
Abstract
The interleukin-1 receptor like ST2 has emerged as a potential drug discovery target since it was identified as the receptor of the novel cytokine IL-33, which is involved in many inflammatory and autoimmune diseases. For the treatment of such IL-33-related disorders, efforts have been made to discover molecules that can inhibit the protein–protein interactions (PPIs) between IL-33 and ST2, but to date no drug has been approved. Although several anti-ST2 antibodies have entered clinical trials, the exploration of small molecular inhibitors is highly sought-after because of its advantages in terms of oral bioavailability and manufacturing cost. The aim of this study was to discover ST2 receptor inhibitors based on its PPIs with IL-33 in crystal structure (PDB ID: 4KC3) using virtual screening tools with pharmacophore modeling and molecular docking. From an enormous chemical space ZINC, a potential series of compounds has been discovered with stronger binding affinities than the control compound from a previous study. Among them, four compounds strongly interacted with the key residues of the receptor and had a binding free energy < − 20 kcal/mol. By intensive calculations using data from molecular dynamics simulations, ZINC59514725 was identified as the most potential candidate for ST2 receptor inhibitor in this study.
Graphical abstract

Similar content being viewed by others


Introduction
Interleukin (IL)-33 is a new member of the IL-1 superfamily that plays important roles in human innate and adaptive immune responses via interaction with its ST2 receptor [1]. IL-33 activates a variety of immune cells involved in type 2 immune responses and allergic inflammations, including T helper (TH2) cells, mastocytes, eosinophils, basophils, type 2 innate lymphoid cells (ILC2s), regulatory T cells (Tregs), dendritic cells and alternatively activated macrophages (AAMs) [2]. ST2 stands for “suppression of tumorigenicity 2” [3] and belongs to the interleukin-1 receptor (IL-1R) family. ST2 was an orphan receptor for many years and has been extensively researched in the context of inflammatory and autoimmune diseases. IL-33 can now be considered a functional and specific ligand of the ST2 receptor. ST2 exists in two major isoforms, including a membrane-bound form (ST2L) and a soluble form (sST2). ST2L is constitutively expressed primarily on the surface of hematopoietic cells and combines with the interleukin-1 receptor accessory protein (IL-1RAcP) to form the transmembrane IL-33 receptor [4, 5].
ST2 release in response to myocardial infarction has long been used as a biomarker for diagnosis [3, 6]. Recently, the IL-33/ST2 pathway has been shown to be strongly involved in inflammatory and autoimmune diseases. ST2 plays a critical part in controlling airway inflammations and relates to TH2-mediated allergic inflammations inducing acute exacerbations in asthmatic patients [7]. In these patients, levels of ST2 and IL-33 were found to increase in sera and tissues [7, 8]. IL-33 acts as an epithelial-mesenchymal cytokine that binds to ST2 receptor and controls airway inflammatory responses; thus, the IL-33/ST2 pathway provides a potential target for asthma treatment [8]. Besides, ST2 is associated with inflammatory bowel diseases such as ulcerative colitis or Crohn’s disease, where the colonic mucosa sST2 level is significant higher in patients with active disease [9, 10]. IL-33 and ST2 receptor were also found to be strongly expressed in the vicinity of β-amyloid plaques and in brain glial cells of patients with Alzheimer’s disease [11]. For autoimmune conditions, ST2 plays a pivotal role in the development of various pathologies such as rheumatoid arthritis, systemic lupus erythematosus and multiple sclerosis [12,13,14].
Recently, when the COVID-19 pandemic raged and infected more than 250 million people with more than five million deaths [15], IL-33 and ST2 were also investigated for their association with this disease. IL-33 is hypothesized to be the cause of the imperfect cytokine storm in COVID-19. The IL-33/ST2 axis might decrease responses of antiviral interferons and induce viral inflammation and thrombosis in severe patients. IL-33 might drive pulmonary fibrosis in patients who survive severe COVID-19 [16].
Currently, no drug that inhibits the ST2 receptor or its IL-33 ligand is approved by the FDA for the treatment of related diseases. However, a number of studies to discover inhibitors of the IL-33/ST2 pathway, including biologics and small-molecule drugs, have been published. For almost a decade, reports seem to focus more on IL-33, including IL-33 neutralizing antibodies, IL-33 trap (complex of recombinant ST2 and IL-1RAcP), anti-IL-33 proteins and small molecules [17,18,19,20,21,22]. Recently, the ST2 receptor has become an attractive target for drug discovery. An sST2-neutralizing monoclonal antibody has been introduced that can reduce inflammation and decrease acute graft-versus-host disease severity and mortality [23]. In particular, another anti-ST2 monoclonal antibody CNTO 7160 has been clinical studied in healthy subjects and patients with asthma or atopic dermatitis [24].
With the increasingly elucidated role of the ST2 receptor, the discovery of inhibitors for this receptor is essential. Small molecule therapy has several advantages over biological therapeutic agents, including easier administration, especially orally, superior tissue penetration, modifiable pharmacokinetic properties, lower production costs and higher uniformity between batches. Ramadan A. M. et al. have identified several small molecular inhibitors for the ST2 receptor with micromolar IC50 by high-throughput screening from a database of 77,701 compounds [25]. In this study, we introduce an attempt to discover novel and stronger small molecular inhibitors for the ST2 receptor from a larger chemical space with millions of compounds using computational methods. By using rapid virtual screening methods including pharmacophore approach and molecular docking combined with intensive bioinformatics methods such as molecular dynamics simulations and MM/GBSA binding free energy calculations, a number of potential compounds have been identified with better binding modes and stronger affinities on the ST2 receptor than control compounds from the previous study.
Materials and methods
Database and virtual screening process
For virtual screening of small molecular inhibitors for the ST2 receptor, a workflow outlined in Fig. 1 was adopted. In this process, the ZINC database [26, 27] was used as a chemical space to search for compounds with the ability to bind to the target. ZINC12 is a large library of more than 22 million commercially available compounds with complete supplier information. Nevertheless, with such a huge number of compounds, searching for biologically active candidates on this library requires an enormous amount of computing resources to generate conformations for each compound in the library. Luckily, Koes D. R. and Carlos J. C. have developed the ZINCPharmer tool for fast and efficient virtual screening through the ZINC library (http://zinc12.docking.org) with more than available 200 million conformations [28]. After screening through the 3D-pharmacophore models, the compounds that satisfied the model were subsequently screened by molecular docking. Ligands were prepared by energy minimization using Molecular Operating Environment (MOE) 2015.10 software [29] with Amber10:EHT forcefield and energy gradient was set to RMS of 0.0001 kcal/mol/A2 to obtain ready-to-dock structures. After being ranked by docking score and binding mode, the most potential compounds bound to the ST2 receptor were explored through molecular dynamics simulated in complex with the protein to investigate their stability of binding and calculate the binding free energy using the MM/GBSA method. Furthermore, in order to better evaluate the potential compounds screened in this in silico study, a control compound iST2-1 was used. iST2-1 is a small molecular inhibitor of ST2 receptor discovered by Ramadan A. M. et al. with experimental IC50 value of 47.7 ± 5.0 μM [25].

Flowchart of virtual screening and computational analysis for discovery process of inhibitors for the ST2 receptor. The database, software and web tools used in each step are shown in parentheses
Pharmacophore approach
In this study, the inhibitors for the ST2 receptor were designed using a structure-based 3D-pharmacophore approach because there is currently no published co-crystallized structure of ST2 with its small molecular ligand. Therefore, 3D-pharmacophore models were constructed based on the interactions between the ST2 receptor with IL-33 in the protein complex with the PDB ID 4KC3 [1] at 3.27 Å resolution. This complex contains the extracellular domain of human ST2L expressed on the membranes of immune cells (Lys19 to Lys231) and the mature IL-33 with cytokine activity (Ser112 to Thr270). The two proteins bind to each other via Site 1 and Site 2 (Fig. 2A). Previous studies have shown that the Site 1 is highly conserved because of the rigidity of the domain 1 and 2. IL-33 binds to the ST2 receptor first at the Site 1 composed of these two domains and leads to a conformational change of the domain 3. As a result, the Site 2 on the domain 3 of the ST2 receptor is more flexible than the Site 1 [1]. Therefore, this study focused on discovery of small molecular inhibitor for the ST2 receptor at Site 1. At this binding interface between the two proteins, the ST2 receptor interacts with IL-33 through residues shown in detail in Table S1. Using surface plasmon resonance (SPR), Liu X. et al. identified the residues that play an important role (hotspot residues) in this protein–protein interaction (PPI). When mutating the residues Glu144, Glu148, Asp149 and Asp244 on IL-33 (isolated and highlighted in Fig. 2B), the binding affinity constant between the ST2 receptor and IL-33 is markedly reduced [1]. Based on these four hotspot residues on IL-33, the seven corresponding key residues on ST2 were identified and are highlighted in Fig. 2B, including Lys22, Arg35, Arg38, Gln39, Tyr119, Thr135 and Agr198.

