
Abstract
This paper presents the thermal properties of highly crosslinked di(methacryloyloxymethyl)naphthalene–divinylbenzene (DMN–DVB) copolymeric microspheres containing polar groups in the structure and their alkyl-bonded derivatives. C8 and C18 alkyl chains were introduced into the aromatic rings of the DMN–DVB porous copolymer by means of the Friedel–Crafts reaction. As a source of C8 and C18 alkyl chains, octyl and octadecyl chlorides were used. It was necessary to check whether the introduction of alkyl chains into the structure of polymeric packing had an impact on its thermal properties. The studies were carried out by thermogravimetry coupled online with FTIR spectroscopy and differential scanning calorimetry in inert atmosphere (helium). It was stated that the modified materials showed 20 and 50% mass losses at higher temperatures than the non-modified one while 1% mass loss was observed at lower temperatures. Moreover, an analysis of volatile decomposition products was performed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alkyl-bonded silicas are the most popular stationary phases used in chromatography. Numerous previous experiments with the polymer packing materials as high efficiency counterparts to alkyl-bonded silicas failed. Unlike silica, most of those polymers were not rigid and compressed at the high eluent linear flow velocities [1]. The other polymers exhibited greater rigidity, but their mass transfer characteristics were poor, and the resulting long analysis times were unacceptable. Additionally, the polymeric materials swell in organic solvents which are particularly troublesome when solvent gradients are used [2, 3]. On the other hand, these materials were stable with the eluents from pH 1–14 and resulted in excellent separations.
To eliminate the above-mentioned disadvantages of polymer stationary phases, it was necessary to increase the degree of polymer crosslinking significantly. As the first Tsyurupa and Davankov applied the hypercrosslinked (HXL) polymers. These materials were obtained via the extensive post-crosslinking of linear polystyrene chains by means of the Friedel–Crafts reaction involving external electrophiles, a synthetic procedure which installed structural bridges between the neighboring aromatics rings [4]. Then Veverka and Jerábek [5] developed further the synthesis of HXL materials via internal electrophile-based chemistry by incorporating vinylbenzyl chloride as a comonomer into the polymer chains and then exploiting the pendent chloromethyl groups in the Friedel–Crafts alkylation reactions.
Our approach consisted in the use of tetrafunctional monomers for the synthesis of polymeric stationary phases [6,7,8,9,10,11,12]. In this study in addition to divinylbenzene (DVB), there was used dimethacrylic derivative of naphthalene, i.e., di(methacryloyloxymethyl)naphthalene (DMN) which resulted in the introduction of polar ester groups into the polymers structure. In this way there were obtained highly crosslinked polymeric microspheres that can be applied as stationary phases for chromatography as well as catalysts and adsorbents. To make them look like alkyl-bonded silica phases, alkyl chains in the Friedel–Crafts reaction were incorporated into the structure of parent polymer. In our case, this reaction meant to introduce alkyl chains instead of forming additional polymer crosslinking. The practical application of such materials requires not only knowledge of their chemical structure and resulting polarity, but also of their thermal resistance. Hence, the aim of this paper was to investigate the thermal stability of the prepared highly crosslinked DMN–DVB microspheres and their alkyl-bonded derivatives using thermogravimetry coupled online with FTIR spectroscopy (TG/FTIR) and differential scanning calorimetry (DSC). Moreover, the influence of modification reaction on the decomposition pathway of the copolymeric microspheres was determined.
Experimental
Materials
DVB, α,α′-azobisisobutyronitrile (98%), decan-1-ol and Aerosol OT-75 were purchased from Sigma-Aldrich (Germany). The DMN monomer being a mixture of 1,4- and 1,5-isomers was synthesized in our laboratory according to the earlier described procedure [13, 14]. The compounds used for modification of the DMN-DVB copolymer, i.e., octyl chloride, octadecyl chloride (Merck, Germany), nitromethane (POCh, Poland) and anhydrous aluminium chloride (Fluka AG, Switzerland), as well as the solvents such as: toluene, methanol, tetrahydrofuran, acetone and hydrochloric acid (POCh, Poland) were analytical reagent grade.
