
Abstract
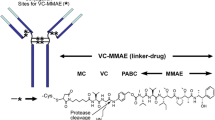
Enfortumab vedotin is an antibody–drug conjugate (ADC) comprised of a Nectin-4–directed antibody and monomethyl auristatin E (MMAE), which is primarily eliminated through P-glycoprotein (P-gp)–mediated excretion and cytochrome P450 3A4 (CYP3A4)–mediated metabolism. A physiologically based pharmacokinetic (PBPK) model was developed to predict effects of combined P-gp with CYP3A4 inhibitor/inducer (ketoconazole/rifampin) on MMAE exposure when coadministered with enfortumab vedotin and study enfortumab vedotin with CYP3A4 (midazolam) and P-gp (digoxin) substrate exposure. A PBPK model was built for enfortumab vedotin and unconjugated MMAE using the PBPK simulator ADC module. A similar model was developed with brentuximab vedotin, an ADC with the same valine-citrulline–MMAE linker as enfortumab vedotin, for MMAE drug–drug interaction (DDI) verification using clinical data. The DDI simulation predicted a less-than-2-fold increase in MMAE exposure with enfortumab vedotin plus ketoconazole (MMAE geometric mean ratio [GMR] for maximum concentration [Cmax], 1.15; GMR for area under the time-concentration curve from time 0 to last quantifiable concentration [AUClast], 1.38). Decreased MMAE exposure above 50% but below 80% was observed with enfortumab vedotin plus rifampin (MMAE GMR Cmax, 0.72; GMR AUClast, 0.47). No effect of enfortumab vedotin on midazolam or digoxin systemic exposure was predicted. Results suggest that combination enfortumab vedotin, P-gp, and a CYP3A4 inhibitor may result in increased MMAE exposure and patients should be monitored for potential adverse effects. Combination P-gp and a CYP3A4 inducer may result in decreased MMAE exposure. No exposure change is expected for CYP3A4 or P-gp substrates when combined with enfortumab vedotin.
ClinicalTrials.gov identifier Not applicable.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Bladder cancer is one of the most common malignancies—among which urothelial carcinoma (UC) is the predominant type—and it is associated with poor long-term prognoses in those with advanced/metastatic disease [1,2,3]. Most (70%) individuals with bladder cancer are older than 65 years and present with age-associated comorbidities [2, 4]. Older individuals taking multiple medications may experience drug–drug interactions (DDIs), which can lead to reduced drug efficacy or serious health risks [5,6,7,8,9]. Therefore, identification of potential DDIs associated with these drugs and their metabolites is essential to support safe and effective prescribing.
Enfortumab vedotin, an antibody–drug conjugate (ADC) directed against Nectin-4, is composed of a fully human anti–Nectin-4 monoclonal antibody (mAb) conjugated to a microtubule-disrupting agent, monomethyl auristatin E (MMAE), via a protease-cleavable linker; the targeted MMAE release in Nectin-4–expressing cells leads to cell-cycle arrest and cell death [10]. Enfortumab vedotin is approved for use in multiple countries for indications including treatment of adults with locally advanced/metastatic UC who have previously received standard of care platinum-based therapy and program death receptor-1/programmed death-ligand 1 (PD-1/L1) inhibitor therapies as well as in patients previously treated with one or more lines of therapy but who are ineligible for cisplatin-containing chemotherapy [10,11,12,13]. The recommended dose of enfortumab vedotin is 1.25 mg/kg (maximum 125 mg) given as an intravenous (IV) infusion over 30 min on days 1, 8, and 15 of every 28-day cycle [10].
The nonclinical pharmacokinetic (PK) data of enfortumab vedotin from an in vivo mass balance study conducted in rats concluded that MMAE delivered by enfortumab vedotin is primarily eliminated in the feces (≤ 15% of dose via urinary excretion) with unchanged MMAE excreted in feces and urine [14]. MMAE undergoes hepatic metabolism, predominantly through cytochrome P450 3A4 (CYP3A4) and is a substrate of P-glycoprotein (P-gp) [10, 14, 15]. Clinical PK data of enfortumab vedotin (EV-101; NCT02091999) showed that mean exposure, as determined by area under the concentration-time curve (AUC) for the 3 analytes measured: ADC (antibody + conjugated drug), total antibody, and MMAE increased with ascending dose [16]. Across the tested dose range (0.5–1.25 mg/kg), maximum ADC concentrations were attained approximately 30 min to 1 h after IV administration, whereas time to reach maximum MMAE concentration was approximately 1.0 to 2.8 days after IV administration [16]. A clinical DDI study with brentuximab vedotin, an ADC featuring the same valine-citrulline–MMAE linker as enfortumab vedotin but a different target (CD30 instead of Nectin-4), showed that, while ADC exposures are unaffected by concomitant rifampin (a CYP3A inducer) or ketoconazole (a CYP3A inhibitor), MMAE exposures were lower with rifampin and higher with ketoconazole. Furthermore, brentuximab vedotin did not affect exposure to midazolam, a sensitive CYP3A substrate [17].