Protein–protein interactions between the ST2 receptor and its ligand IL-33. A X-ray co-crystallized structure of ST2/IL-33 complex (PDB ID: 4KC3) with magnified binding site 1. B Isolated key residues on the binding interface of the ST2 receptor (in magenta) and IL-33 (in green)
The 3D-pharmacophore models were built using the Pharmacophore Query Editor tool in MOE 2015.10 software [29]. The pharmacophore hypothesis for ST2 receptor inhibitor was built based on two different strategies. The first method directly uses PPI between the ST2 receptor and IL-33 in Fig. 2B to generate the corresponding pharmacophore features. The described PPI-based drug design approach has led to the successful discovery for several drugs on the market [30, 31]. The second approach favors a de novo design, in which the pharmacophore model is constructed based on the observation of key residues on the interaction surface of the ST2 receptor and IL-33. This second approach was used to increase the success rate of finding inhibitors for this receptor. The obtained 3D-pharmacophore models were then uploaded to the ZINCPharmer tool (http://zincpharmer.csb.pitt.edu) [28] for high-throughput virtual screening of small molecules with necessary steric and electronic features to ensure the optimal supramolecular interactions with ST2, thereby potentially inhibiting IL-33 activity by competing with this cytokine on the receptor.
Molecular docking
Because the structure of the ST2 receptor in the 4KC3 complex was missing some residues due to weak electron densities, a homology model was built using the I-TASSER Online Server (https://zhanglab.ccmb.med.umich.edu/I-TASSER) [32, 33] to reconstruct the structure of the receptor with the full range of amino acids. The homology structure was built based on the template of the ST2 receptor itself in the 4KC3 complex (chain B) [1]. The obtained homology models were evaluated by the values calculated and provided by I-TASSER including C-score, TM-score, Cluster Density and RMSD values to choose the best structure [34]. In addition, the finest homology model was also evaluated and compared with the crystal structure by the Ramachandran plot [35], Clashscore and MolProbity score calculated by MolProbity [36, 37]. The best homology structure of ST2 receptor was prepared using QuickPrep tool in MOE 2015.10 software including the steps of hydrogen addition, protonate, tether and refinement and saved as *.pdb format. This prepared structure was then fed into the BioSolveIT LeadIT 2.1.8 software [38] to create the binding site. As no co-crystallized ligand with the ST2 receptor has yet been identified, the key residues were used as references to generate the binding site model. They were loaded into the docking program, and a binding site was generated by being expanded with a suitable radius. Finally, a molecular docking model was built and saved as *.fxx format to be applied for virtual screening.
The FlexX program embedded in LeadIT is used for molecular docking. The compounds, after being screened through 3D-pharmacophore models, were gathered into a library of ready-to-dock structures and imported into the LeadIT software. In this study, ligand binding was driven by a hybrid approach: enthalpy (classic triangle matching) and entropy (single interaction scan). The number of poses to keep was Top10, maximum number of solutions per fragmentation was set to 200, and maximum number of solution per iteration was set to 1000 [39].
The receiver operating curve (ROC) and predictiveness curve (PC) were analyzed with active and inactive compounds to evaluate the molecular docking protocol [40, 41]. The active set contains 10 known ST2 inhibitors (their structures and IC50 values are listed in Table S2) [25]. The inactive set includes 500 decoy structures generated from these 10 inhibitors using the Directory of Useful Decoys (DUD-E) server at http://dude.docking.org [42]. Compounds from the two datasets were docked into the docking model of ST2, and the obtained docking scores were introduced into the Screening Explorer web tool (http://stats.drugdesign.fr) for ROC analysis. The area under the ROC curve (ROC AUC) over 0.5 combined with a standardized total gain (\(\overline{\mathrm{TG} }\)) values (calculated from the PC) over 0.25 signifies that docking score variations are relevant in the discrimination of the active compounds [43].
Ligands that successfully docked into the ST2 receptor were ranked based on the docking score. Besides, detailed ligand–receptor interactions were also analyzed, including hydrogen bonds, ionic bonds, arene–arene interactions, cation–arene interactions, arene–H interactions and van der Waals interactions. In particular, because the association between the ST2 receptor and IL-33 is a PPI, the interaction of ligands with the ST2 key residues has been scrutinized to find the most potential inhibitors that compete against the binding of IL-33 to the receptor. The interaction of each ligand with the ST2 receptor was examined using both the PoseView tool of the LeadIT 2.1.8 software and the Ligand Interactions tool of the MOE 2015.10 software. The analysis and statistics of interaction types between ligands with the key residues were performed using the Protein Ligand Interaction Fingerprints (PLIF) tool of the MOE 2015.10 software.
Molecular dynamics simulations
MD simulations were performed with the GROMACS 2020.6 software [44, 45]. To save computation time, only domains 1 and 2 of the ST2 receptor were carried out MD simulations. These are two highly conserved structural domains and make up the binding site with IL-33. The structure of protein was topology generated with the CHARMM-27 force field. Simultaneously, the conformation of the ligands with the best docking results was saved in *.mol2 format. Their topology was generated using the SwissParam online tool (http://www.swissparam.ch) [46] and updated to the topology file of the protein to get the topology of the complex. The complex is placed in a dodecahedron simulation box with a distance of 10 Å between protein and box edges. The system was filled water solvent (TIP3P model) and electrically neutralized by the addition of an appropriate number of Na+ or Cl− ions (salt concentration was 0.15 M). The constructed system was energy minimized for 100 ps using the steepest descent minimization with a maximum force of 10 kJ/mol. The system was then equilibrated by a NVT simulation for 100 ps to a temperature of 300 K using the velocity rescaling thermostat [47], followed by a NPT equilibration for 100 ps to a pressure of 1 bar using the Parrinello–Rahman barostat [48]. Finally, the production of MD was performed using the velocity Verlet algorithm for the ST2 receptor in apoprotein state and complexes with ligands at a temperature of 300 K and a pressure of 1 bar. The LINCS algorithm was used to restrain the hydrogen bonds [49]. Besides, nonbonded interactions were truncated at 12 Å and the particle mesh Ewald method [50] was used to calculate long-range electrostatic interactions. The MD trajectories were saved every 0.01 ns.
After the MD production had been completed, data from the resulting trajectories were used to calculate the RMSD (root mean square deviation), RMSF (root mean square fluctuation), Rg (radius of gyration) and SASA (solvent-accessible surface area) values by GROMACS built-in commands to evaluate the stability of the complexes and the effect of ligands on the receptor when binding occurs. In particular, the occupancy of hydrogen bonds formation was also analyzed using the VMD software [51] to determine the interacting ability of ligands with the key residues. A hydrogen bond was defined by simple geometric criteria: a distance between hydrogen donor (D) and acceptor (A) atoms of < 3.5 Å and an angle D–H⋯A of > 120° [52]. Other interaction types were detected by the PLIF tool of the MOE 2015.10 software using fingerprint schemes included salt bridges, arene interactions (both with minimum thresholds of 0.5 kcal/mol) and surface contacts (within a minimum of 20 Å2).
