since the first isolation of C. campinensis [3], though some of the metabolic pathways and characteristics were revealed, most of them are still unknown due to the lack of whole genome information of the species. Whereas, genome sequence of C. campinensis S14E4C revealed the strain has additional capabilities, such as heavy metal resistance, degradation of aromatic compounds, antibiotic resistance, etc. that enhance its potential for use in biotechnological applications. To predict open reading frames and similarity of the species several annotation and alignment programs were used (PATRIC, RAST, DFAST) and compared to be able to detect genes with high accuracy.
It is indicated that C. campinensis S14E4C adapts and resists to the effect of environmental stresses by functional genes and moreover, genes related to plasmid partitioning and the plasmid initiating (protein RepA) are solid evidence for plasmid existence that perform heavy metal and antibiotic resistance. Through combination of sequencing and comparative genomics our study has provided, for the first time, a comprehensive genomic description of the species Cupriavidus campinensis.
Genomic data of Cupriavidus campinensis S14E4C
The 16S rRNA gene sequence of strain S14E4C (NCBI GenBank accession MK660715) was obtained and BLAST search results based on EzTaxon and GenBank databases both indicated that strain S14E4C belongs to the genus Cupriavidus and it is 100% identical to Cupriavidus campinensis WS2 [3]. The Whole Genome Shotgun project of strain S14E4C has been deposited at DDBJ/ENA/GenBank under the accession VCIZ00000000. The version described in this paper is version VCIZ01000000. The strain S14E4C has been accessioned into the National Collection of Agricultural and Industrial Microorganisms under the accession number NCAIM B.02650.
Genome structure and general features of C. campinensis strain S14E4C
The genome of S14E4C is 6,322,653 bp with a GC content 66.3% after assembly to 52 contigs (contigs shorter than 500 bp were removed) with 78.3 × coverage value. Total of 5968 putative coding sequences (CDSs) were validated by homology and 4460 CDSs that were assigned to one or more function classes (Table 1), whereas, 1508 CDSs are identified as a hypothetical based on function annotation. The draft genome contained 49 tRNA and 7 rRNA genes (including 5S, 16S and 23S rRNA).
Previous studies depicted that known members of the Cupriavidus genus contain 2 large replicons (generally a chromosome and a chromid) and several plasmids [2, 27,28,29,30,31,32]. In the current study the replicons were identified by the PlasmidSPAdes v.3.5 software that the algorithm using contigs’ read coverage information, estimates median coverage, builds assembly graph and generates plasmidic contigs [9]. The analysis resulted in 2 plasmids with the length of 295,460 bp and 50,483 bp, and with the average GC content of 59.9% and 63%, respectively. To validate, the obtained results, the draft genome sequence (52 contigs) and putative chromids/plasmids of S14E4C strain were aligned by Mauve v.2.4 software [33] with whole genome, chromosome and plasmid sequences of the known Cupriavidus species (mainly C. metallidurans) (Supplementary Fig. 1). The alignments clarified the identical genes in Plasmid1 of the S14E4C, exist in both plasmids of the C. metallidurans CH34, whereas only few positions are similar among plasmid 2 and CH34 plasmids (Supplementary Fig. 1, C and D). After the annotation of the plasmid sequences, it is assumed that in the strain S14E4C, the main replicon carried most of the essential housekeeping genes, including those for translation, ribosome production, DNA replication, DNA repair, protein processing, cell component and resistance. Plasmid 1 contains genes mainly encoding mechanisms of heavy metal resistance (cadmium, mercury, copper, zinc, etc.) and membrane cation transport, additionally genes encoding proteins involved in carbohydrate metabolism and c-type cytochrome biogenesis. Whereas, plasmid 2 carries genes for antibiotic resistance (e.g. tetMOPQST) and operon for ribosomal protein synthesis (SSU rRNA, LSU rRNA, 5S rRNA). The existence of the rRNA operon (rrn) on plasmids and chromids was earlier reported on Bacillus and Paracoccus species [34] as well, due to its functional importance.
