
Abstract
The unique value of the neutron activation analysis (NAA) for analytical quality assurance is demonstrated through its contribution to the certification of the reference materials over the time span of 40 years. Ratio primary reference measurement procedures i.e. definitive methods, based on radiochemical NAA, play an important role in confirming the validity of the certified values established in the international intercomparisons. Real analytical situations where the use of NAA was critical to get accurate results are presented. The dangers associated with the possible non-availability of the NAA in the near future are indicated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the contemporary world many important decisions (cf. Fig. 1.) concerning various domains of human activity may depend upon the results provided by various methods of analytical chemistry, including also the inorganic trace analysis. It is obvious that good decisions may be taken only when the results are reliable i.e. they are both accurate and precise. The question arises how reliable were and are now the results delivered by individual laboratories working in the field of inorganic trace analysis. Some examples of drastic discrepancies in results sent by different laboratories during world-wide intercomparisons, initiated by various organizations are reproduced in Table 1 [1,2,3,4,5,6,7,8,9,10,11,12,13]. If we recollect the definition of a measurement accuracy: closeness of agreement between a measured quantity value and a true quantity value of a measurand [14], the data presented in Table 1 clearly show that several laboratories had a tremendous difficulty in determining the true content of certain elements in various matrices. In another words they were making gross errors. As can be seen from Table 1 the range of results reaching in some cases two, three, or even five orders of magnitude is not so rare as one could usually think and the situation is changing rather slowly with time. It should be mentioned that differences in results reaching an order of magnitude occurred also in case when the laboratories were not recruited on a voluntary basis but were specially selected on the basis of their high reputation (shown as “sel.labs.” in Table 1). So, accuracy in trace analysis is still a problem that deserves constant attention. On the other hand one should remember that trace analysis is a difficult domain and there are really many possibilities of making gross errors. These problems have been discussed in detail elsewhere [15, 16] and are summarized in Table 2 [15].

Dependence of various human decisions on results provided by methods of analytical chemistry
The evolution of the position of NAA in the array of methods used in the inorganic trace analysis
Neutron activation analysis (NAA) has already been known for several decades as a well established technique in inorganic trace analysis. When nuclear reactors with their high fluxes of slow neutrons ranging from 1011 up to 1014 n cm−1 s−1 became available for the civil uses, at the end of forties and beginning of fifties of the last century, NAA became all of sudden the technique providing the best detection limits for several elements in comparison to other methods of analytical chemistry being in use at that time. As the time progressed, new sensitive methods for the determination of elements emerged like: atomic absorption spectrometry (AAS), various variants of emission spectroscopy (ES), X-ray fluorescence (XRF) and finally inductively coupled plasma mass spectrometry (ICP-MS), and NAA was gradually losing its unique position. At the same time, partly due to antinuclear hysteria in some countries, the number of research reactors and laboratories using NAA rather decreased than increased in the world. So, it seems worthwhile to look back and realize what was in the past and what is now the position and value of NAA among other methods of inorganic trace analysis, and what are the prospects for the future.
Contribution of NAA to the certification of the candidate reference materials
International world-wide interlaboratory comparisons have been used by various organizations as a tool for accurate determination of several elements in different carefully prepared natural materials to issue (after proper elaboration of results sent by individual laboratories), new certified reference materials (CRMs). The percent share of individual analytical techniques used in selected comparisons over the time span of over 40 years is presented in Figs. 2 and 3.

