
Abstract
The effect of starch-g-copolymers structure on the thermal decomposition course under oxidative conditions by using the simultaneous TG/DSC/FTIR technique has been investigated. To those studies, two types of starch-g-copolymers obtained under the graft copolymerization process of aromatic acrylate monomers such as phenyl acrylate and benzyl acrylate onto starch have been chosen. The TG/DSC investigations confirmed that the course of the decomposition process of all studied copolymers under the heating and in the presence of air was similar and run through three large stages. TG/FTIR studies indicated on the same decomposition mechanism under the first decomposition stage, however, on the completely different decomposition mechanism under the second decomposition stage of the copolymers which was directly dependent on the structure of grafted polymer. In addition, it was also proved that the percent grafting of the copolymers has not influenced on its decomposition course and its decomposition mechanism. The grafting percent influenced only on the Δm in each decomposition stage and on the intensity in the emission of the volatile species.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Meth(acrylate) monomers are generally applied as the building blocks for the production of whole range of various structure synthetic resins. For example, the polymerization of methyl methacrylate leads to the formation of poly(methyl methacrylate). It is linear, amorphous, shiny thermoplastic polymer, and due to its superior properties like excellent weather and chemical resistance, very good optical properties such as high light transmittance (up to 93%) and UV transmittance of ca. 70%, high mechanical strength, low elongation at break, low water absorption capacity, good dimensional stability, favorable rheological properties, low cost, easy handling and processing, it is widely applied in the industry. It is commonly used in optical, automotive, electrical, medical or other industry for the production of panes, for the manufacturing of polymer optical fibers, orthopedic materials, sensors, sunglasses, watch glasses, indicators, lamp covers, packing for tablets, as a thickener in cosmetic formulations, immobilization material, the component of nanocomposites, etc. [1,2,3,4,5,6,7,8,9,10].
The copolymerization of meth(acrylate) monomers with other unsaturated monomers such as styrene, vinyl pyrrolidone, divinylbenzene, acrylamide and acrylonitrile allows obtaining of many novel materials with unique properties which can find their potential applications as a sorbents, modifiers, fillers, porous microspheres, polymers for making the industrial and daily products [11,12,13,14,15,16,17,18,19,20].
Also, meth(acrylate) monomers are widely used in order to modify the structure of polymers having functional groups such as natural polymers. Among them, polysaccharides like starch, cellulose, chitosan, dextrane are the group of biodegradable polymers which are often modified under the graft copolymerization process with series of aliphatic methacrylates, e.g., acrylic acid, methacrylic acid, methyl methacrylate, methyl acrylate, ethyl methacrylate, [21,22,23,24,25,26] or aromatic methacrylates, e.g., phenyl methacrylate, benzyl methacrylate or benzyl acrylate [27,28,29]. The graft modification of polysaccharides enables improving and enhancing their properties and thus broadens their application abilities, especially for biomedical and pharmaceutical applications.
The present paper is a continuous part of the earlier studies on the properties of starch-g-copolymers prepared in the presence of aromatic meth(acrylate) monomers. The aim of this study was to investigate and to compare the thermal properties and the decomposition mechanism under oxidative conditions of two types of starch-g-copolymers which differed in their structure and the grafting percent (%G). To this studies, starch-g-poly(phenyl acrylate) and starch-g-poly(benzyl acrylate) copolymers prepared under a radical graft copolymerization process of potato starch and one of the aromatic acrylate monomers: phenyl acrylate or benzyl acrylate in the presence of potassium persulfate as a initiator, have been chosen. The detailed studies on the thermal behavior and the decomposition mechanism under oxidative conditions of the prepared copolymers by applying the TG/DSC/FTIR-coupled technique have been evaluated.
Experimental
Materials
Potato starch was extracted from the potato flour (Melvit S.A. Poland) according to the method given in Ref. [30]. Benzyl acrylate and phenyl acrylate monomers were obtained in the Department of Polymer Chemistry, Poland, according to the procedure described in Ref. [31]. Potassium persulfate and the solvents: methanol, chloroform and tetrahydrofuran, were delivered by Merck, Germany.