Genome annotation
Metabolic pathways
The genome of C. campinensis S14E4C consists mostly of known genes encoding metabolic modules and various pathways support its growth. The main and common metabolic genes were shown in Fig. 1. Strain S14E4C just as many bacteria has genetic capacity for nitrogen, sulfur, phosphorus and different carbohydrates metabolism. Among them genes of cyanate hydrolysis (cynRXST operon), nitrate and nitrite ammonification (nrf, nar, nit, nat reductase or transport), nitrate reductase (narRKGHJIA, nirVK, norDQBCFE, nosXLYFDZR) and gene clusters responsible for nitrogen metabolism are also present in its genome.

A circular graphical display of the genome (contains chromosome and plasmid contigs) and applicable genes. This includes CDS on the forward strand, CDS on the reverse strand, RNA genes, Transposase, pseudogene, GC content and GC skew. The figure was prepared by CGView circular genome visualization tool [62]. (Color figure online)
The biochemistry of the bacterial sulfur metabolism pathways is quite complex and encoded by soxABXYZDFCRSWH gene cluster. Basically, sulfur oxidation pathways require only five sox genes whose products form three key periplasmic protein complexes: soxYZ, a sulfur carrier protein, soxXA, a c-type cytochrome complex, and soxB, a sulfate thiol hydrolase [28]. In case of organic sulfur assimilation, alkanesulfonate assimilation and utilization occurs by ssuA—alkanesulfonates-binding, ssuB—alkanesulfonate ABC transporter ATP binding, ssuF—organosulfonate utilization, ssuC—alkanesulfonates transport system permease, ssuD—alkanesulfonate monooxygenase, etc. proteins.
S14E4C implements phosphate metabolism with ptsS (putuative periplasmic phosphate binding protein), ptsA (phosphate transport system permease protein), ptsB (phosphate transport ATP binding protein), oprO and oprP (pyrophosphate and phosphate specific outer membrane porins) genes (Fig. 2).

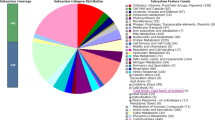
Subsystem coverage and category distribution of whole genome. The pie chart demonstrates the counts for each subsystem feature and the subsystem coverage. Genes for each Subsystem Category were shown in brackets. (Color figure online)
Spectrum of carbohydrate metabolism is broad, but here only few of them are mentioned. Such as, several operons are responsible for maltose and maltodextrin utilization (malEFGKMPRAZ) and mannose metabolism (manYZBCEFGKL, mtpEFGKL). However, based on the annotation results, compared to previously identified Cupriavidus species the metabolism of ketogluconates (some can serve as the sole source of carbon and energy for various bacteria) is quite different in the Cupriavidus campinensis S14E4C.
In C. campinensis strain S14E4C the process proceeds via conversion to 6-phosphogluconate, which is further metabolized through the Entner-Doudoroff (EDP) and/or Pentose Phosphate pathways (PPP). The pathway is encoded by pglDH gene, whereas, there is no gene for 6-phosphogluconate dehydrogenase (6-PGDH) in any earlier sequenced Cupriavidus species [28] (e.g. C. metallidurans, C gilardii, C. necator, etc.).
Genes/gene clusters of heavy metal(loid) resistance (HMR)
Referring to the genome annotation analysis Cupriavidus campinensis strain S14E4C possesses extensive number of heavy metal(loid) resistant genes and gene clusters (Table 2). Comparing to the metal resistant bacterium Cupriavidus metallidurans CH34, strain S14E4C carries many typical metal resistance clusters, such as czcABC, copCBA, etc.[28]. However, for various metal tolerant strains metal resistance mechanisms can differ slightly. For instance, alternate cation-specific mechanisms of Zn2+, Cd2+ and Co2+ resistance and transport encoded by the same genes (czcRDABC) [35].