Synthesis of porous microspheres
The DMN-DVB porous copolymer was synthesized by combined suspension-emulsion polymerization in the presence of pore forming diluents (decan-1-ol and toluene (80:20 v/v)). The DMN and DVB monomers (0.5:0.5 molar ratio) were dissolved in diluents to form an organic phase to which the initiator, α,α′-azobisisobutyronitrile (1 mass% based on monomers), was added. Then the homogeneous organic phase was added to the 180 mL aqueous phase containing as a surfactant Aerosol OT-75 (0.25 mass%) while stirring. Polymerization lasted 20 h at 80 °C. The obtained copolymers in the shape of beads were washed with hot water and then toluene, acetone and methanol in a Soxhlet apparatus. Uniform particles (5–15 μm) were isolated by sedimentation from the methanol-acetone (90:10; v/v) mixture [15].
The C8 and C18 alkyl chains were introduced into the aromatic rings of the DMN-DVB porous copolymer by means of the Friedel–Crafts reaction. In order to bind C8 alkyl chains, the copolymer (6 g), nitromethane (60 mL), octyl chloride (14 g, 0.1 mol) and anhydrous aluminium chloride (8 g) were stirred at 90 °C for 24 h. Released hydrogen chloride was absorbed in water. The same procedure was applied to bind C18 alkyl chains, but the amount of octadecyl chloride was increased to 28 g (0.1 mol). The mixture was then poured into an ice water mixture and the precipitated copolymer washed several times with acetone and methanol in a Soxhlet apparatus. To remove traces of aluminium, the copolymer was additionally washed with tetrahydrofuran-concentrated hydrochloric acid (90:10, v/v). The levels of aluminium determined by the ASA method in C8- and C18-modified copolymers were 0.05 and 0.06%, respectively.
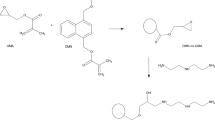
The general scheme of copolymeric microspheres synthesis is presented in Fig. 1.

General scheme of copolymeric microspheres synthesis
Methods of analysis
The microspheres were imaged using a Morphologi G3 optical microscope (Malvern Instruments Ltd, UK) equipped with image analysis software and 987× magnification.
Attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectra were obtained with a Bruker Tensor 27 FTIR spectrometer (Germany). The FTIR spectra were recorded in the spectral range of 600–4000 cm−1 with 32 scans per spectrum with a resolution of 4 cm−1.
Thermogravimetry (TG) was performed with a Netzsch STA 449 F1 Jupiter thermal analyzer (Germany) in the range of 30–1000 °C in helium (flow = 20 cm3 min−1), at the heating rate of 10 °C min−1. All TG measurements were taken in Al2O3 crucibles (mass of 160 ± 1 mg). As a reference, empty Al2O3 crucible was applied. Sample masses of 10.4 ± 0.2 mg were used. The composition of the gas evolved during the decomposition process was analyzed by a Bruker Tensor 27 FTIR spectrometer (Germany) coupled online to a Netzsch STA instrument by Teflon transfer line with 2 mm diameter heated to 200 °C. The FTIR spectra were recorded in the spectral range of 600–4000 cm−1 with 16 scans per spectrum at 4 cm−1 resolution.
Differential scanning calorimetry (DSC) measurements were taken with a Netzsch 204 calorimeter (Germany) operating in the dynamic mode. The dynamic scans were performed at the heating rate of 10 °C min−1 from room temperature to 550 °C in helium atmosphere (flow = 20 cm3 min−1) in two stages. The first scan was performed from room temperature to 120 °C to remove any adsorbed moisture, particularly water. The second scan was conducted from 20 to 550 °C. All DSC measurements were taken in aluminum pans with a pierced lid (mass of 40 ± 1 mg). As the reference the empty aluminum crucible was applied. The sample masses of 7.3 ± 0.2 mg were used.