Physiologically based PK (PBPK) modeling has become an accepted practice for predicting DDI potential when a clinical DDI study is unavailable [18]. Results of PBPK modeling studies assist in developing guidance for clinical trial design as well as to inform drug labeling [18, 19]. Several studies have used the PBPK modeling approach to assess the DDI liability of the MMAE component of ADCs [19, 20]. Although the DDI risk for the mAb component of ADCs is low [19, 21, 22], it is important to assess the DDI potential of mAb because of its potential interaction with Fcγ receptors on the effector cells, its role in inflammatory changes in drug transport proteins, or its immune-mediated changes involving cytochrome enzymes and drug transport proteins [22,23,24,25]. Previous PBPK models for ADCs treated mAb as a small molecule and directly modeled MMAE by dosing MMAE at an equivalent dose as that released by mAb [19, 20]. The simultaneous PBPK modeling of both portions of an ADC—the large mAb and the small cytotoxic molecule—using the ADC module within the Simcyp simulator has not been previously conducted. Previous efforts around ADC modeling using Simcyp did not model the mAb as a biologic or large molecule [19, 20].
The present PBPK analysis was conducted to assess the combination of CYP3A4- and P-gp–mediated DDIs for enfortumab vedotin as both a victim and perpetrator using a model that accounted for large and small molecules. We leveraged the data from clinical DDI studies conducted in brentuximab vedotin for validating our model. The objective of this analysis was to predict the effect of a combined CYP3A4 and P-gp inhibitor (ketoconazole) or a combined CYP3A4 and P-gp inducer (rifampin) on the PK of MMAE when coadministered with enfortumab vedotin. In addition, analysis was performed to predict effects of enfortumab vedotin on the exposure of CYP3A4 substrate (midazolam) and P-gp substrate (digoxin).
Methods
A PBPK model was developed for enfortumab vedotin using clinical data from phase 1 and 2 studies. The phase 1 dose-escalation/dose-expansion study of enfortumab vedotin (EV-101; NCT02091999) [16] was conducted in previously treated patients with Nectin-4–expressing metastatic UC and other malignant solid tumors. Single IV dose data of enfortumab vedotin 1.25 mg/kg were extracted from the study for model building and single IV dose data of enfortumab vedotin 1.0 mg/kg were used for single-dose model verification [16]. In the phase 2 single-cohort study of enfortumab vedotin (EV-201; NCT03219333), 125 patients with locally advanced/metastatic UC who were previously treated with platinum and PD-1/L1 inhibitor therapy received enfortumab vedotin 1.25 mg/kg. Multiple-dose data of enfortumab vedotin 1.25 mg/kg obtained from the study were used for multiple-dose verification [10]. Data from this study were split into a model development and model verification group.
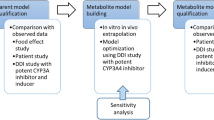
Brentuximab vedotin was used in the present analysis as a baseline comparison to enfortumab vedotin because of the availability of data, including a clinical DDI trial, and similarity in the average drug–antibody ratios between these compounds. Brentuximab vedotin data, which were available from the sponsor’s clinical pharmacology submission to the US Food and Drug Administration, were digitized for model development [11]. The PK data from a single IV dose of brentuximab vedotin 1.8 mg/kg administered over 30 min were used for model building. Data from a single IV dose of brentuximab vedotin 2.7 mg/kg were used for model verification. The clinical DDI study (NCT01026415) with brentuximab vedotin and a combined P-gp and CYP3A inhibitor or inducer was used for DDI verification [17]. For verification of P-gp–mediated interactions by ketoconazole and rifampin, clinical DDI data were obtained from prior publications [12, 13]. Data were extracted from the literature using GetData Graph Digitizer version 2.26. The process for PBPK model construction, verification, and application of enfortumab vedotin is depicted in Fig. 1.

Process for PBPK model construction, verification, and application of enfortumab vedotin. DDI, drug–drug interaction; IV, intravenous; MMAE, monomethyl auristatin E; PBPK, physiologically based pharmacokinetics
Virtual trial designs for the enfortumab vedotin and brentuximab vedotin PBPK models closely resembled the actual clinical studies [16, 17, 26]; however, for enfortumab vedotin, the default numbers of patients in the Simcyp (Certara, Sheffield, UK) simulator (10 trials with 10 patients in each trial) were used. Specifics of the virtual trial design for model construction and verification for enfortumab vedotin and brentuximab vedotin can be found in the Supplementary Methods.