The genome also carries system genes and clusters involved in the transport and resistance of Cd2+, Pb2+, Ni2+, Co2+, such as nccAB, nikABCDEKLMNOQR, cbtACDFGJKLX, ctpD, plasmid-mediated trcD, etc. Unlike the CH34, pbr operon is absent in S14E4C, but previous researches indicate that heavy metal-(Cd, Co, Pb, Zn)-translocating P-type ATPase genes can perform Pb2+ resistance as well [36]. However, the mechanisms of Hg and As resistance is different than Cd, Pb, Zn resistance. Mercuric ions are toxic to bacteria because they bind to sulfhydryl groups and hinder macromolecule synthesis and enzymatic functions. The Hg2+ resistance system at strain S14E4C consisted of merR gene, activates transcription of mer operon (merRTPCADE) in elevated concentrations of mercury, and genes encode the resistance to mercury (Hg) is a well-known property of both Gram-positive and Gram-negative bacteria that generally locate in plasmids [37, 38]. The operon is located on contig 12 and delimited by transposon sequences (Figs. 3, 4). The merC, merT, merE and merP genes function as membrane or periplasmic transport of organic and inorganic Hg2+ (Table 2). The merA and merB genes (merB) responsible for demethylation of organic mercury compounds by cleavage C-Hg bonds, encoding mercuric reductase and the enzyme organomercurial lyase directly followed by genes encoding transport and transcriptional regulators. The other gene encoding organomercury resistance, merD, a secondary regulatory protein, also binds the same region as merR, involved in transcriptional regulation [39, 40].

Mercury resistance gene cluster: The chromosomal region of the focus gene (top) is compared with three similar organisms. The graphic depicts the focus gene, which is red and numbered 1. Sets of genes with similar sequence are grouped with the same number and colour (1- mercuric ion reductase merA; 2- TnpA transposase (left), Transposase Tn3 (right); 3- periplasmic mercury (2 +) binding protein merP; 4- mercuric transport protein merT; 5- transcriptional regulator merR family; 6- mercuric resistance operon coregulator merD; 7- mercuric transport protein merC; 8- mercuric transport protein merE; 9- DNA-invertase). Genes whose relative position is conserved in at least three other species are functionally coupled and share grey background boxes

Phylogenetic relationship of Cupriavidus campinensis S14E4C strain and the species of Cupriavidus based on 16S rRNA gene sequence. Cluster analysis was based upon the neighbour-joining method with Polynucleobacter cosmopolitanus CIP 109840T (AJ550672) as the outgroup root. The MrBayes method were used to generate the tree and its support values (only values above 50% are presented). Bar, 0.02 substitutions per nucleotide position. Tree was visualised by FigTree v1.4.4
In addition to the cluster elements, several ars gene homologs (arsB, arsC, arsR, arsH) were identified in the genome of Cupriavidus campinensis S14E4C for arsenate (AsO43−) resistance and the strategy followed by bacteria depend on the arsenate reductase (ArsC) protein. In the existing operon (arsRBC) the arsC, arsenate reductase is able to transform the arsenate to arsenite and the rest of the process is encoded by arsB, an integral membrane protein, to prohibit arsenic accumulation by expelling out of the cytoplasm [41, 42]. Meanwhile, the metalloregulatory protein ArsR (encoded by arsR) enables the transcription of the operon by attaching the promoter region [43, 44]. The strain can oxidize methyl arsenate compounds (by arsH gene) that contributes to the global biotransformation of arsenic [45]. Among the bacterial species, the arsH is widely distributed in Proteobacteria (not present in Gram-positive) and similarly to other ars genes locates mostly in chromosome [44, 46].
Encoded proteins for the efflux system of cations including chromate (CrO42−) are distributed in the genome of S14E4C as a cluster chrABCEIF, while, available gene clusters, copACDSRZ, cusSARBCF, scsABCD, cutACEF and ycnLKI are encoding proteins that responsible for the uptake, efflux, translocation and periplasmic detoxification of Cu+ and Cu+2 ions. The protein sequences of cusCFBA are similar to the general gene and protein families which pump Cu2+ cations out of cells that had been previously studied. Additionally, except the Cu2+ cations, the cusCBA genes are encoding Ag+ efflux system as well (Table 2). Another gene responsible for Ag+ resistance is putative silver efflux pump (SEP). In the environment Cu2+ is more abundant and less toxic than Cu+ and the presence of protein copA, P-type Cu+ efflux ATPase, controlling the intracellular Cu+ level, whereas, the extracellular periplasmic space is defended by cusCFBA multicomponent efflux transport system [47]. The chemiosmotic carrier cusCFBA RND efflux has been examined in details [47] and the cusF protein displayed as a key periplasmic copper binding protein [48].
It can be concluded that metal(loid) detoxification in C. campinensis S14E4C primarily occurs by efflux system(s): RND (Resistance, Nodulation, cell Division) family of permeases, the CDF (Cation Diffusion Facilitator) family of heavy metal transporters and the P-type ATPase family of ion pumps may operate on a variety of cations [49], including Cd2+, Zn2+, Pb2+, Ni2+, CrO42−, Ag+, Co2+, Cu2+, Cu+, Hg2+, and likely on other heavy metals (Table 2).