Binding free energy calculation and per-residue energy decomposition
The gmx_MMPBSA package was used for calculations based on the single trajectory of GROMACS with CHARMM-27 forcefield [53]. This tool allows free energy calculations using MM/PBSA or GBSA (Molecular Mechanics/ Poisson-Boltzmann or Generalized Born Surface Area) methods. In this work, the snapshots sampled from the MD trajectory of each protein–ligand complex was used to carry out the binding free energy calculation using the MM/GBSA approach because of its low computational resource consumption. The dielectric constant of the solute, temperature and salt concentration were set to 1.0, 298 K and 0.15 M, respectively. The free energy \((\Delta G_{{{\text{bind}}}} )\) for binding of the ligand to ST2 receptor to form the complex was calculated by Eq. (1) [54]:
The Eq. (1) can be decomposed into contributions of different interactions and expressed as:
in which:
where \(\Delta E_{{{\text{MM}}}}\), \(\Delta G_{{{\text{solv}}}}\) and \(- T\Delta S\) are the changes in the gas phase molecular mechanics energy, solvation free energy and conformational entropy upon ligand binding, respectively. \(\Delta E_{{{\text{bond}}}}\), \(\Delta E_{{{\text{angle}}}}\) and \(\Delta E_{{{\text{dihedral}}}}\) are internal bonded energy terms, and they are suppressed for fixed geometries before and after binding. \(\Delta E_{{{\text{vdW}}}}\) and \(\Delta E_{{{\text{ele}}}}\) are the nonbonded van der Waals and electrostatic interaction energy, respectively. \(\Delta G_{{{\text{solv}}}}\) is the sum of the polar contribution \(\Delta G_{{{\text{GB}}}}\) (electrostatic solvation energy) and the nonpolar contribution \(\Delta G_{{{\text{SA}}}}\) between the solute and the continuum solvent. The polar contribution was calculated using GB-OBC1 model [55, 56], while the nonpolar energy was estimated using the solvent-accessible surface area (SASA) [54].
Results and discussion
Pharmacophore model as a rapid virtual screening tool
Two 3D-pharmacophore models were constructed based on PPI of the ST2 receptor with IL-33. The Ph-ST2i-1 model was built by mimicking the properties of hotspot residues on IL-33 to search for small molecular structures that compete against IL-33 for binding to the ST2 receptor. Specifically, the Ph-ST2i-1 model, consisting of five pharmacophore points, accurately mimicked the way that the hotspot residues Glu144, Glu148, Asp149 and Asp244 on IL-33 (Fig. 3A) interact with the ST2 receptor. This model includes 5 features: F1:Ani (anionic), F2:Ani, F3:Ani, F4:Don (hydrogen donor) and F5:Acc (hydrogen acceptor). However, small molecules that are suitable to become drug candidates are often difficult to satisfy a pharmacophore model with too many charged points. Therefore, two anionic points that were far apart in the three-dimensional space F1:Ani and F2:Ani were constrained to “at least one” property.

Two obtained 3D-pharmacophore models generated by A mimicking the hotspot residues of IL-33 and B mimicking the corresponding key residues on the ST2 receptor
In addition, a second 3D-pharmacophore model was built manually as a de novo design, in which the key residues of the ST2 receptor corresponding to the hotspot residues of IL-33 were used, including Lys22, Arg35, Arg38, Gln39, Tyr119, Thr135 and Arg198. Besides, the other residues located in the central of the binding site such as Gln23, Gln39, Met118, Ile136 and Leu138 were also considered for potential interactions. The Pharmacophore Query tool was used to show all potential interactions that can be generated by these residues. Interactions directed to the binding site (where ST2 interacts with IL-33) were selected to create the pharmacophore points according to the “pair-rule” principle. For example, Lys22 has a sidechain − NH2 group that can donor hydrogen bonds (Don), a pharmacophore feature Acc (hydrogen bond acceptor) will be placed on the projection of the nitrogen atom. Using this approach, another 5-point pharmacophore model was built with the features: F1:Acc, F2:Acc, F3:Don, F4:Don, F5:Acc and F6:Acc (Fig. 3B).
These two models were then uploaded to ZINCPharmer [28] to rapidly screen through the ZINC12 library for compounds with appropriate properties to bind the ST2 receptor. A database of more than 200 million conformations was virtually screened through the two pharmacophore hypotheses. This resulted in 1778 compounds satisfying the models, of which 1438 satisfied the Ph-ST2i-1 model (Table S3) and 349 satisfied the Ph-ST2i-2 model (Table S4) (9 compounds satisfied both models). These compounds were further screened by molecular docking as the second funnel of the drug discovery procedure.
Molecular docking model and virtual screening
There were five homology models of ST2 receptor built by I-TASSER Online Server with evaluation parameters presented in Table S5, in which the model HS-1 was assessed as the best structure with C-score, TM-score and cluster density of 0.71, 0.81 ± 0.09 and 0.55, respectively. The ST2 structure in the HS-1 model is superposed with its structure in the crystal complex and shown in Figure S1 with an RMSD of 1.242 Å. The Ramachandra plot of ST2 protein structure in the 4KC3 complex and in the homology model is shown in Figure S2 and Figure S3, respectively. Although the HS-1 model had a lower number of residues in favored regions and in allowed regions (78.5 and 92.5%) than the crystal structure (93 and 99.3%), this model had some better evaluation parameters when analyzed with MolProbity (Table S6) like clashscore and MolProbity score. The clashscore is the number of serious issues (number of overlaps > 0.4 Å per atoms) [36]. The all-atom clashscore of HS-1 model was significantly better than the crystal structure (4.31 compared to 21.18) and was rated 96th percentile ranking versus a PDB sample of structures at similar resolutions by MolProbity. As a combination of the clashscore, percentage Ramachandran not favored and percentage bad sidechain rotamers, MolProbity score is a single number that represents the quality of protein [57]. In this study, the MolProbity score of the HS-1 homology structure was considered to be better than the crystal structure (35th rank versus 9th percentile).
The HS-1 homology structure was selected and prepared using the QuickPrep tool of the MOE 2015.10 software. The binding site was determined by extending a radius of 12 Å from the locations of the key residues Lys22, Arg35, Arg38, Gln39, Tyr119, Thr135 and Arg198. The obtained molecular docking model is illustrated in Fig. 4A. It can be seen that this is a relatively wide and deep binding site with a rich positive charge surface. A small molecule that binds to this cavity and blocks the positively charged residues of the ST2 receptor is expected to potentially inhibit the binding of IL-33 to this receptor. The docking protocol has been implemented for active and inactive datasets. Using ROC and PC analysis, performance of the model was validated with an ROC AUC of 0.754 (> 0.5) and a \(\overline{\mathrm{TG} }\) value of 0.384 (> 0.25). The ROC and predictiveness curves are shown in Figure S4.

A Molecular docking model of ST2 receptor identified from its key residues using LeadIT software. B The top 20 hit compounds suggested by docking results (carbon atoms in gray) are located in the binding site with the presence of the seven key residues (carbon atoms in magenta)
All compounds satisfied the pharmacophore models were structurally prepared and saved to a ligand library in *.sdf format for docking into the aforementioned binding site. As a result, 1755 compounds (98.7%) were successfully docked to the ST2 receptor and 23 compounds (1.3%) were not docked. The list of these compounds and their docking score are presented in Tables S3 and S4. Most of the successfully docked compounds had negative docking scores (1748 compounds) and are shown in the distribution graph in Figure S5A. There are 1253 compounds docked to ST2 with scores < − 19.26 kJ/mol, which was the docking score of the best docking pose of the control compound iST2-1. Notably, 6 ligands strongly bound to the ST2 receptor with docking scores < − 40 kJ/mol and 249 ligands bound to the receptor with docking scores < − 30 kJ/mol (Figure S5B).
When considering the control compound, iST2-1 was identified as the lead inhibitors in the study of Ramadan A. M. et al. (Figure S6). Previous study has shown that iST2-1 can bind to ST2 at various sites [25]. In the binding site determined by our study, iST2-1 also bound at two separate sites when analyzing the 10 docked poses as shown in Figure S6A. This is explained by its small and flexible structure (with molecular weight of 378.43 g/mol and 6 rotatable bonds). iST2-1 in poses 1, 2 and 9 bound to the receptor in the pocket formed by Lys22, Arg38, Gln39 and Tyr119, while the remaining poses are found in the vicinity of Tyr119, Thr135 and Arg198. The two poses having the best binding mode with key residues are shown in Figure S6B and S6C. In particular, iST2-1 in pose 2 accepted two hydrogen bonds from Arg35 and donated a hydrogen bond to Asp137, while in pose 3 it accepted two hydrogen bonds from Arg198 and formed arene-H bonds to Tyr119, Pro134 and Thr135. Notably, most of formed hydrogen bonds are due to the aromatic nitro group of iST2-1.