Model construction
The ADC module provided within the Simcyp simulator version 19 was used for PBPK modeling (Fig. 2). For enfortumab vedotin, the PBPK model was built using the minimal PBPK modeling approach and, for MMAE, the full PBPK approach for small molecules was used (Tables 1 and 2). The neonatal Fc receptor (FcRn) dissociation constant was optimized for enfortumab vedotin based on clinical data because an in vitro experimental value was unavailable. It was determined that the FcRn dissociation constant would be different between brentuximab vedotin and enfortumab vedotin because of the difference in mAb and that the half-life of enfortumab vedotin (~ 3.6 days for the conjugated antibody) was shorter than brentuximab vedotin (~ 4 to 6 days). A lower binding affinity (higher dissociation) to FcRn was required for enfortumab vedotin to capture the ADC elimination time curve and production of MMAE. The source of unconjugated MMAE in the model was through catabolism, additional nonspecific plasma clearance, and deconjugation. Uptake clearance and recycling rate values were unknown for enfortumab vedotin or brentuximab vedotin; due to difficulties in measuring these parameters, default values provided within the Simcyp software were used.

Schematic of (a) Simcyp simulator ADC module (b) linked to full PBPK model for small molecules. Reprinted with permission from Certara UK Limited. ADC, antibody-drug conjugate; IV, intravenous; PBPK, physiologically based pharmacokinetic model
The MMAE compound model was constructed using values obtained from a previous study of PBPK model building of brentuximab vedotin with some modifications [20], because the previous model was built using an older version of Simcyp that did not contain the ADC module. Use of the volume of distribution at steady state and CYP3A4 intrinsic clearance reported in the reference study led to an underestimation of MMAE exposure. Thus, these parameters were optimized for the ADC module and the enfortumab vedotin PBPK model. For the brentuximab vedotin PBPK model, ADC plasma clearance was optimized using clinically observed data from phase 1 studies with brentuximab vedotin administered at 1.8 mg/kg. The same MMAE compound file was utilized for enfortumab vedotin and brentuximab vedotin (Table S1).
Model verification
For enfortumab vedotin, the PBPK model was verified by analyzing the ADC and MMAE concentration time profiles with single-dose (from the first dose within a cycle) enfortumab vedotin 1.0 mg/kg and multiple doses of enfortumab vedotin 1.25 mg/kg from phase 1 and 2 trials, respectively [10, 16]. For brentuximab vedotin, the PBPK model was verified by the ADC and MMAE concentration time profiles with the dose of brentuximab vedotin 2.7 mg/kg from the clinical pharmacology data submitted to the US Food and Drug Administration [26].
Model verification of drug interactions with brentuximab vedotin was conducted with a combined P-gp and CYP3A4 inhibitor or inducer and CYP3A4 substrate. The simulated plasma concentration profiles, ratio of AUC from time 0 extrapolated to infinity (AUCinf), and ratio of maximum serum concentration (Cmax) of ADC and MMAE from brentuximab vedotin 1.2 mg/kg with ketoconazole 400 mg oral daily or brentuximab vedotin 1.8 mg/kg with rifampin 600 mg oral daily or a single IV dose of midazolam 1 mg were compared with clinical PK data from a previously published study [17]. Rifampin simulations were performed with the fold-increase of the P-gp transporter relative activity factor value applied to MMAE, as previously described.
For the cancer population, the distributions of simulated values for age, body weight, plasma albumin value, and hematocrit level were compared with those observed from all patients in the phase 1 study [16] for verification of the Simcyp simulator–provided cancer population modification.
Model for drug–drug interaction simulations
The verified PBPK model was used to evaluate the interaction of enfortumab vedotin with ketoconazole, rifampin, midazolam, and digoxin. For the DDI simulation studies, the clinical brentuximab vedotin DDI data were leveraged to verify the applicability of the brentuximab vedotin PBPK model in predicting DDIs observed clinically. This information was then used for enfortumab vedotin to predict the exposure of MMAE in several DDI simulations.
To analyze effects of the combined P-gp and CYP3A4 inhibitor and inducer on enfortumab vedotin and brentuximab vedotin, the Simcyp simulator–provided PBPK models for ketoconazole and rifampin were slightly modified. An inhibitory constant (Ki) for the P-gp transport for intestine and liver compartments was analyzed and incorporated for both models; for ketoconazole, a P-gp inhibition KI value of 0.67 µM was added to the ketoconazole PBPK compound model file; for rifampin, a KI value of 4.3 µM was added. No other parameters were altered.
To analyze the inhibitory effects of enfortumab vedotin and brentuximab vedotin on CYP3A4 and P-gp substrates, the Simcyp simulator PBPK models for midazolam and digoxin were used for enfortumab vedotin and brentuximab vedotin DDI simulations. The model for midazolam was used without modification. For simulations with rifampin, the relative activity factor for P-gp transport within the digoxin compound file was increased to represent transporter induction. Rifampin simulations were performed twice: once with the increased relative activity value contained within the digoxin compound file, then again with the baseline relative activity value and rifampin dose set to 0. Of note, the state of the simulator at the time of analysis did not support transporter induction for DDI simulation; therefore, the effect of P-gp induction by rifampin was manually implemented.