MIC (Minimum Inhibitory Concentration) results of Cupriavidus campinensis S14E4C
In order to verify metal resistance capability of the strain S14E4C MIC assessment was performed and C. campinensis S14E4C expressed high tolerance/resistance to all four tested heavy metals. The nutrient and Tris-salt mineral medium indicated significant difference in terms of growth inhibition (Table 3). Resistance to heavy metal(loid)s in nutrient media was extremely high most probably due to the adaptation period. Meanwhile, though resistance to HM of the strain S14E4C in low phosphate Tris-salt mineral medium was lower, these values are not considerably high (2 mM Cd2+, 2.4 mM Pb2+, 1.84 mM Hg2+, 1 mM As3+) compared to other Cupriavidus species, though the mercury resistance is noticeable (1.84 mM).
Antibiotic resistance
The Genome Annotation Service in PATRIC uses k-mer-based ABR genes detection method [12] that assigns antibiotic resistance genes and their functions, though, the existence of ABR-related genes in a genome does not directly mean antibiotic resistance phenotypically. A summary of the ABR genes annotated in this genome and related ABR mechanism [51] is given in Table 4. On the basis of genome sequencing results C. campinensis S14E4C is resistant to D-cysteine amino acids and Beta-lactam group antibiotics with around 21 resistance genes. D-cysteine is decomposed into pyruvate, H2S, and NH3 by D-cysteine desulfohydrase. Decomposition of D-cysteine by the desulfohydrase (dcyD gene) produces H2S, which the bacterium can use as a sulfur source. In S14E4C strain the desulfhydrase appears in a cluster with two proteins (CAP, CPP), probably involved in transport processes, and a gene encoding a periplasmic cystine-binding protein (CBP). Additionally, blc, bli, blR, blaI, blaR resistance genes exist in the genome of the S14E4C for resistance of β-lactam group antibiotics. These antibiotics include penicillin, cephalosporin, cephamycin and carbapenem, supply multi-resistance by breaking the antibiotics’ structure. C. campinensis S14E4C carries β-lactam resistance genes that responsible for penicillin binding, repression, hydrolase and regulation [52] to have become effective at their function as antibiotic resistance enzymes. The inner membrane chemiosmotic pump protein acrB and a substitute for the bridging protein acrA are the homologs of Pseudomonas mexA and mexB [53], where C. campinensis S14E4C contains all of these 4 genes. Other ABR genes were shown on Table 4.
The test with 9 antibiotic agents revealed that the strain S14E4C can resist to the number of antibiotics (Table 5). In 2006, it was reported by Baker-Austin and co-workers that heavy metal pollution increases the metal resistance and reduce antibiotic sensitivity due to co-regulation of genes [54, 55]. Hence, we assume that the isolation of S14E4C from the heavy metal polluted industrial city promoted the heavy metal and antibiotic resistance as well.
Metabolism of aromatic compounds
Annotation results identified also the presence of genes encoding enzymes involving in the utilization of various aromatic compounds in C. campinensis S14E4C as a source of carbon and energy. The pathway genes that are found in this strain include, benzoate degradation (benBACDKEF operon) and transport (bt1254 transporters) [56], salicylate ester degradation (salARED, areABCR), additionally, gentisate catabolism and degradation, N-heterocyclic aromatic compounds degradation (OQD, IQOb), etc.
The degradation capacity of our strain was measured connected to phenanthrene and naphthalene and after one week of incubation it was not detectable. It is assumed that the isolation environment (not contaminated by aromatic compounds) of the strain plays direct role in these results, however, the genes for the possible degradation of aromatic compounds could be activated after a long time exposure [57, 58].