Because the 3D-pharmacophore models were built based on the key residues of PPI between the ST2 receptor and IL-33, the number of ligands that interacted with these residues in the binding site was counted and illustrated by the graphs in Figure S7A to further evaluate the performance of the 3D-pharmacophore models. Fingerprints analysis of protein–ligand interaction showed that key residues of ST2 receptor such as Lys22, Arg35, Tyr119, Thr135 and Arg198 formed interactions with a significant proportion with ligands (46, 51, 46, 45 and 71%, respectively). More specifically, these residues formed many hydrogen bonds and salt bridges with the docked ligands (Figures S7B and S7C). These results demonstrated that a large number of ligands bound to the ST2 receptor exactly on the key residues as designed in the 3D-pharmacophore model. The residues Lys22, Arg35 and Arg198 were centers for hydrogen bonding as well as strong ionic interactions. Thus, 3D-pharmacophore models could accelerate the identification of compounds with suitable physicochemical properties for binding to the ST2 receptor from the great database as ZINC.
In an effort to identify the most potential in silico inhibitors for the ST2 receptor, the 20 ligands with the most negative docking scores and satisfying Lipinski’s rule of five [58] were selected for further investigation using MD simulations. For comparison, the two best binding conformations of control compound iST2-1 have also been carried out MD simulations with the same time. The chemical structures of 20 compounds and their properties are presented in Table S7. The docking scores of these 20 top hit compounds and their interactions with receptors in the docking pose are summarized in Table 1 and detailed in Table S8. All 20 of these ligands have docking scores < − 35 kJ/mol and bind to at least three key residues of the ST2 receptor primarily by hydrogen bonds and arene-H bonds. Figure 4B shows all of these ligands neatly bind to the specified binding site (Site 1) on the receptor with the presence of the key residues. Interestingly, all of these 20 potential inhibitors satisfied the 3D-pharmacophore Ph-ST2i-1 model, the one with many anionic features. This suggests that potent and strong inhibitors of ST2 require negatively charged functional groups such as –COO− or –NO2 for interacting with the positively charged surface of the receptor. In particular, similar to the control compound iST2-1, aromatic nitro groups were found in 18 hit compounds. This is actually logical because these ligands were found by a PPI-based pharmacophore model and they are mimetic small molecules of IL-33 with negative charges.
Molecular dynamics simulations and binding free energy calculations help identify the top hit compounds
First, MD simulations were performed with a simulation time of 20 ns for the ST2 receptor in the apoprotein form (without ligand) and in complexes with 20 investigated ligands or the control compound iST2-1. These short MD trajectories were used to analyze the stability of protein, ligands, their interactions and binding free energy, after which the most potential candidates can be selected.
Stability of the receptor–ligand complexes
The RMSD values of the carbon backbone were used to evaluate the overall stability of the proteins (Table 2). The results indicated that most complexes of the ST2 receptor with ligands had average RMSD values smaller than the apoprotein (3.29 ± 1.15 Å) except for ZINC02065976, ZINC08911140, ZINC31554431, ZINC32690934 and ZINC40659148. However, when observing the time graph of RMSD of the complexes versus the apoprotein (Figure S8), all 20 complexes had smaller RMSD fluctuations than did the apoprotein (< 2 Å) after the fluctuating time and then reached equilibrium. The apoprotein appeared to be stable from 0 to 10 ns, but then RMSD values fluctuated strongly toward the end of simulation time (20 ns). For iST2-1, the complex of ST2 receptor with its conformation in docking pose 2 was very stable, but RMSD values of the complex of its pose 3 tended to increase at the end of MD simulations.
In addition, the solvent accessible surface areas (SASA) were calculated to monitor the possible changes in the solvation environment upon ligand binding. SASA of protein has always been considered as a decisive factor in protein folding and stability studies [59]. Generally, the increased value of the protein SASA during simulation indicates structural relaxations and therefore reduced protein stabilities [60]. The mean values of SASA of complexes are shown in Table 2, and a graph showing their change over time is shown in Figure S9. In general, the mean SASA of the ST2 receptor in the form of apoprotein and complexes with 20 ligands did not change much (about 110 nm2). The binding of ligands does not greatly affect the protein structure; it could even stabilize the structure. With the exception of ZINC02065976, ZINC32690934 and ZINC40659148, all ST2 receptor in complexes with the remaining ligands and iST2-1 had smaller SASA than that of the apoprotein (110.40 ± 1.04 nm2).
Furthermore, the radius of gyration (Rg) of the protein was also calculated (the mean is shown in Table 2 and the graph over time in Figure S10). Rg is considered an indicator of protein structure compactness [61]. When a ligand binds to the protein, a conformational change occurs that will alter the radius of gyration. Understanding the Rg will further help in predicting the compactness and binding patterns of the drug and protein [62]. The ST2 receptor in the apoprotein form has an Rg of 19.02 ± 0.35 Å and in its complexes with 20 ligands or iST2-1 had the Rg of approximately or below this value. These results indicated that the protein was stable during 20 ns of MD simulations.
Stability of the binding site and the 20 ligands
The stability of the binding site is determined by the RMSF of the alpha carbons of the residues. RMSF values calculated from all MD trajectories of 20 complexes were overlapped with that of the apoprotein presented in Figure S11A with the key residues in the binding site highlighted. The separate RMSF plot of each complex overlapped with that of the apoprotein is shown in Figure S12. It can be seen that the residues Lys22, Arg35, Arg38, Gln39, Tyr119, Thr135 and Arg198 all have stable fluctuations with an RMSF of < 2 Å, and there is no significant difference in complexes and apoprotein state. Interestingly, however, the binding of the ligands to the protein leads to the stabilization of the β-loop from Tyr50–Gln60 with the RMSF decreasing from about 1–6 Å and slightly increasing the fluctuation of β-loop from Lys107–Leu117 with the RMSF increased from about 0.5–2 Å (Figure S11B).
The average RMSD values of heavy atoms of each ligand in Table 2 and the RMSD–time plot in Figure S13 help identify highly stable ligands during the 20 ns MD simulations. High RMSD values reveal large conformational changes of the ligands during MD simulations. Most of the ligands had mean RMSD values < 2 Å except for ZINC02065976, ZINC02876200, ZINC20527417 and ZINC40659148.
Analysis of MM/GBSA binding free energy and the hydrogen bonds that occurred during the trajectories sampled in the 20 ns MD simulations
Binding free energies between ST2 receptor and the 20 selected ligands were calculated based on data from 201 frames of the 20 ns MD trajectories (from frame 1 to 2001 with interval of 10). The binding capacity of these ligands can be compared by the MM/GBSA \(\Delta G_{{{\text{bind}}}}\) values shown in the graph in Fig. 5. The \(\Delta G_{{{\text{bind}}}}\) values and energy components are presented in Table S9. It is clear that all 20 complexes have negative binding free energy. In particular, the six complexes of ZINC08821844, ZINC08911140, ZINC16933127, ZINC40658091, ZINC59502934 and ZINC59514725 have the remarkable \(\Delta G_{{{\text{bind}}}}\) of < − 20 kcal/mol, suggesting their stable binding to the receptor. Meanwhile, iST2-1 only has the \(\Delta G_{{{\text{bind}}}}\) of pose 2 and pose 3 conformations of − 7.28 ± 3.96 and − 2.97 ± 4.05 kcal/mol, respectively. Evaluation of the \(\Delta G_{{{\text{bind}}}}\) can help unambiguously identify the most potential receptor inhibitors. However, the aim of this study was to discover the small molecules that inhibit the PPI of the IL-33/ST2 complex by binding strongly and specifically at the location of the key residues on the ST2 receptor. Therefore, the interaction of each ligand with these residues should be carefully considered to identify top hit compounds.