Graft copolymerization
Starch graft copolymerization reactions in a glass flask equipped with thermometer, mechanical stirrer and a nitrogen gas inlet have been performed. To obtain the copolymers, firstly, potato starch (the amounts given in Table 1) and water (50 mL) were put into glass flask, heated up to the temperature of 80 °C and stirred for 15 min. After forming a starch gel, initiator (K2S2O8) was added and the glass flask was filled with a gaseous nitrogen and stirred for the next 15 min at 80 °C. Finally, acrylate monomer: phenyl acrylate or benzyl acrylate to the reaction mixture, was added and stirred for the predominant time. The temperature of the grafting reaction was kept on the constant value (80 ± 2 °C). The graft copolymerization process was interrupted by the cooling of the reaction mixture in a water bath to the room temperature and pouring it in methanol (100 mL). Then, the copolymers were isolated from the homopolymers by the purification in CHCl3 and THF, filtered and dried at 60 °C to the constant mass [27,28,29]. The applied grafting parameters allowed obtaining the copolymers with different grafting percent (%G), as given in Table 1, counted using the following equation [32, 33]:
The grafting copolymerization process led to the formation of unsoluble copolymers in organic solvents whose structures based on ATR-FTIR and 13C CP/MAS NMR analyses were confirmed, as given in Tables 2 and 3.
Methodology
The thermal characteristic of the copolymers using a STA 449 F1 Jupiter thermal analyzer produced by Netzsch company (Germany) was made. The TG/DTG/DSC analyses at a heating rate of 10 °C min−1 under the presence of the oxidative conditions: synthetic air with a flow rate of 100 mL min−1 and the temperature range of 40–650 °C, have been performed. All the samples in Al2O3 crucibles were analyzed. The mass of the sample was ca. 10 mg.
The detection of the volatile decomposition products emitted under the decomposition of the copolymers by applying the FTIR analyzer was done. The FTIR analyzer (Tensor 27 Bruker, Germany) in a wavenumber range of 600–4000 cm−1 with a resolution of 4 cm−1 and 16 scans per spectrum was operated.
Results and discussion
TG–DTG–DSC
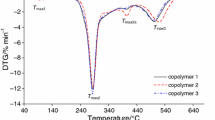
On the basis of the results presented in Fig. 1, up to the temperature of ca. 210–215 °C, the starch-g-poly(phenyl acrylate) copolymers were thermally stable, and no mass loss was indicated. However above 210 °C, the beginning of the decomposition process of the studied materials was observed. It was well visible that the applied oxidative atmosphere caused their decomposition in three stages. The first decomposition stage happened from ca. 210 °C to ca. 330–335 °C with T max1 282–286 °C, as given in Table 4. As it was clearly visible, the mass loss under the first stage was directly dependent on the %G and ranged from 45.6% for copolymer 1 (%G = 10.2) to 29.6% for copolymer 3 (%G = 41.8). Meanwhile, under the second decomposition stage (from ca. 330–335 °C to ca. 415–430 °C), the mass losses were changed from 25.5% (copolymer 1) to 51.5% (copolymer 3). When the copolymers were heated above 415–430 °C, the third mass loss up to temperature of ca. 560 °C with T max3 491–508 °C was appeared. The mass loss was from 27.8% for copolymer 1 up to 15% for copolymer 3 during this stage.