Phylogenetic analysis
Phylogenetic analysis based on 16S rRNA gene sequence suggested that C. campinensis S14E4C strain is a member of Cupriavidus genus and its closest relative was C. campinensis LMG 1195T (AF312020) (Fig. 5). Besides, strain S14E4C phylogenetically close to C. gilardii and C. pampae with 98% similarity value. Additionally, some other core genes (e.g. gyrB, rpoD, recA, etc.) were concatenated with the 16S rRNA gene and analysed on PATRIC [12]. In fact, the precise phylogenetic position of C. campinensis S14E4C was placed and depicted in Fig. 5 and these results supported by the tree generated on TYGS database (based on genome signatures) that depicted similar results (Fig. 6) [59]. The genome-wide Average Nucleotide Identity (gANI) value between strain S14E4C and C. metallidurans CH34 was identified [60] as 81.98% to confirm the genomic relatedness. This method suggests firm resolution amidst closely linked genomes (80–100% ANI) [61].

Phylogenomic tree of the Cupriavidus campinensis S14E4C based on concatenation of 16S rRNA gene with core genes (e.g. gyrB, rpoD, recA, etc.). Tree was built on PATRIC online pipeline

Phylogenomic tree predicted on TYGS database. Genomic G + C content (63.53–68.47%), δ values (0.08–0.2), overall genome sequence length (5,783,696–9,185,558 bp), number of proteins (5142–7932). Values increase based on the colour range (from white to black) [59]
References
Tchounwou PB, Yedjou CG, Patlolla AK, Sutton DJ (2013) Molecular, clinical and environmental toxicology: v.2: clinical toxicology. Choice Rev 47(10):5683
Hong KW, Thinagaran D, Gan HM, Yin WF, Chan KG (2012) Whole-genome sequence of cupriavidus sp. Strain BIS7, A heavy-metal-resistant bacterium. J Bacteriol 194(22):6324
Goris J et al (2001) Classification of metal-resistant bacteria from industrial biotopes as Ralstonia campinensis sp. nov., Ralstonia metallidurans sp. nov. and Ralstonia basilensis Steinle et al. 1998 emend. Int J Syst Evol Microbiol 51(5):1773–1782
Han L, Zhao D, Li C (2015) Isolation and 2, 4-D-degrading characteristics of Cupriavidus campinensis BJ71. Braz J Microbiol 441:433–441
Szuróczki S et al (2019) Arundinibacter roseus gen. nov., sp. nov., a new member of the family Cytophagaceae. Int J Syst Evol Microbiol 69:2076–2081
Yoon SH et al (2017) Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int J Syst Evol Microbiol 67(5):1613–1617
States D, Gish W (1994) Combined use of sequence similarity and codon Bias for coding region identification. J Comput Biol 1(1):39–50
Bankevich A et al (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19(5):455–477
Antipov D, Hartwick N, Shen M, Raiko M, Pevzner PA (2016) plasmidSPAdes : assembling plasmids from whole genome sequencing data, pp 1-7
Gurevich A, Saveliev V, Vyahhi N, Tesler G (2013) QUAST : quality assessment tool for genome assemblies. Bioinformatics 29(8):1072–1075
Aziz RK et al (2008) The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:1–15
Wattam AR et al (2017) Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res 45(D1):D535–D542
Tanizawa Y, Fujisawa T, Nakamura Y (2018) DFAST: a flexible prokaryotic genome annotation pipeline for faster genome publication. Bioinformatics 34(6):1037–1039
Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K (2017) KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 45(D1):D353–D361
Tatusov EV, Galperin RL, Natale MY, Koonin DA (2000) The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28(1):33–36
Di Dong C, Chen CF, Chen CW (2012) Determination of polycyclic aromatic hydrocarbons in industrial harbor sediments by GC-MS. Int J Environ Res Public Health 9(6):2175–2188
Hong-Wen C (2004) Determination of polycyclic aromatic hydrocarbons in water by solid phase microextraction and liquid chromatography. Anal Sci 20(2):1383–1388
Rawa-Adkonis M, Wolska L, Przyjazny A, Namieśnik J (2006) Sources of errors associated with the determination of PAH and PCB analytes in water samples. Anal Lett 39(11):2317–2331