MM/GBSA binding free energies calculated from 20 ns MD trajectories for the investigated 20 ligands as they bound to the ST2 receptor
The number of hydrogen bonds formed between the 20 ligands and the receptor was calculated from the trajectories of 20 ns MD simulations using the VDM software (Figure S14). Their mean values in Table S10 indicate that the five ligands that interacted most strongly with the receptor are ZINC08911140, ZINC16933127, ZINC20527417, ZINC40658091 and ZINC59514725. This result contributes to the explanation for their good binding free energy as mentioned above. As the object of this study was to find compounds that interact with the key residues of IL-33/ST2 PPIs, the percentage occupancy of hydrogen bonds was also investigated (Table S10). With the exception of ZINC31554431, most ligands act as hydrogen bond acceptors from ST2 receptor residues. By comparing the frequency of hydrogen bond forming, the four ligands ZINC08911140, ZINC16933127, ZINC40658091 and ZINC59514725 were identified as the most potent inhibitors of the ST2 receptor because they strongly interacted with key residues by hydrogen bonding at very high occupancy. The iST2-1 formed hydrogen bonds with the key residues with low frequency (< 10%) in both conformations even though they did in molecular docking. This suggests that iST2-1 may have moved out of the binding cavity during the simulation. Meanwhile, ZINC08911140 interacted with Arg198 and Tyr119 by hydrogen bond acceptor atoms with a total frequency of 275.48 and 18.23%, respectively. ZINC16933127 received hydrogen bonding from Arg35 with a total frequency of 187.17% and with other key residues such as Lys22, Thr135 and Arg198 with a frequency of 40.86, 55.65 and 19.78%, respectively. ZINC40658091 formed hydrogen bonds with Lys22, Arg35 and Tyr119 with a frequency of 61.84, 225.32 and 47.05%, respectively. In particular, ZINC59514725 exhibited potential as a strong inhibitor of the ST2 receptor when interacting with five over the seven key residues Lys22, Arg35, Agr38, Gln39 and Thr135 with hydrogen bond forming frequencies of 98.10, 259.45, 36.36, 83.47 and 19.73%, respectively. These four ligands also have stable protein binding free energy with a \(\Delta {G}_{bind}\) of − 20.79 ± 4.56, − 20.92 ± 6.11, − 22.67 ± 8.96 and − 36.92 ± 8.89 kcal/mol, respectively.
A rapid assessment for the physicochemical and ADMET properties of the 20 ligands was carried out by using the ADMETlab 2.0 web tool [63], and the predicted results are presented in Table S11. The four compounds mentioned above have potential for medicinal use because of their compliance with the Lipinski and Pfizer rules, no PAINS (Pan assay interference compounds) alert, good or moderate oral absorption, no serious toxicity and ease of synthesis. Therefore, 100 ns MD simulations were performed and analyzed to more closely investigate the binding processes of these top hit compounds. This process was also performed for the docking pose 2 of the control compound iST2-1 due to its better 20 ns MD simulation results (compared to pose 3).
Identify of the top hit compounds by long MD simulations
Similar to the 20 ns MD simulations, RMSD and RMSF profiles were also used to evaluate protein stability in four complexes. As illustrated in Fig. 6A, both the apoprotein and the four complexes have RMSD carbon backbone fluctuations less than 2 Å. However, it can be seen that the complex of ZINC40658091 has a higher volatility than the other three complexes. Especially, complexes of ZINC16933127 and ZINC59514725 have a really low protein RMSD fluctuation (< 1 Å). These results were in perfect agreement with the protein RMSF plot (Fig. 6B), as these two ligands helped stabilize the loops on the secondary structure at domains 1 and 2 of the ST2 receptor. When observing the stability of each hit compounds through the RMSD values of their heavy atoms (Fig. 6C), the ligands reached high stability after about 40 ns. ZINC08911140, ZINC16933127 and ZINC59514725 have RMSD ligands of just 0.5 Å. ZINC40658091 again exhibited instability at the end of the simulation time point although it has reached a prolonged equilibrium over a period of 40 − 90 ns. For the complex of iST2-1 with the receptor, the protein and ligand RMSD values fluctuated greatly (Fig. 6A, C) and revealed multiple translocations of this ligand in throughout 100 ns of MD simulations. The results of hydrogen bond analysis from the data of the last 50 ns of MD simulations in Table 3 and Fig. 6D show that the ligands formed an average of three to four hydrogen bonds with the protein. The maximum number of hydrogen bonds they have formed with the ST2 receptor ranges from 7 to 10, while iST2-1 could only form up to four in a few frames.

A Protein carbon backbone RMSD and B carbon alpha RMSF values of the ST2 receptor in apoprotein form and in complexes with the top four hit compounds and the control compound iST2-1 calculated using data of 100 ns trajectories of MD simulations. C Ligand heavy atom RMSD profiles estimated from the same MD trajectories. D Number of hydrogen bonds and E MM/GBSA binding free energy variation over time of the four ligands and iST2-1 when binding to the receptor calculated using the last 50 ns trajectories of 100 ns MD simulations
The data from MD trajectories of the last half simulation time were used for further analyses including interaction frequency, binding free energy and per-residues energy decomposition. The ability to interact with key residues was considered as an important criterion for assessing hit compounds in this study. The percentage of hydrogen bond occupancy of each ligand with the ST2 receptor was analyzed by the VMD software and is detailed in Table 3. Furthermore, because the PPI of IL-33/ST2 is formed mainly by salt bridges between the hotspot residues, ionic interactions between the ST2 receptor with the four small molecular ligands were of high interest to be investigated. Other types of interactions such as ionic interactions, arene interactions and surface contacts were analyzed from 500 frames extracted from the last 50 ns of 100 ns MD simulations trajectories using the PLIF tool in the MOE 2015.10 software and are detailed in Table 4. The results showed that all four ligands exhibit strong interactions with the key residues mainly by hydrogen bonding and salt bridges, while the interaction of iST1-1 is negligible. The two-dimensional diagrams in Fig. 7 more visually illustrate the interaction of ligand atoms with the ST2 residues. For easy tracking, the atoms of each ligand are numbered by their atom index in the MD simulations data. Some residues have a > 100% interaction forming frequency because they have multiple interactions of a single type with the same ligand atom. For example, the arginine side chain has four hydrogen bond donors that can all hydrogen bond to a single acceptor of ligand as shown in Table 3.

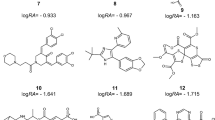
Schematic of detailed ligand atom interactions with the ST2 receptor residues
ZINC08911140 interacted with the key residue Arg198 by only its carboxylate group (–COmainO4−) via hydrogen and ionic bonds with a total frequency of 184.5 and 100.0%, respectively (Table 4 and Fig. 7A). In addition, this compound also interacted with Lys27 by two similar interactions at the carboxylate group position with the frequency of 93.3 and 96.2%, respectively. At the other end of the molecule, the nitro group (–N+O5O1−) formed weak hydrogen bonds with Lys130 and contacted by arene-arene stacking with Tyr132 with a frequency of 27.7 and 34.9%, respectively. The result suggests that ZINC08911140 seems to bind to the ST2 receptor by anchoring mainly on a local site of Lys127 and Arg198. Similarly, ZINC40658091 (Fig. 7C) also binds to the receptor on a side of the molecule by accepting hydrogen bonds from Lys22, Arg35, and Arg38 on the same carboxylate group (–COmainO4−) with frequencies of 96.4, 93.3 and 78.4%, respectively; hydrogen bonds donor with Ser21 by the –N2H group with a frequency of 93.3%; and weak π-alkyl interaction with Lys19 with a frequency of 6.1%.
ZINC16933127 bound to the target more consistently and is demonstrated in its protein RMSD, RMSF and ligand RMSD plots (Fig. 6). The binding mode of this ligand is also more balanced at both sides of the molecule (Fig. 7B). This compound formed both hydrogen and ionic bonds with the two key residues Lys22 and Arg38 with high frequency. The structural core of molecule ZINC16933127 is (Z)-2-amino-5-benzylidenethiazol-4(5H)-one. At the carboxylate group (–CO1O4−) of the 1-carboxylatoethoxy side branch, the ligand accepted the hydrogen bonding of Arg38 and Arg96 with a frequency of 257 and 44.6%, respectively. This carboxylate group also formed ionic interactions with these two residues with a frequency of 99.2 and 37.4%, respectively (Table 4). At the 5-hydroxybenzoate ring, the oxygen atoms of the carboxylate group (–COmainO3−) accepted hydrogen bonds and formed salt bridges with Lys22 with a frequency of 32.2 and 35.4%, respectively, the − O5H group donated hydrogen bond with Gln23 with a frequency of 39.8%, and the phenyl ring formed a weak π- alkyl contact with Lys22 with a frequency of 21.4%. In addition, the key residue Gln39 also interacts with the central part of the molecule by hydrogen bonds with O2 and O6 atoms with a frequency of 21.1 and 53.9%, respectively, and a π-alkyl interaction with phenyl ring with an occupancy of 23.4%.