TG/DTG/DSC curves for starch-g-poly(phenyl acrylate) copolymers in air atmosphere
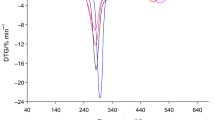
The similar course of the TG/DTG/DSC curves for starch-g-poly(benzyl acrylate) copolymers in air as compared to those observed for starch-g-poly(phenyl acrylate) copolymers has been observed, as shown in Fig. 2. The starch-g-poly(benzyl acrylate) copolymers also exhibited three main, non-well separated degradation stages. The first decomposition stage between the temperatures of 200–215 °C and 320–325 °C with maximum rate peak (T max1) at 285–287 °C was indicated. The second one was visible from the temperature of 320–325 °C up to the temperature of 410–420 °C with T max2 at 366–380 °C. The last, third decomposition stage was appeared between the temperatures of 410–420 °C and 585–610 °C with T max3 above 499 °C, as given in Table 4. Under the first decomposition stage, the mass loss was from 45.4% for copolymer 4 to 27.2% for copolymer 6. The second mass loss was changed between 20.2% (copolymer 4) and 48.0% (copolymer 6). However, the third mass loss was from 28.6% for copolymer 4 to 21.0% for copolymer 6, as given in Table 4.

TG/DTG/DSC curves for starch-g-poly(benzyl acrylate) copolymers in air atmosphere
By comparing the thermal degradation behavior of the copolymers, it was concluded that their degradation profile in each decomposition stage changed with the grafting percent (%G). The mass loss under the first decomposition stage decreased as the %G was increased, the mass loss under the second decomposition stage increased as the %G was increased, and finally, the mass loss under the third decomposition stage decreased as the %G was increased. In addition, the heating of the copolymers to ca. 600–650 °C led to their full decomposition under oxidative atmosphere because no residual mass after the heating process was observed.
On the DSC curves, as shown in Figs. 1 and 2, two types of the thermal signals were clearly visible. Small endothermic peaks which appeared at the temperature below 130 °C were connected with the evaporation of moisture from the copolymers. However, the endothermic peaks visible at higher temperatures were responsible for the breaking of the bonds in the structure of the copolymers. Meanwhile, the occurrence of some, high-intensity, exothermic signals on the DSC curves was directly connected with the presence of an oxygen under the heating of the copolymers which caused the oxidation reactions of the intermediates and/or residue formed as a result of the cleavage of the bonds in the structure of the copolymers [35,36,37,38]. As a result, under the oxidation processes, the creation of another gaseous decomposition products as compared to those formed in inert conditions was expected.
TG–FTIR
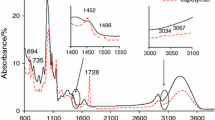
The heating of the starch-g-poly(phenyl acrylate) and starch-g-poly(benzyl acrylate) copolymers under the conditions of synthetic air caused their decomposition at three main stages which was connected with the emission of different structure gaseous products as it is shown in Figs. 3–5. Analyzing of the FTIR data obtained for the starch-g-poly(phenyl acrylate) copolymers led to the following conclusion: under the first decomposition stage, the creation of the decomposition products of starch such as CO2 (two bands at 670 cm−1 and at 2330–2365 cm−1), CO (two bands at 2000 and 2200 cm−1), H2O (bands at 3500–3900 cm−1), furane [bands at 742–754 cm−1, 825 cm−1, 905 cm−1 (δ C–H), 1508–1560 cm−1 (ν ring C=C)], alkane [bands at 2870–2970 cm−1 (ν C–H), 1340–1480 cm−1 (δ C–H)], acid [bands at 1780 cm−1 (ν C=O), 1163 cm−1 (ν C–O), above 3500 cm−1 (ν OH)], aldehyde [bands at 1745 cm−1 (ν C=O), 2728 cm−1, 2800 cm−1 (ν C–H), 1105 cm−1 (ν C–O)] and alcohol (bands at 1081 cm−1 (ν C–O), above 3500 cm−1 (ν OH) fragments) was clearly indicated [39,40,41]. Meanwhile, when the temperature achieved ca. 330 °C, the emission of the following volatile decomposition products: aromatic: phenyl alcohol [bands at 748–887 cm−1 (δ CAr–H), 1068 cm−1 (ν C–O), two bands at 1489 and 1600 cm−1 (ν C=CAr), 3052 cm−1 (ν CAr–H) and above 3500 cm−1 (ν OH)], ketone (mainly cyclohexanone), acids and anhydride fragments [bands at 3647 cm−1 (ν OH), 1182–1260 cm−1 (ν C–O), 1332–1450 cm−1 (δ C–H), 1756–1783 cm−1 (ν C=O) and small bands at 2860–2970 cm−1 (ν C–H)] and CO2, CO and H2O, was indicated, as shown in Fig. 3. In addition, on the basis of the results shown in Fig. 3, under the third decomposition stage, mainly the evolution of CO2, CO and H2O from the FTIR spectra as a result of the oxidation process of the residue was observed.