Yilmaz P et al (2014) The SILVA and ‘all-species Living Tree Project (LTP)’ taxonomic frameworks. Nucleic Acids Res 42(D1):643–648
Ronquist F et al (2012) Mrbayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61(3):539–542
Patrício AR et al (2012) Global phylogeography and evolution of chelonid fibropapilloma-associated herpesvirus. J Gen Virol 93(5):1035–1045
Ondov BD et al (2016) Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol 17(1):1–14
Davis JJ et al (2016) PATtyFams: protein families for the microbial genomes in the PATRIC database. Front Microbiol 7:1–12
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32(5):1792–1797
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313
Stamatakis A, Hoover P, Rougemont J (2008) A rapid bootstrap algorithm for the RAxML web servers. Syst Biol 57(5):758–771
Amadou C et al (2008) Genome sequence of the β-rhizobium. Genome Res 18:1472–1483
Janssen PJ et al (2010) The complete genome sequence of Cupriavidus metallidurans strain CH34, a master survivalist in harsh and anthropogenic environments. PLoS ONE 5(5):e10433
Poehlein A, Kusian B, Daniel R, Bowien B (2011) Complete genome sequence of the type strain Cupriavidus necator N-1. J Bacteriol 193(18):3912181
Wang X et al (2015) Genome sequence analysis of the naphthenic acid degrading and metal resistant bacterium cupriavidus gilardii CR3. PLoS ONE 10(8):1–21
Cuadrado V, Gomila M, Merini L, Giulietti AM, Moore ERB (2010) Cupriavidus pampae sp. nov., a novel herbicide-degrading bacterium isolated from agricultural soil. Int J Syst Evol Microbiol 60(11):2606–2612
Moriuchi R, Dohra H, Kanesaki Y, Ogawa N (2019) Complete genome sequence of 3-chlorobenzoate-degrading bacterium Cupriavidus necator NH9 and reclassification of the strains of the genera cupriavidus and ralstonia based on phylogenetic and whole-genome sequence analyses. Front Microbiol 10:1–21
Darling ACE, Mau B, Blattner FR, Perna NT (2004) Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14:1394–1403
Anda M et al (2015) Bacterial clade with the ribosomal RNA operon on a small plasmid rather than the chromosome. Proc Natl Acad Sci USA 112(46):14343–14347
Chizzola R, Mitteregger US (2005) Cadmium and zinc interactions in trace element accumulation in chamomile. J Plant Nutr 28(8):1383–1396
Naik MM, Dubey SK (2013) Lead resistant bacteria: lead resistance mechanisms, their applications in lead bioremediation and biomonitoring. Ecotoxicol Environ Saf 98:1–7
Gupta A, Phung LT, Chakravarty L, Silver S (1999) Mercury resistance in Bacillus cereus RC607: transcriptional organization and two new open reading frames. J Bacteriol 181(22):7080–7086
Nascimento AMA, Chartone-Souza E (2003) Operon mer: bacterial resistance to mercury and potential for bioremediation of contaminated environments. Genet Mol Res 2(1):92–101
Brown NL, Stoyanov JV, Kidd SP, Hobman JL (2003) The MerR family of transcriptional regulators. FEMS Microbiol Rev 27(2–3):145–163
Naguib MM, El-Gendy AO, Khairalla AS (2018) Microbial diversity of mer operon genes and their potential rules in mercury bioremediation and resistance. Open Biotechnol J 12(1):56–77
Yang HC, Fu HL, Lin YF, Rosen BP (2012) Pathways of arsenic uptake and efflux. Elsevier, Amsterdam
Zhu Y-G, Yoshinaga M, Zhao F-J, Rosen BP (2014) Earth abides arsenic biotransformations. Annu Rev Earth Planet Sci 42(1):443–467
Busenlehner LS, Pennella MA, Giedroc DP (2003) The SmtB/ArsR family of metalloregulatory transcriptional repressors: structural insights into prokaryotic metal resistance. FEMS Microbiol Rev 27(2–3):131–143
Ben Fekih I et al (2018) Distribution of arsenic resistance genes in prokaryotes. Front Microbiol 9:1–11
Yang HC, Rosen BP (2016) New mechanisms of bacterial arsenic resistance. Biomed J 39(1):5–13
Páez-Espino AD, Durante-Rodríguez G, de Lorenzo V (2015) Functional coexistence of twin arsenic resistance systems in Pseudomonas putida KT2440. Environ Microbiol 17(1):229–238
Gudipaty SA, Larsen AS, Rensing C, Mcevoy MM (2012) Regulation of Cu(I)/Ag(I) efflux genes in Escherichia coli by the sensor kinase CusS. FEMS Microbiol Lett 330(1):30–37