In particular, ZINC59514725 (Fig. 7D) can be considered the best hit compound in this virtual screening study and could be a potential candidate for future ST2 receptor inhibitors. ZINC59514725 is the diamide of 5-pivalamidoisophthalic acid with two molecules of 3-aminobenzoic acid. It bound stably to the receptor on three key residues, including Lys22, Arg35 and Arg38. Looking back at the PPI between IL-33 and the ST2 receptor in Fig. 2, the interaction with these residues may enable ZINC59514725 to competitively bind to hotspot residues on IL-33 such as Glu148, Glu144 and Asp244. These residues have been experimentally demonstrated that their mutant form leads to an apparent change in binding affinity between ST2 and IL-33 (Fig. 2B). The two carboxylate groups at the ends of the ZINC59514725 molecule show similarities to the two acidic and negatively charged residues Glu144 and Glu148 of IL-33. The 5-pivalamidoisophthalic acid center played as a linker and appropriate spacer that helped the two groups of acids to have suitable spatial positions to interact with the basic and positively charged residues of the ST2 receptor. The –CO1O5− carboxylate group accepted three hydrogen bonds from Lys22 and Arg35 with a frequency of 96.5 and 107.9%, respectively, and also formed salt bridges with these residues with a frequency of 100.0 and 99.2%, respectively. In the remaining carboxylate group (–COmainO4−), ZINC59514725 accepted four hydrogen bonds from Arg38 and Arg96 with a frequency of 196.3 and 38.0%, respectively, and also formed ionic interactions with these two residues with a frequency of 99.6 and 27.7%, respectively. Arg38 also formed a weak π-alkyl interaction with the phenyl group with a frequency of 28.9%. The results suggested that ZINC59514725 is a very promising inhibitor for the ST2 receptor. The very low fluctuations of ZINC59514725 (Fig. 6C) and its complex with the protein shown in Figs. 6A, B indicate a high stability of this compound in binding to the ST2 receptor.
Our 100-ns MD simulation study for iST2-1 compound has repeated results of Ramadan A. M. et al. [25] when two other binding sites of were found. Initially, the control compound bound at the ST2/IL-33 interaction interface. But it has moved quickly to Pocket 1 and stayed there for 20 ns, then continued to move away from the protein for the next 50 ns and finally bound stably in Pocket 2. These events are illustrated in Figure S15A. The flexibility of iST2-1 has resulted in its low frequency of interaction forming with specific residues (Tables 3, 4).
Binding mode of the four top hit compounds at the end time of 100 ns MD simulations is illustrated in Fig. 8. Because the binding surface of the ST2 receptor is quite wide, it can be seen that ZINC08911140 binds at a different location than the other three ligands (Figure S15B). It bound to ST2 at the location with the presence of key residues such as Tyr119, Thr135 and Arg198. However, this ligand only interacted strongly with Arg198 by hydrogen bonds and salt bridges (Fig. 8A). The remaining ligands had almost the same binding pattern. They bound to the receptor at the location defined by the key residues such as Lys22, Arg35, Arg38 and Gln39. As a result, they have nearly identical binding modes at the end of the MD simulations as illustrated in Figure S8B, C and D. However, as discussed above, ZINC40658091 only interacted with key residues at one side of the molecule and the opposite structural part has become very flexible. ZINC16933127 and ZINC59514725 further proved to be more stable when their entire structures were fixed onto the ST2 receptor by strong hydrogen and ionic bonds. They have not moved away from the originally defined binding site and were becoming more tightly bound to the key residues as shown in Figure S15C and D.

Visualization of binding mode of the four top hits at the end of the 100 ns MD simulations in the presence of residues within a radius of 5 Å. Only residues that interact with the ligand are labeled
MM/GBSA binding free energy of the four top hit compounds was calculated using data of the last 50 ns of 100 ns MD trajectories. The \(\Delta {G}_{bind}\) of the ligands still had negative values (Table 5), and their variation over time is illustrated in Fig. 6E, in which ZINC16933127 and ZINC59514725 had stable binding free energy and possessed the most negative mean values of − 28.93 ± 6.66 and − 26.84 ± 5.07 kcal/mol, respectively. Even, the \(\Delta {G}_{bind}\) of the two top hit compounds tends to decrease at the end of the simulation and reflects an increase in binding affinity. The ligands ZINC08911140 and ZINC40658091 have higher \(\Delta {G}_{bind}\) values, suggesting weaker binding. iST2-1 had a \(\Delta {G}_{bind}\) value close to 0 kcal/mol from 50 to 70 ns because it flew out of the protein and only reached negative value when returning to Pocket 2 from 70 to 100 ns of MD trajectory.
To determine more clearly the contribution of residues in the interaction between the ST2 receptor and the ligands, the per-residue energy decomposition was carried out; the results are presented in Table 6. As analyzed above, ZINC08911140 interacted mainly by hydrogen bonds and salt bridges with Arg198, leading to a large contribution of this residue in the binding free energy (− 7.11 kcal/mol). In addition, Tyr132 also contributed a significant amount of free energy (− 3.52 kcal/mol). Similarly, Lys22, Arg35, Arg38 and Tyr119 also contributed for ZINC40658091 and ST2 receptor binding with free energies of − 6.11, − 3.91, − 2.56 and − 1.87 kcal/mol, respectively. In the case of ZINC16933127, Arg38 contributed a remarkable free energy of − 11.26 kcal/mol, along with other key residues such as Lys22, Arg35 and Gln39 with energies of − 1.99, − 2.05 and − 1.99 kcal/mol, respectively. Finally, ZINC59514725 still exhibited excellent binding to the ST2 receptor as the key residues Lys22, Arg35 and Arg38 contributed significant free energy levels of − 5.03, − 5.88 and − 8.92 kcal/mol, respectively. Moreover, the two key residues Gln39 and Thr135 also contributed a part of the energy with values of − 0.65 and − 0.44 kcal/mol, respectively.
Thus, ZINC59514725 was identified as the best hit compound within the scope of this study by analyzes such as stability of complexes and ligand, frequency of interactions occupancy, MM/GBSA binding free energy and per-residues energy decomposition. These results strongly suggested a promising lead compound for an inhibitory effect on the interaction between the ST2 receptor and IL-33. Further in vitro and in vivo experiments need to be performed to confirm the effect of this compound in the near future.
Conclusion
The association of the cytokine IL-33 with its ST2 receptor on the surface of blood cells leads to a cascade of immune responses and is implicated in many inflammatory and autoimmune diseases. PPIs between the two proteins have become an interesting target for new drug discovery projects to treat related diseases, even for COVID-19. However, the successful discovery of small molecule drugs that inhibit PPIs has never been easy because of its novelty and complexity. X-ray crystallography and mutational studies have helped to elucidate the nature of PPIs between IL-33 and the ST2 receptor. These biological data became the basis for medicinal chemist to begin research on inhibitors of IL-33 activity.
The drug discovery process is always time-consuming and very expensive. In this study, rational drug design methods helped to discover several molecules that have the ability to inhibit the ST2 receptor in silico. High-performance screening methods such as pharmacophore modeling and molecular docking have become effective funnels for the initial search of compounds capable of binding on the target. Subsequently applied computational biology methods such as MD simulations and MM/GBSA binding free energy calculations supported the ranking and identification the most potential inhibitors for the ST2 receptor. In the era of big data, the huge ZINC12 database with more than 22 million chemical structures corresponding to more than 200 conformations has been applied to search for compounds with desired activity. A series of compounds have exhibited better in silico binding ability to the ST2 receptor than the control inhibitor iST2-1. ZINC08911140, ZINC16933127, ZINC40658091 and ZINC59514725 were identified as the four most potential hits demonstrated by their strong and stable interactions with the ST2 receptors. Calculations based on long MD simulations data revealed ZINC59514725 to be the most promising drug candidate with binding free energy of − 26.84 kcal/mol and abundant interactions with many key residues in PPIs of IL-33/ST2. These potential compounds will soon enter the next stages of drug discovery process. Successful discovery of the ST2 receptor inhibitors will open a new hope for the treatment of inflammatory, autoimmune, infectious and neurodegenerative diseases with a completely new mechanism.