The difference between enfortumab vedotin and brentuximab vedotin populations was hypothetically linked to the included cancer population (solid tumor vs. blood cancer) within the clinical studies [16, 26]. The Simcyp cancer population model was used without modification for all brentuximab vedotin simulations. For simulations of enfortumab vedotin, the population was slightly modified and the influence of the tissue–volume scaling factor for plasma on the ADC plasma concentrations was investigated. For digoxin simulations, the Simcyp simulator–containing PBPK model for the healthy volunteer population was used without modifications.
Application of the drug–drug interaction simulation and sensitivity analysis
The verified enfortumab vedotin PBPK model was employed to simulate interactions between enfortumab vedotin and rifampin, ketoconazole, midazolam, and digoxin using the virtual trial design (Table S2). Sensitivity analysis was conducted to evaluate the uncertainty of P-gp (biliary) versus CYP3A4 contribution on elimination of MMAE. The drug interactions between enfortumab vedotin and ketoconazole or rifampin were used to analyze the effect of the elimination pathway on the GMRs of MMAE Cmax and AUC from time 0 to last quantifiable concentration (AUClast; information regarding times for AUClast appears in Table S2). Simulations were run for a sufficient time duration for the AUClast ratio to be equivalent to the AUCinf ratio.
Statistical analysis
Summary statistics of simulated Cmax, AUClast, AUCinf, AUC from time 0 to day 7 (AUCd0–7), AUC from time 0 to day 14 (AUCd0–14), Cmax ratio, and AUCinf/last ratio were calculated by noncompartmental analysis. For DDI simulations involving rifampin, midazolam, and digoxin, geometric mean ratios (GMRs) were calculated for Cmax and AUClast (reported instead of AUCinf due to limitations in calculating AUCinf for some simulations). The GMR was calculated from the 2 simulations using R-Studio (R-Studio, Boston, MA).
Results
Enfortumab vedotin
The simulation reproduced the observed plasma concentration-time profile and PK parameters for enfortumab vedotin (ADC) and MMAE from the phase 1 clinical study for enfortumab vedotin (Table 3) [16]. Simulated concentration-time profiles aligned with observed concentration-time profiles for a single dose of enfortumab vedotin 1.0 and 1.25 mg/kg at all time points for enfortumab vedotin and from days 2 to 7 for MMAE (Fig. 3). Similarly, for multiple doses of enfortumab vedotin 1.0 and 1.25 mg/kg, the simulation reproduced the observed concentration-time profile (Fig. 4) and PK parameters for enfortumab vedotin (ADC) and MMAE (Table 4). Thus, for enfortumab vedotin, the PBPK model reasonably characterized the observed PK.

Simulated versus observed semilog plasma concentration curves of (a) conjugated enfortumab vedotin and (b) MMAE following IV administration of enfortumab vedotin 1.0 mg/kg and 1.25 mg/kg. IV, intravenous; MMAE, monomethyl auristatin E

Simulated versus observed semilog plasma concentration curves of (a) conjugated enfortumab vedotin and (b) MMAE following multiple IV administration of 1.25 mg/kg. IV, intravenous; MMAE, monomethyl auristatin E
Brentuximab vedotin
For brentuximab vedotin 1.8 and 2.7 mg/kg, the simulation aligned with the observed concentration-time profile for a single dose (Figure S1) and PK parameters for brentuximab vedotin (ADC) and MMAE (Table S3). The DDI simulation of brentuximab vedotin with ketoconazole adequately reproduced the observed fold-change in AUC and Cmax (Table 5). The simulated DDI fold-change in AUClast when brentuximab vedotin was dosed with rifampin or midazolam was slightly overestimated; however, the difference was deemed acceptable. No additional optimization was deemed necessary for the MMAE elimination pathway or CYP3A4 inhibition constant, and the MMAE CYP3A4 and P-gp pathway was verified.
Cancer population
The base cancer population within the Simcyp simulator was modified for enfortumab vedotin simulations by changing the tissue–volume scaling factor for plasma from 1.2 to 1.0 due to underestimation of Cmax in conjugated enfortumab vedotin plasma concentrations. The simulated population closely resembled the observed patient population, and the population demographics were generally similar (Table S4). The model was determined to be biologically plausible for combined P-gp and CYP3A4 drug-interaction simulations because it successfully re-created clinically observed plasma–concentration data (Table 6) and because similar results were observed in clinical DDI studies performed with brentuximab vedotin, which has the same cytotoxic agent and drug linker as enfortumab vedotin.