Percent share of results by various analytical methods in the IAEA intercomparisons in the years 1975–1980

Percent share of results by various analytical methods in the INCT and MODAS intercomparisons in the years 1987–2015
From the seventies and up to first half of the nineties of the previous century the domination of NAA is clearly visible. The contribution of NAA in the IAEA intercomparisons starting from the year 1975 was the highest from among all methods used and most often exceeded 50% (cf. Fig. 2). AAS was on the second place, significant contributions were due to ES and XEF methods, with only negligible contribution from mass spectrometry (MS). Similar situation was observed in the INCT intercomparisons (CTA-AC-1, CTA-FFA-1, CTA-OTL-1 and CTA-VTL-2) up to the middle of nineties of the XXth century although the share of NAA was smaller than in the seventies and barely exceeded 40% (cf. Fig. 3). However, at the break of a century NAA lost its leading role and was overtaken by AAS. In the intercomparisons INCT-TL-1, and INCT-MPH-2, considerable share of results was provided by ES and MS (ICP-MS) with minor contribution from XRF method. Ten years later, the four leading methods i.e. ICP-MS, AAS, NAA and XRF provided almost equal share of results in the INCT-OBTL-5 intercomparison. However, after only 5 years more, the leading role of ICP-MS was already beyond discussion and the share of NAA dropped to 12–18% as can be seen from M-2 BotSed and M-3 HerTis intercomparisons (see Fig. 3). The leading role of ICP-MS is not surprising and reflects the current trend in the contemporary trace analysis, but constantly decreasing share of NAA results is regrettable and if this tendency is going to be continued it may become dangerous in the long run for quality assurance, at least in some special cases as will be shown later in this paper.
The eminent role of NAA in the process of certification of the reference materials was emphasized also by several authors describing the production of NBS/NIST and BCR CRMs [17,18,19,20].
The dominant place of NAA in the earlier world-wide intercomparisons is especially worth highlighting if we consider that the share of NAA in the global number of analyses performed in various domains of science and technology was rather moderate. As an illustration can serve the statistics on relative usage of analytical methods used in exploration geochemistry in North America in 1971, based on a survey conducted by the Association of Exploration Geochemists [21], which is shown in Table 3. One can see that NAA is not directly listed in Table 3, what means that it is hidden in the “Others” and its share most probably did not exceed 1%. The great share of NAA results in world-wide laboratory intercomparisons, intended for certification of the candidate reference materials, seem to demonstrate that NAA specialists were generally confident that their results were accurate and they were not afraid to show their results to the public view. The same conclusion can be drawn from the survey of the laboratories which published their results on the analysis of NBS/NIST CRMs before 1980, as compiled by Gladney [22]. For NBS 1571 Orchard Leaves (1972), NBS 1573 Tomato Leaves (1976), NBS 1632 Trace Elements in Coal (1974) and NBS 1633 Trace Elements in Coal Fly Ash (1975) the sum of results published in chemical literature by analysts using AAS, XRF, ES and MS methods was in each case smaller than the number of results published by NAA specialists alone.
A case illustrating the problems with the accurate determination of scandium
In the certification process of the candidate CRMs, the usual intention of the organizers of an intercomparison is to get for each element a good number of results by various methods and to see (after rejection of outliers) whether the means of results by various analytical techniques are in agreement. In the case of positive answer the established certified value is considered to be built on a solid foundation. Sometimes however, the results by only one method remain after outlier rejection and if there are good reasons to do so, the certification is performed using the results by a single method. In the beginning of the era of CRMs with the biological matrix, the detection limits for some rare earth elements (REE), Hf etc. apparently were too high for most of the methods except NAA or these methods might have been prone to systematic errors. Several examples of elements which were certified on the basis of NAA results alone are shown in Table 4. It is worth noting that Sc from the early times till the present moment is almost always within these elements.
When ICP-MS method with its excellent detection limits became available to many laboratories, one could presume that the problems with the determination of low concentrations of all elements also in biological matrices will suddenly disappear. In reality, at least in the case of scandium this does not seem to be the case. In the recent M-3 HerTis intercomparison (cf. Table 1) the eleven results for Sc (in ng g−1) provided by the participating laboratories were as follows: (1) 2.74(NAA); (2) 2.93(NAA); (3) 3.09(NAA); (4) 3.30(NAA); (5) 3.50(NAA); (6) 3.58(NAA); (7) 5.92(ICP-MS); (8) 6.39(NAA); (9) 10.4(ICP-MS); (10) 42.5(ICP-MS); (11) 154 (ICP-MS).