Results and discussion
The highly crosslinked porous microspheres of DMN-DVB with diameters in the range of 10–50 μm were obtained via the radical suspension-emulsion polymerization. In order to modify their chemical structure, the Friedel–Crafts reaction was performed. In the course of the aromatic substitution, the C8 and C18 alkyl chains were introduced into the aromatic rings of the DMN-DVB copolymer and DMN–DVB–C8 and DMN-DVB-C18 materials are produced. Figure 2 presents the optical images of the obtained microspheres.

Optical images of a DMN-DVB-C8 and b DMN-DVB-C18 microspheres
The chemical structure of the starting copolymer and modified ones was confirmed by the ATR-FTIR analysis. As can be seen in the spectra presented in Fig. 3, in the structure of the parent DMN-DVB copolymer the ester, aliphatic and aromatic fragments are present. It is evidenced by the following absorption bands at: 1726 cm−1 (C=O), 1165, 1124 cm−1 (C–O–C), 1602, 1452, 833–709 cm−1 (Ar), 2958–2875 cm−1 (CH3, CH2). Furthermore, a low intense absorption band at 1638 cm−1 originated from the unreacted C=C double bonds is visible. In turn, in the spectra of modified copolymers the intensity of absorption bands typical of ester groups decreased significantly. Moreover, the absorption band of ester carbonyl group is shifted to lower wavenumbers (1702 cm−1). At the same time the intensity of absorption bands characteristic of methyl and methylene groups increased. This points out that the alkyl chains were successfully incorporated into the structure of the parent copolymer.

ATR-FTIR spectra of copolymers
Thermal properties of the obtained copolymers were determined in inert atmosphere using the TG and DSC methods. Table 1 compiles characteristic temperatures for the decomposition process of the copolymers, i.e., T1, T20, T50 and Tmax, that refer to the temperatures of 1, 20, 50% mass loss and of the maximum rate of mass loss as well as the residual masses, whereas the thermogravimetric (TG) and differential TG (DTG) curves are shown in Fig. 4. The FTIR spectra of volatile decomposition products of copolymers are given in Figs. 5–8. In turn, Fig. 9 shows the DSC curves obtained for these materials.

TG and DTG curves of copolymers

3D plots of FTIR spectra of volatile products obtained during the thermal decomposition of copolymers
According to the data presented in Table 1, the modified materials (DMN-DVB-C8 and DMN-DVB-C18) exhibit higher T20 and T50 than the non-modified one (DMN-DVB), while the T1 values are lower. On this basis it could be concluded that the introduction of alkyl chains into the structure of the polymeric packing deteriorates its thermal stability only in the initial decomposition stage. Taking the residual mass into account, one can state that the modified copolymers decomposed to a lower extent in comparison with the non-modified one.