Prediction of drug–drug interaction with enfortumab vedotin and sensitivity analysis
The GMRs for Cmax and AUClast of MMAE from enfortumab vedotin in the presence of ketoconazole were 1.15 (15% increase) and 1.38 (38% increase) respectively. (Table 5). The GMRs for Cmax and AUClast of MMAE in the presence of rifampin were 0.72 (28% decrease) and 0.47 (53% decrease), respectively, indicating moderate impact on MMAE plasma exposure. The GMRs of midazolam in the presence of enfortumab vedotin were 1.00 for Cmax and 1.14 for AUClast and the GMRs of digoxin in the presence of enfortumab vedotin were 1.00 for both Cmax and AUClast, predicting no effect on digoxin exposure in the presence of enfortumab vedotin, thus indicating that enfortumab vedotin has minimal impact on CYP3A4 or P-gp substrates. These findings were similar to the fold-difference clinically observed and simulated for brentuximab vedotin.
Considering the potential range of biliary excretion, sensitivity analyses indicated that a 10% difference in the assumed fraction clearance through the bile had limited impact on the magnitude of DDI simulation results when enfortumab vedotin 1.25 mg/kg was coadministered with ketoconazole 400 mg or rifampin 600 mg (Table S5).
Discussion
Physiologically based PK is a mechanistic modeling framework that has become an important tool for PK and DDI prediction for drug development [18, 28]. In this study, a PBPK model was built to predict the MMAE-based DDI for enfortumab vedotin and effect of enfortumab vedotin on CYP3A4 and P-gp substrates; the feasibility of using the Simcyp ADC module for PBPK modeling was also demonstrated. Based on the results of this study, patients receiving enfortumab vedotin with combination P-gp and CYP3A4 inhibitors may experience increases in MMAE exposure. The predicted increase in AUC with concomitant administration of enfortumab vedotin and ketoconazole was less than 2-fold. At the recommended clinical dose of enfortumab vedotin 1.25 mg/kg, there was minimal interpatient variability in enfortumab vedotin exposure. However, the interpatient variability in MMAE exposure was greater than 2-fold; therefore, a less-than-2-fold increase in plasma exposure can be considered within interpatient variability. Regardless, patients should be monitored for adverse reactions. Patients may also receive concurrent treatment with enfortumab vedotin and combined P-gp and strong CYP3A4 inducers may experience a decrease in exposure to MMAE. The predicted decrease in unconjugated MMAE AUC with concomitant administration of rifampin was greater than 50% but less than 80%, suggesting a moderate impact of rifampin on the plasma exposure of MMAE.
With regards to enfortumab vedotin being a perpetrator of drug interactions of CYP3A or P-gp substrates, the predicted increase in midazolam AUC and no change in digoxin AUC indicated that enfortumab vedotin has minimal impact on these substrates. Hence, no dose adjustments would be required for CYP3A or P-gp substrates when concomitantly used with enfortumab vedotin.
In the present study, PBPK modeling was based on the advanced Simcyp “ADC” module feature, which simultaneously simulates the large mAb and the cytotoxic small molecule. Unlike previous studies, the PBPK model used in this study considered the effect of P-gp on the resulting DDIs, whereas previous studies did not [17, 19, 20]. The PBPK model using the Simcyp “ADC” module feature allows researchers to study specific populations of virtual patients to assess their likely response to a specific ADC. In addition, this PBPK approach facilitates mechanism-driven modeling, characterization, and simulation studies of ADCs and DDIs. The present work was used in lieu of a dedicated clinical study. Similar results to the clinical DDI studies for brentuximab vedotin were demonstrated based on predicted/observed ratios reported in Table 5. In addition, the model successfully re-created clinically observed plasma concentration data (Table 6). Extrapolation of the model for brentuximab vedotin to enfortumab vedotin was rational because both ADCs contain MMAE and the same linker; thus, the model was highly biologically plausible for the combined P-gp and CYP3A DDI simulations.
In terms of model limitations, because of limited data, assumptions were made when creating the conjugated mAb portion of the enfortumab vedotin model. At the time this study was conducted, antibody conjugated MMAE measurements were unavailable. Moreover, MMAE concentration-time profiles displayed large variability that could not be fully captured by the model. Association with conjugation and changes in FcRn binding has been reported in the literature and there are instances where conjugation increases or decreases FcRn binding [29, 30]. Binding of brentuximab vedotin to FcRn was obtained from the literature and experimental values [31]. Because an in vitro experimental value for the binding affinity of enfortumab vedotin to the FcRn was unknown at the time of modeling, values were optimized based on observed clinical data; however, this limitation did not affect the ability of the model to evaluate the drug interaction potential of enfortumab vedotin. Considering the similarity in the linker and MMAE, it was assumed that the deconjugation rate and additional plasma clearance were the same as brentuximab vedotin. A worst-case scenario, with the assumption that the excretion of MMAE through transport was limited only by the clearance of P-gp, was used for the passive diffusion of MMAE across the hepatocytes prior to P-gp export. Due to lack of available data, target-mediated drug disposition (TMDD) could not be included in the present model and no interpretations of the role of TMDD on enfortumab vedotin distribution and elimination could be made. Although it would be expected that MMAE exposure in tumors should be greater than in healthy tissue given the greater expression of Nectin-4 in tumors versus healthy tissue [32,33,34], predictions of drug interactions as a result of target exposure in tissues expressing Nectin-4 (i.e., MMAE concentration in healthy tissue vs. tumors) were not studied.