The results (7)–(11) were rejected as outliers in the process of evaluation of results used in our Laboratory [23, 24]. Scandium was certified on the basis of results (1)–(6) as: 3.2 ± 0.4 ng g−1 (H. Polkowska-Motrenko et al. private communication).
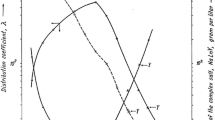
In the recent work by Dybczyński et al. [25], NAA and ICP-MS were used to compare the results of REE determination in the two CRMs: Tea Leaves INCT-TL-1 and Chinese Stream Sediment NCS DC73316. The method for the effective and quantitative separation of REE as group was also devised. While the results for most of the lanthanides by the two methods were in a good agreement, and in agreement with the certified value, the results for scandium in INCT-TL-1 by direct ICP-MS were systematically much too high (cf. Fig. 4). However after pre-separation of REE as a group the results by ICP-MS agreed with the certified value and the INAA| results. Scandium has only one stable isotope: 45Sc and presumably the formation of molecular ions from the matrix elements e.g. 12C16O12H+, 28Si16O1H+, 29Si16O+, 14N162O1H+, 13C16O2+ may be the reason for systematic errors when determining this element by ICP-MS [25]. As can be seen from Fig. 4, after elimination of the matrix the problem disappeared. On the other hand NAA is very well suited for the determination of trace amounts of scandium in various matrices. The reaction 45Sc(n,γ)46Sc has a good activation cross-section for thermal neutrons: 26.5 barns and a convenient half-life of the product nuclide (83.8 d). The two gamma lines of 46Sc: 889 and 1121 keV can be easily measured and in the case of need, long cooling time can be used to get rid of interfering activities due to short-lived and medium half-life radionuclides. So, the determination of Sc in various matrices is as a rule highly reliable and accurate.

Comparison of results for Sc in INCT-TL-1 by INAA, direct ICP-MS and ICP-MS after preliminary isolation of REE group by ion exchange procedure. Numerical data were taken from [25]
Ratio primary reference measurement procedures (RPRMP): definitive methods
Two most important means of achieving accurate results in trace analysis are:
- 1.
The use of certified CRMs with the type of matrix and concentration level of the analyte possibly closely matching those in the test samples
- 2.
Application of the methods with “guaranteed accuracy”
The prominent role of NAA in the process of certification of the candidate reference materials has been discussed in the preceding paragraphs of the present paper.
The possession of methods that would give truly accurate results has always been a dream of people working in the field of trace analysis, where, as was indicated earlier, the chances of making gross errors are both real and multiple. Various names such as e.g. “definitive” and “primary” etc. have been associated in the past with such methods of “guaranteed accuracy”. In the Institute of Nuclear Chemistry and Technology (INCT) the work on “definitive methods by radiochemical NAA” started more than 30 years ago. The principal idea is the combination of neutron activation with selective and quantitative isolation of the desired radionuclide by column chromatography followed by γ-ray spectrometric measurement. Several rules which should be obeyed when devising such methods were formulated [26,27,28]. The definition of “definitive methods” formulated by Uriano and Gravatt [29] sounded: “Definitive methods of chemical analysis are those that have a valid and well described theoretical foundation, have been experimentally evaluated so that reported results have negligible systematic errors, and have high levels of precision.”. In the real sense this definition is very close to the more exact definition formulated by CCQM (5th Meeting, 1999) [30] namely: “A primary method of measurement in the SI is a method having the highest metrological qualities whose model (mathematical equation)and realization are completely described and understood in terms of SI units.
The use of a primary direct method results in a value of an unknown quantity without reference to a standard of the same quantity.
The use of a primary ratio method results in a value of the ratio of two values of the same quantity without reference to a standard of the same quantity.
In both cases, the results must be accompanied by a complete uncertainty statement.”