From the course of DTG curves (Fig. 4), it follows that DMN-DVB copolymer decomposed in three stages, whereas the DMN-DVB-C8 and DMN-DVB-C18 ones in four stages. On the curve of DMN-DVB copolymer, there can be seen one small and broad peak with the maximum at 190 °C and a small one visible as a shoulder with the maximum at 500 °C, both connected with ~ 2% mass loss. There is also a large intense peak in the wide range of ~ 290–480 °C (with the maximum at 403 °C) related to 83% mass loss. However, on the DSC curve (Fig. 8) in this temperature range two endothermic peaks can be seen with the maxima at 349 and 414 °C. This suggests that the main decomposition of the parent copolymer proceeds in two steps. On the basis of the FTIR spectra of volatile decomposition products (see Fig. 6), it can be stated that at both 349 and 414 °C temperatures the decomposition is associated with the evolution of the same substances, i.e., water (bands at ~ 4000–3500 cm−1, associated with the stretching vibrations and at ~ 1800–1300 cm−1—with the bending vibrations), carbon dioxide (bands at 2359–2310 cm−1, attributed to the asymmetric stretching vibrations and at 669 cm−1, associated with the degenerate bending vibrations), carbon monoxide (bands at 2181 and 2114 cm−1, related to the stretching vibrations), unsaturated compounds (at 3095–2886 cm−1, attributed to the C–H stretching vibrations of methylene and methyl groups and at 960 and 910 cm−1, related to the C–H out-of-plane deformation vibrations of vinyl group) and carbonyl compounds, including esters (bands at 1772–1750 cm−1, characteristic of the C=O stretching vibrations and at 1124 cm−1, connected with the C–O stretching vibrations). The differences occur only in the amounts of the generated products (different peak intensities). From the spectrum collected at 190 °C (Figs. 5 and 6), it arises that at the beginning of the decomposition the emission of water and carbon dioxide takes place. This can be due to the degradation of ester bonds. In the last decomposition step (at 500 °C), the evolution of mainly water and carbon dioxide was recorded (Fig. 6). Moreover, in this FTIR spectrum the presence of aromatic compounds was detected (bands at 3015 cm−1, attributed to the C–H stretching vibrations and at 783 and 753 cm−1, associated with the C–H out-of-plane deformation vibrations). Referring to the DSC curve, it should be noted that besides the endothermic peaks, the exothermic one with the maximum at 178 °C is also visible, which is associated with the post-crosslinking. This is a typical reaction of highly crosslinked polymers that possess unreacted C=C double bonds in their structure [9, 16, 17].

FTIR spectra of volatile products obtained at the maximum rate of mass loss and additionally at 349 °C of the thermal decomposition of DMN-DVB copolymer
The course of DMN-DVB-C8 and DMN-DVB-C18 materials decomposition differs significantly from that of the parent copolymer. Four peaks are observed on DTG curves for both modified copolymers (Fig. 4). The first one with the maximum at ~ 60 °C is connected with the evaporation of water molecules absorbed in the microsphere pores (see FTIR spectra in Figs. 7 and 8) [17,18,19]. The next one with the maximum at 183 °C for DMN-DVB-C8 and 249 °C for DMN-DVB-C18 is mainly associated with the decomposition of the aliphatic chains attached to the microspheres. The FTIR spectra (Figs. 7 and 8) exhibit the absorption bands typical of aliphatic carbonyl compounds (at 2966–2877, 1766 and 1123 cm−1), carbon dioxide and water. As follows from the TG curves, a higher mass loss in this step is observed for the former copolymer (11 vs. 5%). Such difference is most probably caused by the efficiency of the alkyl Friedel–Crafts reaction. Although the molar feed of C8 and C18 alkyl chlorides was the same, this substitution reaction proceeds easier in the case of C8 alkyl chloride due to a steric hindrance. The following decomposition step with the highest mass loss of ~ 48% for both copolymers is observed in the temperature range of 285–500 °C. It is connected with the decomposition of the core crosslinked network similarly to that of the parent DMN-DVB copolymer. However, the maximum of the corresponding DTG peak is shifted to higher temperature (from 403 to 418 °C). It can be assumed that this is a result of the alkyl chains decomposition. The formed solid products deposited onto the microsphere surface and inside large macropores delay the breakdown of ester linkages and clog the evolution of volatile