References
Liu X, Hammel M, He Y, Tainer JA, Jeng US, Zhang L, Wang S, Wang X (2013) Structural insights into the interaction of IL-33 with its receptors. Proc Natl Acad Sci USA 110:14918–14923. https://doi.org/10.1073/pnas.1308651110
Cayrol C, Girard J-P (2018) Interleukin-33 (IL-33): a nuclear cytokine from the IL-1 family. Immunol Rev 281:154–168. https://doi.org/10.1111/imr.12619
Villacorta H, Maisel AS (2016) Soluble ST2 testing: a promising biomarker in the management of heart failure. Arq Bras Cardiol 106:145–152. https://doi.org/10.5935/abc.20150151
Liew FY, Girard J-P, Turnquist HR (2016) Interleukin-33 in health and disease. Nat Rev Immunol 16:676–689. https://doi.org/10.1038/nri.2016.95
Kakkar R, Lee RT (2008) The IL-33/ST2 pathway: therapeutic target and novel biomarker. Nat Rev Drug Discov 7:827–840. https://doi.org/10.1038/nrd2660
Weinberg EO, Shimpo M, De Keulenaer GW, MacGillivray C, Tominaga S-i, Solomon SD, Rouleau J-L, Lee RT (2002) Expression and regulation of ST2, an Interleukin-1 receptor family member, in cardiomyocytes and myocardial infarction. Circulation 106:2961–2966. https://doi.org/10.1161/01.cir.0000038705.69871.d9
Oshikawa K, Kuroiwa K, Tago K, Iwahana H, Yanagisawa K, Ohno S, Tominaga S-I, Sugiyama Y (2001) Elevated soluble ST2 protein levels in sera of patients with asthma with an acute exacerbation. Am J Respir Crit Care Med 164:277–281. https://doi.org/10.1164/ajrccm.164.2.2008120
Oboki K, Nakae S, Matsumoto K, Saito H (2011) IL-33 and airway inflammation. Allergy Asthma Immunol Res 3:81–88. https://doi.org/10.4168/aair.2011.3.2.81
Beltran CJ, Nunez LE, Diaz-Jimenez D, Farfan N, Candia E, Heine C, Lopez F, Gonzalez MJ, Quera R, Hermoso MA (2010) Characterization of the novel ST2/IL-33 system in patients with inflammatory bowel disease. Inflamm Bowel Dis 16:1097–1107. https://doi.org/10.1002/ibd.21175
Latiano A, Palmieri O, Pastorelli L, Vecchi M, Pizarro TT, Bossa F, Merla G, Augello B, Latiano T, Corritore G, Settesoldi A, Valvano MR, D’Incà R, Stronati L, Annese V, Andriulli A (2013) Associations between genetic polymorphisms in IL-33, IL1R1 and risk for inflammatory bowel disease. PLoS ONE 8:e62144. https://doi.org/10.1371/journal.pone.0062144
Xiong Z, Thangavel R, Kempuraj D, Yang E, Zaheer S, Zaheer A (2014) Alzheimer’s disease: evidence for the expression of interleukin-33 and its receptor ST2 in the brain. J Alzheimers Dis 40:297–308. https://doi.org/10.3233/JAD-132081
Pei C, Barbour M, Fairlie-Clarke KJ, Allan D, Mu R, Jiang HR (2014) Emerging role of interleukin-33 in autoimmune diseases. Immunology 141:9–17. https://doi.org/10.1111/imm.12174
Matsuyama Y, Okazaki H, Tamemoto H, Kimura H, Kamata Y, Nagatani K, Nagashima T, Hayakawa M, Iwamoto M, Yoshio T, Tominaga S, Minota S (2010) Increased levels of interleukin 33 in sera and synovial fluid from patients with active rheumatoid arthritis. J Rheumatol 37:18–25. https://doi.org/10.3899/jrheum.090492
Mok MY, Huang FP, Ip WK, Lo Y, Wong FY, Chan EY, Lam KF, Xu D (2010) Serum levels of IL-33 and soluble ST2 and their association with disease activity in systemic lupus erythematosus. Rheumatology 49:520–527. https://doi.org/10.1093/rheumatology/kep402
WHO Coronavirus (COVID-19) Dashboard. https://covid19.who.int/. Accessed 24 Nov 2021
Zizzo G, Cohen PL (2020) Imperfect storm: is interleukin-33 the Achilles heel of COVID-19? Lancet Rheumatol 2:e779–e790. https://doi.org/10.1016/s2665-9913(20)30340-4
Park SB, Kim SJ, Cho SW, Choi CY, Lee S (2020) Blocking of the IL-33/ST2 signaling axis by a Single-Chain Antibody Variable Fragment (scFv) specific to IL-33 with a defined epitope. Int J Mol Sci. https://doi.org/10.3390/ijms21186953
Kim YH, Yang TY, Park CS, Ahn SH, Son BK, Kim JH, Lim DH, Jang TY (2012) Anti-IL-33 antibody has a therapeutic effect in a murine model of allergic rhinitis. Allergy 67:183–190. https://doi.org/10.1111/j.1398-9995.2011.02735.x
Liu X, Li M, Wu Y, Zhou Y, Zeng L, Huang T (2009) Anti-IL-33 antibody treatment inhibits airway inflammation in a murine model of allergic asthma. Biochem Biophys Res Commun 386:181–185. https://doi.org/10.1016/j.bbrc.2009.06.008
Holgado A, Braun H, Van Nuffel E, Detry S, Schuijs MJ, Deswarte K, Vergote K, Haegman M, Baudelet G, Haustraete J, Hammad H, Lambrecht BN, Savvides SN, Afonina IS, Beyaert R (2019) IL-33trap is a novel IL-33–neutralizing biologic that inhibits allergic airway inflammation. J Allergy Clin Immunol 144:204–215. https://doi.org/10.1016/j.jaci.2019.02.028
Osbourn M, Soares DC, Vacca F, Cohen ES, Scott IC, Gregory WF, Smyth DJ, Toivakka M, Kemter AM, le Bihan T, Wear M, Hoving D, Filbey KJ, Hewitson JP, Henderson H, Gonzàlez-Cìscar A, Errington C, Vermeren S, Astier AL, Wallace WA, Schwarze J, Ivens AC, Maizels RM, McSorley HJ (2017) HpARI protein secreted by a helminth parasite suppresses interleukin-33. Immunity 47:739-751.e5. https://doi.org/10.1016/j.immuni.2017.09.015
Le MT, Mai TT, Huynh PNH, Tran TD, Thai KM, Nguyen QT (2020) Structure-based discovery of interleukin-33 inhibitors: a pharmacophore modelling, molecular docking, and molecular dynamics simulation approach. SAR QSAR Environ Res 31:883–904. https://doi.org/10.1080/1062936x.2020.1837239
Zhang J, Ramadan AM, Griesenauer B, Li W, Turner MJ, Liu C, Kapur R, Hanenberg H, Blazar BR, Tawara I, Paczesny S (2015) ST2 blockade reduces sST2-producing T cells while maintaining protective mST2-expressing T cells during graft-versus-host disease. Sci Transl Med 7:308ra160. https://doi.org/10.1126/scitranslmed.aab0166
Nnane I, Frederick B, Yao Z, Raible D, Shu C, Badorrek P, van den Boer M, Branigan P, Duffy K, Baribaud F, Fink D, Yang T-Y, Xu Z (2020) The first-in-human study of CNTO 7160, an anti-interleukin-33 receptor monoclonal antibody, in healthy subjects and patients with asthma or atopic dermatitis. Br J Clin Pharmacol 86:2507–2518. https://doi.org/10.1111/bcp.14361
Ramadan AM, Daguindau E, Rech JC, Chinnaswamy K, Zhang J, Hura GL, Griesenauer B, Bolten Z, Robida A, Larsen M, Stuckey JA, Yang CY, Paczesny S (2018) From proteomics to discovery of first-in-class ST2 inhibitors active in vivo. JCI Insight. https://doi.org/10.1172/jci.insight.99208
Irwin JJ, Shoichet BK (2005) ZINC—a free database of commercially available compounds for virtual screening. J Chem Inf Model 45:177–182. https://doi.org/10.1021/ci049714+
Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG (2012) ZINC: a free tool to discover chemistry for biology. J Chem Inf Model 52:1757–1768. https://doi.org/10.1021/ci3001277
Koes DR, Camacho CJ (2011) Pharmer: efficient and exact pharmacophore search. J Chem Inf Model 51:1307–1314. https://doi.org/10.1021/ci200097m
Molecular Operating Environment (MOE) (2015) Version 2015.10. Chemical Computing Group Inc., Montreal