The above definement was included into the definition of the “Primary reference measurement procedure” as given in VIM 3 [14, 31]. Our definitive methods can now be termed: Ratio primary reference measurement procedures (RPRMP) [28, 31]. In the high accuracy RNAA methods devised in the INCT i.e. definitive (RPRMP) methods, the mass fraction of an element being determined is described by a mathematical equation [31]. The methods provide the results with very low levels of uncertainty, traceable to SI units. The expanded uncertainty (k = 2) is usually of the order of 2.3–2.8% (e.g. for Cd, Co, Cr and Fe determination). Only in the case of RPRMP for molybdenum, the expanded uncertainty is greater (3.4–5.2%) [27]. While the other RPRMPs were devised as single element methods, the separation scheme and the whole method for Mo had to include the determination of uranium, because the same radionuclide which is used for the determination of Mo by NAA i.e. 99Mo–99mTc, may be also formed in U(n,f)99Mo reaction [32, 33].
Our definitive (RPRMP) methods can be an option or alternative to ID-MS methods, which however cannot be used for monoisotopic elements. Generally, the availability of two primary methods based on various physicochemical principles could be an ideal solution for the certification of certain elements in the candidate reference materials. It should be remembered that our RPRMPs were elaborated, so far, only for biological materials, because in this case it can be safely assumed that the decomposition and dissolution of the test samples will be complete. During a quarter of a century the definitive methods were elaborated for As, Cd, Co, Cu, Fe, Mo, Ni, Se and U. Recently, new RPRMP for chromium was published by Zuba and Polkowska-Motrenko [34]. It should be remembered that even the primary method has only potential to give the results of the highest metrological quality and the execution of the method is as important as the method itself. Therefore in our definitive methods we use a series of criteria [27, 28, 31] which must be simultaneously fulfilled before the result can be acknowledged as being obtained by RPRMP. The specialists in the ID-MS also issued a warning to potential users of their method writing: “ID-MS is not a “magic bullet”. Like any other technique, failure to understand all sources of error and how to deal with them may lead to a significant bias in the results” [35].
The examples of the use of definitive methods elaborated in the INCT for the verification of the certified values established as a result of interlaboratory comparisons are shown in Fig. 5.

Examples of the use of RPRMP (definitive methods) for the confirmation of the validity of certified values established in the world-wide intercomparisons. a Comparison of the certified value for Mo in INCT Corn Flour (INCT-CF-3) with the results of the definitive method, on the background of the original range of results submitted by the participants [11]. b Comparison of the certified value for Cd in MODAS-3 Herring Tissue (M-3 HerTis) with the results of the RPRMP (definitive method) on the background of the total range of laboratory averages and the range of “accepted laboratory averages) i.e. those that were left after rejection of outliers (Z. Samczyński private communication)
The hypothetical imminent danger of non-availability of NAA and its consequences
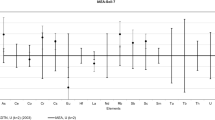
As can be seen from Figs. 2 and 3, the share of NAA in the results sent in by the participants of the world-wide intercomparisons dropped abruptly in the recent 20 years. On one hand it is a natural process because of the increased availability of the ICP-MS method, which features excellent detection limits for most of elements and is a truly multielement technique. On the other hand it is the effect of diminishing number of nuclear centers with well equipped NAA laboratories as well as the diminishing number of researchers specializing in NAA and especially in RNAA. So the future in which there will be almost no NAA or at least no NAA available for the