compounds. In the FTIR spectra (Figs. 7, 8), the absorption bands characteristic of carbon dioxide, carbon monoxide, water and carbonyl compounds, including esters, is visible. Moreover, the spectra exhibit the bands pointing to the formation of alcohols (at 3582, 1013 cm−1, related to the O–H and C–OH stretching vibrations, respectively) and methane (at ~ 3100–3016 and 1305–1200 cm−1, attributed to the stretching and deformation vibrations, respectively [20, 21]). The peak at 3016 cm−1 can also originate from the unsaturated compounds (C–H stretching vibrations). Formation of unsaturated products is also evidenced by the bands at 1632 cm−1 (C=C stretching vibrations) and 943 and 910 cm−1 (C–H out-of-plane deformation vibrations of vinyl group). The last peak on the DTG curves with the maximum at ~ 505 °C relates to 8% mass loss. The FTIR spectra of volatile products obtained at this temperature display bands originating from carbon dioxide, carbon monoxide, methane, carbonyl and aromatic compounds, water and alcohols. In both C8- and C18-modified copolymers, the residual mass is about 3 times higher than that of the non-modified one. This can be caused by the secondary reactions taking place inside microspheres, as a result of which the nonvolatile products are generated. In summary, the FTIR analyses of volatile decomposition products performed simultaneously confirmed our assumptions about the decomposition pathway of the copolymers. Considering the TG/FTIR data, it can be stated that microspheres of both types mainly decomposed according to the random chain scission mechanism starting from the cleavage of ester bonds. Furthermore, in the case of modified microspheres the carbon–carbon cleavage within the alkyl chains probably occurred [22, 23]. With regard to the DSC analysis, one can notice the exothermic peaks associated with the post-crosslinking processes on the curves obtained for the modified copolymers, just as in the case of non-modified one (Fig. 9). They are followed by the endothermic peaks with the maxima at 238 and 412 °C (for DMN-DVB-C8) and at 241 and 410 °C (for DMN-DVB-C18) corresponding to the decomposition of aliphatic chains and crosslinked network, respectively.

FTIR spectra of volatile products obtained at the maximum rate of mass loss of the thermal decomposition of DMN-DVB-C8 copolymer

FTIR spectra of volatile products obtained at the maximum rate of mass loss of the thermal decomposition of DMN-DVB-C18 copolymer

DSC curves of copolymers
Conclusions
On the basis of the thermogravimetric data obtained, it can be concluded that the parent DMN–DVB copolymer is thermally stable up to 219 °C, whereas the modified ones up to 139 °C (DMN–DVB–C8) and 166 °C (DMN–DVB–C18), as measured at the temperature of 1% mass loss. The modification process had an influence on the thermal decomposition of the prepared materials, particularly in the initial stage. Although the introduction of alkyl chains into the structure of DMN–DVB copolymer deteriorated its thermal stability, it is sufficient to apply these modified materials as adsorbents in different separation techniques. The investigations also show that the thermal decomposition process of the copolymers proceeded in three (for the non-modified one) or four (for the modified ones) stages. In all cases, the main decomposition took place in the penultimate stage. Moreover, the DSC analysis shows that the decomposition of the parent and modified copolymers is an endothermic process, while the observed post-crosslinking is an exothermic one. The basic volatile products obtained during the thermal decomposition of the copolymers were carbon dioxide, carbon monoxide, aromatic and unsaturated compounds, water, carbonyl compounds, including esters, and in the case of DMN–DVB–C8 and DMN–DVB–C18 also alcohols and methane. One common conclusion for the studied copolymers is that these materials decomposed according to the random chain scission mechanism.
References
Benson JR, Woo DJ. Polymeric columns for liquid chromatography. J Chromatogr Sci. 1984;22:386–99.
Yang Y-B, Regnier FE. Coated hydrophilic polystyrene-based packing materials. J Chromatogr. 1991;544:233–47.
Xiao WT, Zhang ZJ. Fabrication of porous polymers. Prog Chem. 2009;21:1299–303.
Tsyurupa MP, Davankov VA. Porous structure of hypercrosslinked polystyrene: state-of-the-art mini-review. React Funct Polym. 2006;66:768–79.
Veverka P, Jerábek K. Mechanism of hypercrosslinking of chloromethylated St-DVB copolymers. React Funct Polym. 1999;41:21–5.