Conclusion
Based on the results of the PBPK simulation for enfortumab vedotin, no dose adjustment is required for concomitant administration of enfortumab vedotin when used in combination with a P-gp and inhibitors or inducers of CYP3A4. However, patients receiving enfortumab vedotin with combined P-gp and CYP3A4 inhibitors may experience increases in MMAE exposure and should be monitored for signs of adverse events. No change in exposure is expected with CYP3A4 or P-gp substrates for patients concomitantly receiving these substrates with enfortumab vedotin. This work underpins the applicability of PBPK modeling to predict ADC drug interactions and demonstrates that MMAE associated with enfortumab vedotin has limited potential for causing clinically relevant DDIs.
Data availability
Researchers may request access to anonymized participant level data, trial level data and protocols from Astellas sponsored clinical trials at www.clinicalstudydatarequest.com. For the Astellas criteria on data sharing see: https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Astellas.aspx. Researchers may request access to the Simcyp files used to support this article by contacting the corresponding author.
References
Hepp Z, Shah SN, Liang SY, Tan K, Balakrishna S (2021) Real-world outcomes in locally advanced or metastatic urothelial carcinoma following platinum and PD-1/L1 inhibitor therapy. Future Oncol 17(32):4343–4353. https://doi.org/10.2217/fon-2021-0573
National Cancer Institute Surveillance E, and End Results Program (2022) Cancer stat facts: bladder cancer. https://seercancer.gov/statfacts/html/urinbhtml. Accessed on August 26, 2022.
Wong MCS, Fung FDH, Leung C, Cheung WWL, Goggins WB, Ng CF (2018) The global epidemiology of bladder cancer: a joinpoint regression analysis of its incidence and mortality trends and projection. Sci Rep 8(1):1129. https://doi.org/10.1038/s41598-018-19199-z
Guancial EA, Roussel B, Bergsma DP, Bylund KC, Sahasrabudhe D, Messing E, Mohile SG, Fung C (2015) Bladder cancer in the elderly patient: challenges and solutions. Clin Interv Aging 10:939–949. https://doi.org/10.2147/CIA.S74322
Ramsdale E, Mohamed M, Yu V, Otto E, Juba K, Awad H, Moorthi K, Plumb S, Patil A, Vogelzang N, Dib E, Mohile S (2022) Polypharmacy, potentially inappropriate medications, and drug-drug interactions in vulnerable older adults with advanced cancer initiating cancer treatment. Oncologist 27(7):e580–e588. https://doi.org/10.1093/oncolo/oyac053
Laatikainen O, Sneck S, Bloigu R, Lahtinen M, Lauri T, Turpeinen M (2016) Hospitalizations due to adverse drug events in the elderly-a retrospective register study. Front Pharmacol 7:358. https://doi.org/10.3389/fphar.2016.00358
van Leeuwen RWF, Jansman FGA, van den Bemt P, de Man F, Piran F, Vincenten I, Jager A, Rijneveld AW, Brugma JD, Mathijssen RHJ, van Gelder T (2015) Drug-drug interactions in patients treated for cancer: a prospective study on clinical interventions. Ann Oncol 26(5):992–997. https://doi.org/10.1093/annonc/mdv029
Hines LE, Murphy JE (2011) Potentially harmful drug-drug interactions in the elderly: a review. Am J Geriatr Pharmacother 9(6):364–377. https://doi.org/10.1016/j.amjopharm.2011.10.004
Elmeliegy M, Vourvahis M, Guo C, Wang DD (2020) Effect of p-glycoprotein (p-gp) inducers on exposure of p-gp substrates: review of clinical drug-drug interaction studies. Clin Pharmacokinet 59(6):699–714. https://doi.org/10.1007/s40262-020-00867-1
Rosenberg JE, O’Donnell PH, Balar AV, McGregor BA, Heath EI, Yu EY, Galsky MD, Hahn NM, Gartner EM, Pinelli JM, Liang SY, Melhem-Bertrandt A, Petrylak DP (2019) Pivotal trial of enfortumab vedotin in urothelial carcinoma after platinum and anti-programmed death 1/programmed death ligand 1 therapy. J Clin Oncol 37(29):2592–2600. https://doi.org/10.1200/JCO.19.01140
Brentuximab Vedotin (Adcetris) (2011) Clinical Pharmacology and Biopharmaceutics Review(s). Center for Drug Evaluation and Research. 25 Feb 2011.