Franke S, Grass G, Rensing C, Nies DH (2003) Molecular analysis of the copper-transporting efflux system CusCFBA of Escherichia coli. J Bacteriol 185(13):3804–3812
Hu N, Zhao B (2006) Key genes involved in heavy-metal resistance in Pseudomonas. FEMS Microbiol Lett 267:1–6
Monsieurs P et al (2011) Heavy metal resistance in Cupriavidus metallidurans CH34 is governed by an intricate transcriptional network. Biometals 24(6):1133–1151
McArthur AG et al (2013) The comprehensive antibiotic resistance database. Antimicrob Agents Chemother 57(7):3348–3357
Massova I, Mobashery S (1998) Kinship and diversification of bacterial penicillin-binding proteins and β-lactamases.pdf. Antimicrob. Agents Chemother 42(1):1–17
Morita Y, Tomida J, Kawamura Y (2012) Mexxy multidrug efflux system of Pseudomonas aeruginosa. Front Microbiol 3:1–13
Baker-Austin C, Wright MS, Stepanauskas R, McArthur JV (2006) Co-selection of antibiotic and metal resistance. Trends Microbiol 14(4):176–182
Seiler C, Berendonk TU (2012) Heavy metal driven co-selection of antibiotic resistance in soil and water bodies impacted by agriculture and aquaculture. Front Microbiol 3:1–10
Denef VJ et al (2004) Biphenyl and benzoate metabolism in a genomic context: outlining genome-wide metabolic networks in Burkholderia xenovorans LB400. Appl Environ Microbiol 70(8):4961–4970
Yan S, Wu G (2017) Reorganization of gene network for degradation of polycyclic aromatic hydrocarbons (PAHs) in Pseudomonas aeruginosa PAO1 under several conditions. J Appl Genet 58(4):545–563
Berthoumieux S et al (2013) Shared control of gene expression in bacteria by transcription factors and global physiology of the cell. Mol Syst Biol 9(634):1–11
Meier-Kolthoff JP, Göker M (2019) TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat Commun 10(1):1–10
Yoon SH, Ha SM, Lim J, Kwon S, Chun J (2017) A large-scale evaluation of algorithms to calculate average nucleotide identity, Antonie van Leeuwenhoek. Int J Gen Mol Microbiol 110(10):1281–1286
Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S (2018) High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun 9(1):1–8
Stothard P, Wishart DS (2005) Circular genome visualization and exploration using CGView. Bioinformatics 21(4):537–539
Acknowledgements
Open access funding provided by Eötvös Loránd University (ELTE). The study was funded by the ELTE Institutional Excellence Program supported by the National Research, Development and Innovation Office (NKFIH-1157-8/2019-DT). We are thankful to Zsuzsa Kéki for the test of aromatic compounds degradation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11033_2020_5490_MOESM1_ESM.png
Supplementary Figure 1. Syntheny plot analysis of the S14E4C sequence vs known Cupriavidus species and their replicons. (A) Syntheny plot of C. campinensis S14E4C vs C. gilardii CR3, C. necator N-1, C. metallidurans CH34 (PNG 433 kb)
11033_2020_5490_MOESM2_ESM.png
Supplementary Figure 1. Syntheny plot analysis of the S14E4C sequence vs known Cupriavidus species and their replicons. (B) Chromosome from C. metallidurans CH34 vs C. campinensis S14E4C whole genome (PNG 433 kb)
11033_2020_5490_MOESM3_ESM.png
Supplementary Figure 1. Syntheny plot analysis of the S14E4C sequence vs known Cupriavidus species and their replicons. (C) Megaplasmid from C. Metallidurans CH34 vs C. campinensis S14E4C whole genome (PNG 433 kb)
11033_2020_5490_MOESM4_ESM.png
Supplementary Figure 1. Syntheny plot analysis of the S14E4C sequence vs known Cupriavidus species and their replicons. (D) Plasmid pMOL28 and pMOL30 from C. metallidurans vs plasmid 1 and plasmid 2 from C. campinensis S14E4C (PNG 433 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Abbaszade, G., Szabó, A., Vajna, B. et al. Whole genome sequence analysis of Cupriavidus campinensis S14E4C, a heavy metal resistant bacterium. Mol Biol Rep 47, 3973–3985 (2020). https://doi.org/10.1007/s11033-020-05490-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-020-05490-8