certification of the CRMs may be not far from reality. The danger associated with such hypothetical situation will be visualized on the example of the case of iron in Oriental Tobacco Leaves (INCT-OBTL-5) [12, 13]. In total there were 63 laboratory averages (302 individual determinations) sent by the participating laboratories, only 3 averages were identified as outliers in the general population by the method being used by us during data evaluation [23, 24]. The number of results by individual techniques were as follows: AAS (21), ES (21), ICP-MS (6) NAA (15). The hypothetical situation when there would not be at all results by NAA is shown in Fig. 6a. The confidence intervals calculated for the three different methods i.e. AAS, ES and ICP-MS overlap as shown in Fig. 6a and the organizer could have been tempted to establish a “recommended” or “certified” value for iron which would amount then to 1401 ± 58 mg kg−1 (k = 2). In fact, however the NAA results were provided in this intercomparison and are shown in Fig. 6b. As can be seen the results for NAA were quite different from those of the other three methods, the confidence interval is far away from the confidence intervals of the other methods. Because of the drastic disagreement between the methods, the iron in the INCT-OBTL-5 could not be certified and only information value (1491 mg kg−1) was given [13]. A year later a new RPRMP (definitive method) for iron was devised [36] and the results by this method for Fe in INCT-OBTL-5 were also inserted into Fig. 6b. One can easily note that the results by RPRMP fully confirm that only NAA provided Fe results for OBTL-5 intercomparison which could be named accurate. In this follow up action [36] the results for Fe by RPRMP: 1750 ± 46 mg kg−1 and those by INAA with long cooling time: 1730 ± 51 mg kg−1 were combined to propose a tentative certified value (TCV) for Fe in the INCT-OBTL-5 as 1740 ± 43 mg kg −1. The reason for erroneous results for Fe in OBTL-5 and also in another material based on oriental tobacco leaves (CTA-OTL-1) [36] by the methods requiring dissolution of the sample, could be the high silicon content in these materials as discussed previously [36, 37]. That is why in our definitive methods by RNAA, the decomposition of the analyzed biological material is always carried out with the use of HNO3 + HF mixture to remove silica, followed by evaporation with the addition of H3BO3 to get rid of fluorides.

Illustration of the danger of false certification of Fe content in the absence of NAA results. a Hypothetical situation if in the OBTL-5 intercomparison there were no NAA results. b Real situation in the OBTL-5 intercomparison plus the result by RPRMP (definitive method)
Conclusions
For several decades NAA played an important role in quality assurance providing dominant share or results in the process of certification of the candidate reference materials. As a nuclear method, the NAA, if performed properly, is generally an accurate method, less dependent than other methods of trace analysis on the matrix effects. NAA has the advantage that in many cases the analysis may be performed purely instrumentally and is essentially free from the blank problems. On the other hand, elaborated in the INCT Ratio primary reference measurement procedures (definitive methods) by RNAA, are together with ID MS methods an important instruments for verification of accuracy of other methods used in inorganic trace analysis. Diminishing number of good NAA laboratories around the world raises the concern that some scientific competences may be lost in the near future with the obvious harm to the problems of quality assurance in inorganic trace analysis. It should be in the interest of the whole scientific community, not only radioanalytical chemists, to keep this method alive still for many years to come.
References
von Lehmden DJ, Jungers RH, Lee RE Jr (1974) Determination of trace elements in coal, fly ash, fuel oil, and gasoline—a preliminary comparison of selected analytical techniques. Anal Chem 46:239–245