Gaseous FTIR spectra for starch-g-poly(phenyl acrylate) copolymers
Figure 4 shows the FTIR spectra for the starch-g-poly(benzyl acrylate) copolymers gathered at T max1, T max2 and T max3 under air atmosphere. On the basis of Fig. 4, one can clearly see the emission of the same volatile decomposition products under the first decomposition stage (T max1) as compared to those observed for the starch-g-poly(phenyl acrylate) copolymers. This was confirmed by the presence of the following bands on the FTIR spectra gathered at T max1 which were responsible for the following inorganic species: CO2 (at 670 and 2330–2365 cm−1), CO (2000–2200 cm−1), H2O (3500–3900 cm−1) and some organic species. The presence of organic species such as furanes [bands at 740–900 cm−1 (out of plane δ C–H), 1510–1580 cm−1 (ν ring C=C)], acid [bands at 1787 cm−1 (ν C=O), 1162 cm−1 (ν C–O), above 3500 cm−1 (ν OH)], aldehyde [bands at 1745 cm−1 (ν C=O), 2720–2800 cm−1 (ν C–H), 1100 cm−1 (ν C–O)] and alcohol (bands at 1080 cm−1 (ν C–O) and above 3500 cm−1 (ν OH) [39,40,41]) has been indicated. It confirmed that the decomposition mechanism of starch-g-copolymers under the first decomposition stage was independent on the type of aromatic acrylate monomer used for the grafting process. Under this decomposition stage, the cleavage of the glycosidic bonds of starch, dehydration and thermal condensation of hydroxyl groups were the most probable processes which were in accordance with other studies [39, 42, 43].

Gaseous FTIR spectra for starch-g-poly(benzyl acrylate) copolymers
However, the differences in the emission of volatiles under the second decomposition stage for the starch-g-poly(phenyl acrylate) and starch-g-poly(benzyl acrylate) copolymers have been clearly observed. In Fig. 5, the exemplary 3D FTIR spectra for two chosen copolymers (copolymer 2 and copolymer 5) were presented. In the case of the starch-g-poly(phenyl acrylate) copolymers, high emission of both inorganic and organic species was observed. However, what is interesting is that the higher emission of inorganic species (CO2, CO, H2O) than organic species under the second decomposition stage of starch-g-poly(benzyl acrylate) has been indicated. It may be the result of the oxidation processes of some organic volatile decomposition products formed under the cleavage of the bonds in the structure of grafted polymer. Regarding the structure of starch-g-poly(benzyl acrylate) copolymers and the previous study performed for the copolymers in inert conditions [27], the cleavage of the main and side chains of graft polymer was the two main processes happened under the heating. It led to the formation of aromatic aldehyde, aromatic alcohol, alkene, cyclic ketone, acid anhydride and monomer [27] as a primary decomposition products. However, it can be suspected that under the influence of oxygen some of the initially formed volatiles generated under the decomposition of poly(benzyl acrylate) can be oxidized leading to the formation of higher amounts of inorganic species. The formation of some volatile organic species was confirmed by the presence of the following absorption bands on the FTIR spectra: above 3500 cm−1 (ν OH), at 3031 and 3064 cm−1 (CAr–H and ν = C–H), at 2870–2930 cm−1 (ν C–H), at 2720 and 2790 cm−1 (ν C–H in CHO), at 1716–1778 cm−1 (ν C=O), at 1018–1200 cm−1 (ν C–O) and below 900 cm−1 (out of plane δ = C–H and CAr–H) [39].