Voet A, Banwell EF, Sahu KK, Heddle JG, Zhang KYJ (2013) Protein interface pharmacophore mapping tools for small molecule protein: Protein interaction inhibitor discovery. Curr Top Med Chem 13:989–1001. https://doi.org/10.2174/1568026611313090003
Voet A, Zhang KYJ (2012) Pharmacophore modelling as a virtual screening tool for the discovery of SMPPIIs. Curr Pharm Des 18:4586–4598. https://doi.org/10.2174/138161212802651616
Zhang Y (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinform 9:40. https://doi.org/10.1186/1471-2105-9-40
Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y (2015) The I-TASSER Suite: protein structure and function prediction. Nat Methods 12:7–8. https://doi.org/10.1038/nmeth.3213
Roy A, Kucukural A, Zhang Y (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5:725–738. https://doi.org/10.1038/nprot.2010.5
Ramachandran GN, Ramakrishnan C, Sasisekharan V (1963) Stereochemistry of polypeptide chain configurations. J Mol Biol 7:95–99. https://doi.org/10.1016/S0022-2836(63)80023-6
Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB 3rd, Snoeyink J, Richardson JS, Richardson DC (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35:W375–W383. https://doi.org/10.1093/nar/gkm216
Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB, Jain S, Lewis SM, Arendall WB 3rd, Snoeyink J, Adams PD, Lovell SC, Richardson JS, Richardson DC (2018) MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci 27:293–315. https://doi.org/10.1002/pro.3330
LeadIT version 2.1.8. http://www.biosolveit.de/LeadIT/. Accessed 10 Dec 2019
Ngo T-D, Tran T-D, Le M-T, Thai K-M (2016) Computational predictive models for P-glycoprotein inhibition of in-house chalcone derivatives and drug-bank compounds. Mol Divers 20:945–961. https://doi.org/10.1007/s11030-016-9688-5
Lasko TA, Bhagwat JG, Zou KH, Ohno-Machado L (2005) The use of receiver operating characteristic curves in biomedical informatics. J Biomed Inform 38:404–415. https://doi.org/10.1016/j.jbi.2005.02.008
Empereur-mot C, Guillemain H, Latouche A, Zagury J-F, Viallon V, Montes M (2015) Predictiveness curves in virtual screening. J Cheminform 7:52. https://doi.org/10.1186/s13321-015-0100-8
Mysinger MM, Carchia M, Irwin JJ, Shoichet BK (2012) Directory of Useful Decoys, Enhanced (DUD-E): better ligands and decoys for better benchmarking. J Med Chem 55:6582–6594. https://doi.org/10.1021/jm300687e
Empereur-Mot C, Zagury J-F, Montes M (2016) Screening explorer—an interactive tool for the analysis of screening results. J Chem Inf Model 56:2281–2286. https://doi.org/10.1021/acs.jcim.6b00283
Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2:19–25. https://doi.org/10.1016/j.softx.2015.06.001
Lindahl E, Abraham MJ, Hess B, Spoel Dvd (2020) GROMACS 2020.6 Documentation (Release 2020.6). Zenodo. https://doi.org/10.5281/zenodo.4576060
Zoete V, Cuendet MA, Grosdidier A, Michielin O (2011) SwissParam: a fast force field generation tool for small organic molecules. J Comput Chem 32:2359–2368. https://doi.org/10.1002/jcc.21816
Bussi G, Donadio D, Parrinello M (2007) Canonical sampling through velocity rescaling. J Chem Phys 126:014101. https://doi.org/10.1063/1.2408420
Parrinello M, Rahman A (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys 52:7182–7190. https://doi.org/10.1063/1.328693
Hess B, Bekker H, Berendsen HJC, Fraaije JGEM (1997) LINCS: a linear constraint solver for molecular simulations. J Comput Chem 18:1463–1472. https://doi.org/10.1002/(SICI)1096-987X(199709)18:12%3c1463::AID-JCC4%3e3.0.CO;2-H
Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG (1995) A smooth particle mesh Ewald method. J Chem Phys 103:8577–8593. https://doi.org/10.1063/1.470117
Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14:33–38. https://doi.org/10.1016/0263-7855(96)00018-5
Rungrotmongkol T, Nunthaboot N, Malaisree M, Kaiyawet N, Yotmanee P, Meeprasert A, Hannongbua S (2010) Molecular insight into the specific binding of ADP-ribose to the nsP3 macro domains of chikungunya and venezuelan equine encephalitis viruses: Molecular dynamics simulations and free energy calculations. J Mol Graph Model 29:347–353. https://doi.org/10.1016/j.jmgm.2010.09.010
Valdés-Tresanco MS, Valdés-Tresanco ME, Valiente PA, Moreno E (2021) gmx_MMPBSA: a new tool to perform end-state free energy calculations with GROMACS. J Chem Theory Comput. https://doi.org/10.1021/acs.jctc.1c00645
Wang E, Sun H, Wang J, Wang Z, Liu H, Zhang JZH, Hou T (2019) End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem Rev 119:9478–9508. https://doi.org/10.1021/acs.chemrev.9b00055
Onufriev A, Bashford D, Case DA (2004) Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 55:383–394. https://doi.org/10.1002/prot.20033
Amber 2020 Reference Manual. https://ambermd.org/Manuals.php. Accessed 18 May 2021
Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66:12–21. https://doi.org/10.1107/s0907444909042073
Lipinski CA (2000) Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods 44:235–249. https://doi.org/10.1016/S1056-8719(00)00107-6
Ali SA, Hassan MI, Islam A, Ahmad F (2014) A review of methods available to estimate solvent-accessible surface areas of soluble proteins in the folded and unfolded states. Curr Protein Pept Sci 15:456–476. https://doi.org/10.2174/1389203715666140327114232
Borjian Boroujeni M, Shahbazi Dastjerdeh M, Shokrgozar M, Rahimi H, Omidinia E (2021) Computational driven molecular dynamics simulation of keratinocyte growth factor behavior at different pH conditions. Inform Med Unlocked 23:100514. https://doi.org/10.1016/j.imu.2021.100514
Lobanov MY, Bogatyreva NS, Galzitskaya OV (2008) Radius of gyration as an indicator of protein structure compactness. Mol Biol 42:623–628. https://doi.org/10.1134/S0026893308040195
Sneha P, Doss CGP (2016) Chapter seven—Molecular dynamics: new frontier in personalized medicine. In: Donev R (ed) Advances in protein chemistry and structural biology. Academic Press, pp 181–224. https://doi.org/10.1016/bs.apcsb.2015.09.004
Dong J, Wang NN, Yao ZJ, Zhang L, Cheng Y, Ouyang D, Lu AP, Cao DS (2018) ADMETlab: a platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. J Cheminform 10:29. https://doi.org/10.1186/s13321-018-0283-x
Acknowledgements
This work was supported by the Vietnam National Foundation for Science and Technology Development (NAFOSTED) [108.05-2018.15]. Tan Thanh Mai was funded by Vingroup Joint Stock Company and supported by the Domestic Master/PhD Scholarship Programme of Vingroup Innovation Foundation (VINIF), Vingroup Big Data Institute (VINBIGDATA), code VINIF.2020.TS.128.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mai, T.T., Nguyen, P.G., Le, MT. et al. Discovery of small molecular inhibitors for interleukin-33/ST2 protein–protein interaction: a virtual screening, molecular dynamics simulations and binding free energy calculations. Mol Divers 26, 2659–2678 (2022). https://doi.org/10.1007/s11030-021-10359-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-021-10359-4