Dybczyński R, Veglia A, Suschny O (1980) Milk powder (A-11)—a new IAEA reference material for trace and other element analysis. In: Brätter P, Schramel P (eds) Trace element analytical chemistry in medicine and biology. W. de Gruyter, New York
Pszonicki L, Hanna AN (1985) Report on intercomparison IAEA/V-10 of the determination of trace elements in Hay Powder, IAEA/RL/123. International Atomic Energy Agency, Vienna
Byrne AR, Camara Rica C, Cornelis R, De Goeij JJM, Iyengar GV, Kirkbright G, Knapp G, Parr RM, Stoeppler M (1987) Results of a co-ordinated programme to improve the certification of IAEA Milk Powder A-11 and animal muscle H-4 for eleven “difficult” trace elements. Fresenius Z Anal Chem 326:723–729
Zeiller E, Strachnov V, Dekner R (1990) Intercomparison study IAEA-155 on the determination of inorganic constituents in whey powder, IAEA/AL/034. IAEA, Vienna
Dybczyński R, Polkowska-Motrenko H, Samczyński Z, Szopa Z (1998) Virginia tobacco leaves (CTA-VTL-2)—new Polish CRM for inorganic trace analysis including microanalysis. Fresenius J Anal Chem 360:384–387
Dybczyński R, Polkowska-Motrenko H, Samczyński Z, Szopa Z (1997) Preparation and certification of the Polish reference material Virginia Tobacco Leaves (CTA-VTL-2) for inorganic trace analysis including microanalysis, Raporty IChTJ. Seria A nr 3/97, Institute of Nuclear Chemistry and Technology, Warszawa, ISSN 1425-7343
Dybczyński R, Danko B, Kulisa K, Chajduk-Maleszewska E, Polkowska-Motrenko H, Samczyński Z, Szopa Z (2004) Final certification of two new reference materials for inorganic trace analysis. Chem Anal (Warsaw) 49:143–158
Dybczyński R, Danko B, Kulisa K, Maleszewska E, Polkowska-Motrenko H, Samczyński Z, Szopa Z (2002) Preparation and certification of the Polish reference material: Tea Leaves (INCT-TL-1) for inorganic trace analysis, Raporty IChTJ. Seria A nr 3/2002, Institute of Nuclear Chemistry and Technology, Warszawa, ISSN 1425-7343
Polkowska-Motrenko H, Dybczyński R, Chajduk E, Danko B, Kulisa K, Samczyński Z, Sypuła M, Szopa Z (2006) Polish reference material: Soya Bean Flour (INCT-SBF-4) for inorganic trace analysis—preparation and certification, Raporty IChTJ. Seria A nr 4/2006
Polkowska-Motrenko H, Dybczyński RS, Chajduk E, Danko B, Kulisa K, Samczyński Z, Sypuła M, Szopa Z (2007) New Polish certified reference materials for inorganic trace analysis: corn flour (INCT-CF-3) and soya bean flour (INCT-SBF-4). Chem Anal (Warsaw) 52:361–376
Samczyński Z, Dybczyński RS, Polkowska-Motrenko H, Chajduk E, Pyszynska M, Danko B, Czerska E, Kulisa K, Doner K, Kalbarczyk P (2011) Preparation and certification of the new Polish reference material: Oriental Basma Tobacco Leaves (INCT-OBTL-5) for inorganic trace analysis. Institute of Nuclear Chemistry and Technology, Warszawa. ISBN 978-83-929013-9-6
Samczyński Z, Dybczyński RS, Polkowska-Motrenko H, Chajduk E, Pyszynska M, Danko B, Czerska E, Kulisa K, Doner K, Kalbarczyk P (2012) Two new reference materials based on tobacco leaves: certification for over a dozen of toxic and essential elements. Sci World J. https://doi.org/10.1100/2012/216380
ISO/IEC Guide 99:2007 (2007) International vocabulary of metrology—basic and general concepts and associated terms (VIM3). ISO, Geneva
Dybczyński R (2002) Considerations on the accuracy of determination of some essential and/or toxic elements in biological materials. Chem Anal (Warsaw) 47:325–334
Dybczyński RS, Polkowska-Motrenko H (2015) Certified reference materials in inorganic trace analysis. In: Baranowska I (ed) Handbook of Trace Analysis. Springer, Heidelberg
Greenberg RR (1987) The role of neutron activation in the certification of NBS standard reference materials. J Radioanal Nucl Chem 113:233–247
Lindstrom RM (1987) Nuclear analytical methods in standards certification. In: Comparison of nuclear analytical methods with competitive methods. IAEA-TECDOC-435 International Atomic Energy Agency, Vienna
Zeisler R, Paul RL, Oflaz Spatz R, Yu LL, Mann JL, Kelly WR, Lang BE, Leigh SD, Fagan J (2011) Elemental analysis of a single-wall carbon nanotube candidate reference material. Anal Bioanal Chem 399:509–517