3D FTIR spectra for copolymer 2 and copolymer 5
Finally, under the third decomposition stage of starch-g-poly(benzyl acrylate), the presence of the same decomposition products (CO2, CO and H2O) as for starch-g-poly(phenyl acrylate) was indicated.
Conclusions
TG/DSC/FTIR-coupled technique was applied to study the thermal behavior and the decomposition mechanism under oxidative conditions of starch-g-copolymers prepared in the presence of two aromatic acrylate monomers: phenyl acrylate and benzyl acrylate which differed in their structure and grafting percent. On the basis of the conducted investigations, the following conclusions could be drawn:
-
all the copolymers decomposed in three, large stages which happened in the similar temperature range under oxidative conditions;
-
the first decomposition stage was happened between the temperatures of 210 °C and ca. 330–335 °C [for starch-g-poly(phenyl acrylate) copolymers] and 200–215 °C and 320–325 °C [for starch-g-poly(benzyl acrylate) copolymers], and it was attributed to the decomposition of starch from the copolymers and thus with the emission of CO2, CO, H2O, furane, alkane, acid, aldehyde and alcohol fragments as volatiles;
-
the second decomposition stage was observed from ca. 330–335 °C to ca. 415–430 °C [for starch-g-poly(phenyl acrylate) copolymers] and 320–325 °C up to 410–420 °C [for starch-g-poly(benzyl acrylate) copolymers], and it was mainly associated with the decomposition of grafted polymer and the evolution of the mixture of various volatile species which type was directly dependent on the type of grafted polymer. The decomposition of poly(phenyl acrylate) chains led to the formation of mainly phenyl alcohol, ketone (mainly cyclohexanone), acids and anhydride fragments, CO2, CO and H2O. However, the decomposition of poly(benzyl acrylate) chains was connected with the creation of higher amounts of inorganic species (CO2, CO, H2O) than organic species (aromatic aldehyde, aromatic alcohol, alkene, cyclic ketone, acid anhydride and monomer) which was connected with the oxidation processes of some volatile products under the decomposition. The firstly created volatiles were more susceptible on the oxidation processes than volatiles formed as a result of the decomposition process of poly(phenyl acrylate);
-
the third decomposition stage for all copolymers happened between 415–430 °C and 560 °C [for starch-g-poly(phenyl acrylate) copolymers] and 410–420 °C and 585–610 °C, and it was due to the oxidation processes of some residues formed under the previous decomposition stages connecting with the emission of CO2, CO and H2O; and
-
it was also found that the grafting percent (%G) did not influence on the decomposition course and the decomposition mechanism of the copolymers; it influenced only on the intensity in the emission of the volatiles.
References
Zaharieva J, Milanova M, Todorovsky D. Poly(methyl-methacrylate) as immobilization matrix for europium b-diketonates-morphology and fluorescent properties. Appl Surf Sci. 2011;257:6858–66.
Harlin A, Myllymaki H, Grahn K. Polymeric optical fibres and future prospects in textile integration. Autex Res J. 2002;2:132–43.
Arica MY, Yavuz H, Patir S, Denizli A. Immobilization of glucoamylase onto spacer-arm attached magnetic poly(methyl methacrylate) microspheres: characterization and application to a continuous flow reactor. J Mol Catal B Enzym. 2000;11:127–38.
Mergo P, Gil M, Podkościelny W, Worzakowska M. Physical sorption and thermogravimetry as the methods used to analyse linear polymeric structure. Adsorption. 2013;19:851–9.
Lewis S, Haynes V, Wheeler-Jones R, Sly J, Perks RM, Piccirillo L. Surface characterization of poly(methylmethacrylate) based nanocomposite thin films containing Al2O3 and TiO2 nanoparticles. Thin Solid Films. 2010;518:2683–7.