Gawdzik B. Chemical modification of highly cross-linked di(methacryloyloxymethyl)naphthalene–divinylbenzene copolymer for HPLC. Chromatographia. 1993;35:548–54.
Gawdzik B, Osypiuk J. Reversed-phase high-performance liquid chromatography on porous copolymers of different chemical structure. J Chromatogr A. 2000;898:13–21.
Maciejewska M. Synthesis and thermal properties of parent and modified DMN–co-GMA copolymers. J Therm Anal Calorim. 2018;133:969–80.
Grochowicz M, Gawdzik B. Preparation and characterization of porous crosslinked microspheres of new aromatic methacrylates. J Porous Mater. 2013;20:339–49.
Podkościelna B. The highly crosslinked dimethacrylic/divinylbenzene copolymers. Characterization and thermal studies. J Therm Anal Calorim. 2011;104:725–30.
Gawdzik B, Podkościelna B, Bartnicki A. Synthesis, structure and properties of new methacrylic derivatives of naphthalene-2,3-diol. J Appl Polym Sci. 2006;102:1886–95.
Podkościelna B, Bartnicki A, Podkościelny P. New ion exchangers based on copolymers: 2,3-(2-hydroxy-3-methacryloyloxy-propoxy)naphthalene–styrene. Sep Sci Tech. 2014;49:1672–8.
Matynia T, Gawdzik B. Influence of synthesis conditions of porous copolymers of 1,4-di(methacryloyloxymethyl)naphthalene with divinylbenzene on their structure. 1. Angew Makromol Chem. 1987;147:123–32.
Gawdzik B, Matynia T. Influence of synthesis conditions of porous copolymers of 1,4-di(methacryloyloxymethyl)naphthalene with divinylbenzene on their structure. 2. Angew Makromol Chem. 1987;152:33–9.
Gawdzik B, Gawdzik J, Czerwińska-Bil U. Copolymer of di(methacryloyloxymethyl)naphthalene and divinylbenzene as a column packing for high performance liquid chromatography. Chromatographia. 1988;26:399–407.
Grochowicz M. Investigation of the thermal behavior of 4-vinylpyridine–trimethylolpropane trimethacrylate copolymeric microspheres. J Therm Anal Calorim. 2014;118:1603–11.
Maciejewska M. Thermal properties of TRIM–GMA copolymers with pendant amine groups. J Therm Anal Calorim. 2016;126:1777–85.
Grochowicz M, Pączkowski P, Gawdzik B. Investigation of the thermal properties of glycidyl methacrylate–ethylene glycol dimethacrylate copolymeric microspheres modified by Diels-Alder reaction. Therm Anal Calorim. 2018;133:499–508.
Maciejewska M. Influence of the filler on thermal properties of porous VP-TRIM copolymers. J Therm Anal Calorim. 2015;119:507–13.
The website http://webbook.nist.gov/chemistry/.
Lu R, Purushothama S, Yang X, Hyatt J, Pan WP, Riley JT, Lloyd WG. TG/FTIR/MS study of organic compounds evolved during the co-firing of coal and refuse-derived fuels. Fuel Process Technol. 1999;59:35–50.
Billaud F, Chaverot P, Berthelin M, Freund E. Thermal decomposition of aromatics substituted by a long aliphatic chain. Ind Eng Chem Res. 1988;27:1529–36.
Zámostný P, Bělohlav Z, Starkbaumová L, Patera J. Experimental study of hydrocarbon structure effects on the composition of its pyrolysis products. J Anal Appl Pyrolysis. 2010;87:207–16.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gawdzik, B., Rogulska, M., Grochowicz, M. et al. Studies of thermal properties of di(methacryloyloxymethyl)naphthalene–divinylbenzene (DMN–DVB) copolymer and its alkyl-bonded derivatives. J Therm Anal Calorim 138, 4385–4393 (2019). https://doi.org/10.1007/s10973-019-08150-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-019-08150-7