Kramer KJM, de Haan E, Dorten WS, Kramer GN, Quevauviller Ph (2001) bcr information reference materials. The certification of the contents (mass fractions) of Ce, Dy, Er, Eu, Gd, Ho, La, Lu, Nd, Pr, Sc, Sm, Tb, Tm,Y,Yb, Th and U in Aquatic plant BCR-670. EUR 19499 EN ISBN 92-894-1487-1 European Communities, Brussels
Levinson AA (1974) Introduction to exploration geochemistry. Applied Publishing Co., Calgary
Gladney ES (1980) Elemental concentrations in NBS biological and environmental standard reference materials. Anal Chim Acta 118:385–396
Dybczyński R, Polkowska-Motrenko H, Samczyński Z, Szopa Z (1991) Two new polish geological-environmental reference materials: apatite concentrate (CTA-AC-1) and fine fly ash (CTA-FFA-1). Geostand Newsl 15:163–185
Polkowska-Motrenko H, Dybczyński R, Chajduk E (2010) Certification of reference materials for inorganic trace analysis, the INCT approach. Accred Qual Assur 15:245–250
Dybczyński RS, Samczyński Z, Bartosiewicz I, Kulisa K, Polkowska-Motrenko H, Pyszynska M, Zuba I (2017) Two new separation schemes for group isolation of rare earth elements (REE) from biological and other matrices and their determination by ICP-MS, NAA and chromatographic methods. Nukleonika 62(3):199–211. https://doi.org/10.1515/nuka-2017-0030
Dybczyński R, Wasek M, Maleszewska H (1989) A definitive method for the determination of small amounts of copper in biological materials by neutron activation analysis. J Radioanal Nucl Chem 130:365–388
Dybczyński RS, Danko B, Polkowska-Motrenko H, Samczyński Z (2007) RNAA in metrology: a highly accurate (definitive) method. Talanta 71:529–536
Dybczyński RS, Polkowska-Motrenko H, Chajduk E, Danko B, Pyszynska M (2014) Recent advances in ratio primary reference measurement procedures (definitive methods) and their use in certification of reference materials and controlling assigned values in proficiency testing. J Radioanal Nucl Chem 302:1295–1302. https://doi.org/10.1007/s10967-014-3607-y
Uriano GA, Gravatt CC (1977) The role of reference materials and reference methods in chemical analysis. Crit Rev Anal Chem 6:361–364
BIPM, Consultative Committee for Amount of Substance (CCQM)—5th Meeting (February 1999)
Dybczyński RS (2015) 50 years of adventures with neutron activation analysis with the special emphasis on radiochemical separations. J Radioanal Nucl Chem 303:1067–1090. https://doi.org/10.1007/s10967-014-3822-6
Danko B, Dybczyński R (1995) Radiochemical scheme for the determination of Mo and U in biological materials by NAA. J Radioanal Nucl Chem 192(1):117–129
Danko B, Dybczyński R (1997) Determination of molybdenum and uranium in biological materials by radiochemical neutron activation analysis. J Radioanal Nucl Chem 216:51–57
Zuba I, Polkowska-Motrenko H (2019) Ratio primary reference measurement procedure (RPRMP) for the certification of chromium content in biological materials. Radiochim Acta 107(2):141–147
Harte R, Newman G, Sargent M (2004) Achieving traceable chemical measurements: inter-laboratory evaluation of a simplified technique for isotope dilution mass spectrometry (IDMS). Part 1: methodology for high accuracy analysis of trace metals. Accred Qual Assur 9:33–38
Dybczyński RS, Danko B, Pyszynska M, Polkowska-Motrenko H (2012) Ratio primary reference measurement procedure (RPRMP) for the determination of iron in biological materials by RNAA. Radiochim Acta 100:409–416
Bulska E, Danko B, Dybczyński RS, Krata A, Kulisa K, Samczyński Z, Wojciechowski M (2012) Inductively coupled plasma mass spectrometry in comparison with neutron activation and ion chromatography with UV/VIS detection for the determination of lanthanides in plant materials. Talanta 97:303–311
Acknowledgements
Thanks are due to dr Z. Samczyński for fruitful discussions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Dybczyński, R.S. The role of NAA in securing the accuracy of analytical results in the inorganic trace analysis. J Radioanal Nucl Chem 322, 1505–1515 (2019). https://doi.org/10.1007/s10967-019-06675-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-019-06675-7