Ali U, Karim KJ, Buang NA. A review of the properties and applications of poly(methyl methacrylate) (PMMA). Polym Rev. 2015;55:678–705.
Zhao L, Guo Z, Ran S, Cao Z, Fang Z. The effect of fullerene on the resistance to thermal degradation of polymers with different degradation processes. J Therm Anal Calorim. 2014;115:1235–44.
Li Y, Zhao B, Xie S, Zhang S. Synthesis and properties of poly(methyl methacrylate)/montmorillonite (PMMA/MMT) nanocomposites. Polym Int. 2003;52:892–8.
Sutherland HJ, Lingle R. An acoustic characterization of polymethyl methacrylate and three epoxy formulations. J Appl Phys. 1972;43:4022.
Webb JCJ, Spencer RF. The role of polymethylmethacrylate bone cement in modern orthopaedic surgery. J Bone Joint Surg. 2007;89-B:851–7.
Fu J, Wang L, Zhang A. Synthesis of EPDM-graft-methyl methacrylate and styrene and its toughening effect on MS resin. Polym Bull. 2008;60:405–16.
Maeda H, Egawa H. Studies of selective adsorption resins. XXV. Preparation of macroreticular chelating resins containing aminomethylphosphonic acid groups from methyl methacrylate/divinylbenzene copolymer beads and their adsorption capacity. J Appl Polym Sci. 1987;33:1275–81.
Atia AA, Donia AM, El-Eneim SA, Yousif AM. Effect of chain length of aliphatic amines immobilized on a magnetic glycidyl methacrylate resin towards the uptake behavior of Hg(II) from aqueous solutions. Sep Sci Technol. 2007;42:403–20.
Mert EH, Yıldırım H. Porous functional poly(unsaturated polyester-co-glycidyl methacrylate-co-divinylbenzene) polyHIPE beads through w/o/w multiple emulsions: preparation, characterization and application. e-Polymers. 2014;14:65–73.
Gokmen MT, Du Prez FE. Porous polymer particles—a comprehensive guide to synthesis, characterization, functionalization and applications. Prog Polym Sci. 2012;37:365–405.
Donia AM, Atia AA, El-Boraey HA, Mabrouk DH. Adsorption of Ag(I) on glycidyl methacrylate/N,N′-methylene bis-acrylamide chelating resins with embedded iron oxide. Sep Purif Technol. 2006;48:281–7.
Kucharski M, Lubczak R. Copolymerization of hydroxyalkyl methacrylates with acrylamide and methacrylamide I. Determination of reactivity ratios. J Appl Polym Sci. 1997;64:1259–65.
Dickens SH, Stansbury JW, Choi KM, Floyd CJE. Photopolymerization kinetics of methacrylate dental resins. Macromolecules. 2003;36:6043–53.
Warson H, Finch CA. Applications of synthetic resin latices, latices in diverse applications, vol. 3. New York: Wiley; 2001.
Leung D, Spratt DA, Pratten J, Gulabivala K, Mordan NJ, Young AM. Chlorhexidine-releasing methacrylate dental composite materiale. Biomaterials. 2005;26:7145–53.
Çankaya N. Synthesis of graft copolymers onto starch and its semiconducting properties. Results Phys. 2016;6:538–42.
Kumar Thakur V. Cellulose-based graft copolymers. Structure and chemistry. New York: CRC Press, Taylor and Francis Group; 2015.
Wang S, Wang Q, Fan X, Xu J, Zhang Y, Yuan J, Jin H, Cavaco-Paulo A. Synthesis and characterization of starch-poly(methyl acrylate) graft copolymers using horseradish peroxidase. Carbohydr Polym. 2016;136:1010–6.
Gautam J, Kumar Pal M, Singh B, Bhatnagar U. Thermal and electrical properties of acrylic acid grafted onto starch by ceric ammonium nitrate and potassium permanganate initiator. Int J Plast Technol. 2011;15:188–98.