Larsen UL, Hyldahl Olesen L, Guldborg Nyvold C, Eriksen J, Jakobsen P, Ostergaard M, Autrup H, Andersen V (2007) Human intestinal P-glycoprotein activity estimated by the model substrate digoxin. Scand J Clin Lab Invest 67(2):123–134. https://doi.org/10.1080/00365510600986084
Greiner B, Eichelbaum M, Fritz P, Kreichgauer HP, von Richter O, Zundler J, Kroemer HK (1999) The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J Clin Invest 104(2):147–153. https://doi.org/10.1172/JCI6663
Center for Drug Evaluation and Research (2019) Application Number: 761137Orig1s000. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/761137Orig1s000MultiDiscliplineR.pdf. Accessed February 1, 2023.
Chang HP, Cheung YK, Shah DK (2021) Whole-body pharmacokinetics and physiologically based pharmacokinetic model for monomethyl auristatin E (MMAE). J Clin Med 10(6). https://doi.org/10.3390/jcm10061332
Rosenberg J, Sridhar SS, Zhang J, Smith D, Ruether D, Flaig TW, Baranda J, Lang J, Plimack ER, Sangha R, Heath EI, Merchan J, Quinn DI, Srinivas S, Milowsky M, Wu C, Gartner EM, Zuo P, Melhem-Bertrandt A, Petrylak DP (2020) EV-101: a phase I study of single-agent enfortumab vedotin in patients with nectin-4-positive solid tumors, including metastatic urothelial carcinoma. J Clin Oncol 38(10):1041–1049. https://doi.org/10.1200/JCO.19.02044
Han TH, Gopal AK, Ramchandren R, Goy A, Chen R, Matous JV, Cooper M, Grove LE, Alley SC, Lynch CM, O’Connor OA (2013) CYP3A-mediated drug-drug interaction potential and excretion of brentuximab vedotin, an antibody-drug conjugate, in patients with CD30-positive hematologic malignancies. J Clin Pharmacol 53(8):866–877. https://doi.org/10.1002/jcph.116
Sager JE, Yu J, Ragueneau-Majlessi I, Isoherranen N (2015) Physiologically based pharmacokinetic (PBPK) modeling and simulation approaches: a systematic review of published models, applications, and model verification. Drug Metab Dispos 43(11):1823–1837. https://doi.org/10.1124/dmd.115.065920
Samineni D, Ding H, Ma F, Shi R, Lu D, Miles D, Mao J, Li C, Jin J, Wright M, Girish S, Chen Y (2020) Physiologically based pharmacokinetic model-informed drug development for polatuzumab vedotin: label for drug-drug interactions without dedicated clinical trials. J Clin Pharmacol 60(Suppl 1):S120–S131. https://doi.org/10.1002/jcph.1718
Chen Y, Samineni D, Mukadam S, Wong H, Shen BQ, Lu D, Girish S, Hop C, Jin JY, Li C (2015) Physiologically based pharmacokinetic modeling as a tool to predict drug interactions for antibody-drug conjugates. Clin Pharmacokinet 54(1):81–93. https://doi.org/10.1007/s40262-014-0182-x
Liu SN, Li C (2021) Clinical pharmacology strategies in supporting drug development and approval of antibody-drug conjugates in oncology. Cancer Chemother Pharmacol 87(6):743–765. https://doi.org/10.1007/s00280-021-04250-0
López-Matencio JMS, NC, Baladrón AM, Santos Castañeda S (2018) Drug interactions of monoclonal antibodies-clinical perspective. J Immunol Sci 2(2):4–7. https://doi.org/10.29245/2578-3009/2018/2.1126
Ferri N, Bellosta S, Baldessin L, Boccia D, Racagni G, Corsini A (2016) Pharmacokinetics interactions of monoclonal antibodies. Pharmacol Res 111:592–599. https://doi.org/10.1016/j.phrs.2016.07.015
Bournazos S, DiLillo DJ, Ravetch JV (2015) The role of Fc-FcgammaR interactions in IgG-mediated microbial neutralization. J Exp Med 212(9):1361–1369. https://doi.org/10.1084/jem.20151267
Zhou H, Mascelli MA (2011) Mechanisms of monoclonal antibody-drug interactions. Annu Rev Pharmacol Toxicol 51:359–372. https://doi.org/10.1146/annurev-pharmtox-010510-100510
Brentuximab Vedotin (Adcetris). Pharmacology Review(s). 125388Orig1s000. Center for Drug Evaluation and Research. 25 Feb 2011. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/125388Orig1s000Approv.pdf
Hedrich WD, Fandy TE, Ashour HM, Wang H, Hassan HE (2018) Antibody-drug conjugates: pharmacokinetic/pharmacodynamic modeling, preclinical characterization, clinical studies, and lessons learned. Clin Pharmacokinet 57(6):687–703. https://doi.org/10.1007/s40262-017-0619-0
U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (2020) The Use of Physiologically Based Pharmacokinetic Analyses — Biopharmaceutics Applications for Oral Drug Product Development, Manufacturing Changes, and Controls: Guidance for Industry, Draft Guidance. https://www.fda.gov/media/142500/download. Accessed February 1, 2023.