Kumar Pati M, Nayak P. Grafting vinyl Monomers onto chitosan: IV: Graft copolymerized of acrylic acid onto chitosan using ceric ammonium nitrate as the initiator-characterization and antimicrobial activities. Mater Sci Appl. 2011;2:1741–8.
Hemalatha T, Yadav S, Krithiga G, Sastry TP. Chitosan as a matrix for grafting methyl methacrylate: synthesis, characterization and evaluation of grafts for biomedical applications. Polym Bull. 2016;73:3105–17.
Worzakowska M. Thermal behavior, decomposition mechanism and some physicochemical properties of starch-g-poly(benzyl acrylate) copolymers. J Therm Anal Calorim. 2016;126:531–40.
Worzakowska M, Grochowicz M. Effect of some parameters on the synthesis and the physico-chemical properties of new amphiphilic starch-g-copolymers. Carbohydr Polym. 2015;130:344–52.
Worzakowska M. Starch-g-poly(benzyl methacrylate) copolymers. Characterization and thermal properties. J Therm Anal Calorim. 2016;124:1309–18.
Lim ST, Lee JH, Shin DH, Lim HS. Comparison of protein extraction solutions for rice starch isolation and effects of residual protein content on starch pasting properties. Starch/Stärke. 1999;51:410–5.
Grochowicz M, Gawdzik B. Preparation and characterization of porous crosslinked microspheres of new aromatic methacrylates. J Porous Mater. 2013;20:339–49.
Athawale VD, Rathi SC. Role and relevance of polarity and solubility of vinyl monomers in graft polymerization onto starch. React Funct Polym. 1997;34:11–7.
Fakhru’l-Razi A, Qudsieh IYM, Yunus WMZM, Ahmad MB, Rahman MZA. Graft copolymerization of methyl methacrylate onto sago starch using ceric ammonium nitrate and potassium persulfate as redox initiator system. J Appl Polym Sci. 2001;82:1375–81.
Sokrates G. Infrared and Raman characteristic group frequencies, tables and chartes. New York: Wiley; 2001.
Krongauz VV, Ling MTK. Photo-crosslinked acrylates degradation kinetics. J Therm Anal Calorim. 2009;96:715–25.
Wang C, Yang Y, Tsai YT, Deng J, Shu ChM. Spontaneous combustion in six types of coal by using the simultaneous thermal analysis-Fourier transform infrared spectroscopy technique. J Therm Anal Calorim. 2016;126:1591–602.
Yang J, Chen H, Zhao W, Zhou J. Combustion kinetics and emission characteristics of peat by using TG–FTIR technique. J Therm Anal Calorim. 2016;124:519–28.
Soi K, Ye L, Li G. Thermal oxidative aging behavior and stabilizing mechanism of highly oriented polyamide 6. J Therm Anal Calorim. 2016;126:795–805.
NIST Chemistry Webbook, NIST Standard Reference Data, 2011, http://webbook.nist.gov.
Liu L, Zhao X, Ma C, Chen X, Li S, Jiao C. Smoke suppression properties of carbon black on flame retardant thermoplastic polyurethane based on ammonium polyphosphate. J Therm Anal Calorim. 2016;126:1821–30.
Plis A, Kotyczka-Morańska M, Kopczyński M, Łabojko G. Furniture wood waste as a potential renewable energy source. A thermogravimetric and kinetic analysis. J Therm Anal Calorim. 2016;125:1357–71.
Janković B. Thermal characterization and detailed kinetic analysis of Cassava starch thermo-oxidative degradation. Carbohydr Polym. 2013;95:621–9.
Liu X, Ma H, Yu L, Chen L, Tong Z, Chen P. Thermal-oxidative degradation of high amylose corn starch. J Therm Anal Calorim. 2014;115:659–65.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Worzakowska, M. The effect of starch-g-copolymers structure on the oxidative behavior studied by the TG/DSC/FTIR-coupled method. J Therm Anal Calorim 129, 367–376 (2017). https://doi.org/10.1007/s10973-017-6182-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-017-6182-y