Hsu Y, Carroll J, Ramsay J, McDermott L (2021) Correlation of Discreet Drug-linker Variation on Antibody Drug Conjugates (ADC) to Binding Activity Using Surface Plasmon Resonance. https://www.sigmaaldrich.com/deepweb/assets/sigmaaldrich/marketing/global/documents/769/863/adc-poster-ps7609en-ms.pdf. Accessed February 1, 2023.
Acchione M, Kwon H, Jochheim CM, Atkins WM (2012) Impact of linker and conjugation chemistry on antigen binding, Fc receptor binding and thermal stability of model antibody-drug conjugates. MAbs 4(3):362–372. https://doi.org/10.4161/mabs.19449
Brachet G, Respaud R, Arnoult C, Henriquet C, Dhommee C, Viaud-Massuard MC, Heuze-Vourc’h N, Joubert N, Pugniere M, Gouilleux-Gruart V (2016) Increment in drug loading on an antibody-drug conjugate increases its binding to the human neonatal Fc receptor in vitro. Mol Pharm 13(4):1405–1412. https://doi.org/10.1021/acs.molpharmaceut.6b00082
Derycke MS, Pambuccian SE, Gilks CB, Kalloger SE, Ghidouche A, Lopez M, Bliss RL, Geller MA, Argenta PA, Harrington KM, Skubitz AP (2010) Nectin 4 overexpression in ovarian cancer tissues and serum: potential role as a serum biomarker. Am J Clin Pathol 134(5):835–845. https://doi.org/10.1309/AJCPGXK0FR4MHIHB
Nishiwada S, Sho M, Yasuda S, Shimada K, Yamato I, Akahori T, Kinoshita S, Nagai M, Konishi N, Nakajima Y (2015) Nectin-4 expression contributes to tumor proliferation, angiogenesis and patient prognosis in human pancreatic cancer. J Exp Clin Cancer Res 34(1):30. https://doi.org/10.1186/s13046-015-0144-7
Challita-Eid PM, Satpayev D, Yang P, An Z, Morrison K, Shostak Y, Raitano A, Nadell R, Liu W, Lortie DR, Capo L, Verlinsky A, Leavitt M, Malik F, Avina H, Guevara CI, Dinh N, Karki S, Anand BS, Pereira DS, Joseph IB, Donate F, Morrison K, Stover DR (2016) Enfortumab vedotin antibody-drug conjugate targeting Nectin-4 is a highly potent therapeutic agent in multiple preclinical cancer models. Cancer Res 76(10):3003–3013. https://doi.org/10.1158/0008-5472.CAN-15-1313
Acknowledgements
Medical writing and editorial support were provided by Arpita Kulshrestha, PhD, and Cathy R. Winter, PhD, from Peloton Advantage, an OPEN Health company, and were funded by the study sponsors.
Funding
This study is sponsored by Astellas Pharma, Inc., and Seagen Inc.
Author information
Authors and Affiliations
Contributions
Peter Bonate, Mary P. Choules, Amit Garg, Mei Tang, Yukio Otsuka, Peiying Zuo conceptualized the manuscript, prepared the original draft, and reviewed and edited all drafts. Mary P. Choules developed the methodology and completed the formal analysis and investigation. Supervision was provided by Peter Bonate and Mei Tang. All authors made substantial contributions to the conception or design of the work; or the acquisition, analysis, or interpretation of data; or the creation of new software used in the work; drafted the work or revised it critically for important intellectual content; approved the version to be published; and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Competing interests
Mary P. Choules is an employee of Astellas Pharma, Inc. Peiying Zuo was an employee of Astellas at the time of the analysis; any meeting attendance to support this analysis was paid for by Astellas. They report an unpaid role as Secretary and steering committee member of Mathematical and Computational Sciences (MCS) special interest group in International Society of Pharmacometrics (ISoP). Yukio Otsuka is an employee of Astellas Pharma, Inc. and reports stock or stock options with Astellas Pharma, Inc. Amit Garg is an employee of Seagen, Inc. Mei Tang is an employee of Astellas Pharma, Inc. Peter Bonate is an employee of Astellas Pharma, Inc.
Research involving human participants and/or animals
This PBPK DDI study utilized data from clinical trials conducted in accordance with the Declaration of Helsinki and International Council for Harmonisation guidelines for good clinical practice.
Informed consent
This PBPK DDI study utilized data from clinical trials in which written/signed informed consent was obtained from patients before screening.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Choules, M.P., Zuo, P., Otsuka, Y. et al. Physiologically based pharmacokinetic model to predict drug–drug interactions with the antibody–drug conjugate enfortumab vedotin. J Pharmacokinet Pharmacodyn (2023). https://doi.org/10.1007/s10928-023-09877-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10928-023-09877-5