
Abstract
Continual reduction of landfill space along with rising CO2 levels and environmental pollution, are global issues that will only grow with time if not correctly addressed. The lack of proper waste management infrastructure means gloablly commodity plastics are disposed of incorrectly, leading to both an economical loss and environmental destruction. The bioaccumulation of plastics and microplastics can already be seen in marine ecosystems causing a negative impact on all organisms that live there, ultimately microplastics will bioaccumulate in humans. The opportunity exists to replace the majority of petroleum derived plastics with bioplastics (bio-based, biodegradable or both). This, in conjunction with mechanical and chemical recycling is a renewable and sustainable solution that would help mitigate climate change. This review covers the most promising biopolymers PLA, PGA, PHA and bio-versions of conventional petro-plastics bio-PET, bio-PE. The most optimal recycling routes after reuse and mechanical recycling are: alcoholysis, biodegradation, biological recycling, glycolysis and pyrolysis respectively.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Although the large scale production of plastics only dates back to the 1950’s, they have become vital materials used in a huge variety of everyday life applications [1]. The plastic industry has grown exponentially due to both, the variety of plastics available and the relatively cheap production from petroleum. In general, plastics offer excellent mechanical and barrier properties with a low bulk density and inertness, making them superior materials for a wide number of applications. A 2014 report estimated that an astonishing 311 MT of plastic was generated in a single year, which consumed 6% of world oil production as feedstock for polymer synthesis [2]. Of the total amount of plastic, 26% by volume was used in packaging applications and only 5% of which was recycled for subsequent use, deriving in an economic loss of £62–92 billion [2]. Annual plastic production is estimated to double by 2034, and by 2050 is estimated to reach 1124 MT which would consume 20% of the world oil production [2, 3].
The proportion of different polymers that make up global non-fibre plastic production is as follows: polyethylene (PE) (36%), polypropylene (PP) (21%), polyvinylchloride (PVC) (12%), polyurethane (PUR), polyethylene terephthalate (PET), and polystyrene (PS) (< 10% each). Fibre production consists primarily of polyester, polyamide and polyacrylic fibres, with PET accounting for 70%. These six non-fibre groups along with the fibres account for 92% of all plastics ever made [1]. Packaging applications alone have consumed 42% of all non-fibre plastics (mostly PP, PE, and PET), the next largest consuming sector is building and construction which has consumed 69% of all PVC and 19% of the total non-fibre plastics [1].
From 2010 to 2025 and estimated 100 MT of plastic waste will have entered the oceans [4, 5]. Once plastic enters the ocean it slowly degrades and fragments into increasingly smaller sizes until they become microplastics. Microplastics cause widespread contamination of marine ecosystems as they are ingested by zooplankton and phytoplankton (algae) causing a negative impact on their function and health [3, 6, 7]. Microplastics have shown to inhibit the growth of algae and considering that approximately 70% of the world’s oxygen is produced from photosynthesizing marine plants this could have huge consequence for climate change and global warming [7, 8]. Ultimately, microplastics bioaccumulate in humans and once inside the body they release constituent monomers as well as additives and toxins, causing physiological harm ranging from oxidative stress to carcinogenic behavior [9]. It was estimated that annual microplastic consumption in humans ranges from 74,000 to 121,000 particles [10].
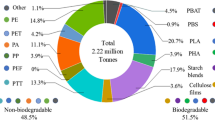
To remedy such concerns, the bioplastic industry is emerging as a promising solution to replace fossil fuel-based polymers. Biopolymer precursors are generated from biomass making them more renewable materials. Biomass growth consumes CO2 via photosynthesis. Therefore, overall production of bioplastics have a smaller carbon footprint than petro-plastics [11, 12]. In addition, some bioplastics possess very high biodegradability also making them more environmentally sustainable materials. In 2018 the global production for biopolymers was 2.11 MT, and it is predicted to reach 2.62 MT by 2023 [13]. Despite this rapid market growth, bioplastics still represent less than 1% of the total plastic production [14]. The more expensive production and typically inferior mechanical properties of bioplastics, compared to petro-plastics, are the main reasons for their limited application [4, 11].
The present review will cover the most promising biopolymers (based on market share value) PLA, PGA, PHA and bio-versions of petro-plastics bio-PET and bio-PE, identifying their main potential recycling routes. Polylactic acid (PLA) makes up a growing 10% of the bioplastic market, it has mechanical properties similar to PS and could replace it in its applications as a more sustainable material [13]. Polyglycolic acid (PGA) production is minuscule by comparison < 1%, nevertheless, PGA dominates the biomaterial suture market (valued at £1.1 billion) mainly because of its good mechanical properties, excellent biodegradation and biocompatibility [13, 15]. Polyhydroxyalkanoates (PHA) likewise only makes up a small percentage of the bioplastic market 1.4% but their production is set to quadruple by 2023 [13]. This is in part because of their excellent barrier property’s similar to PET, and because of their good mechanical properties similar to LDPE. Additionally, their monomers are highly variable which allows for tailoring of the final polymer properties. PE and PET are both very important polymers making up 32% and 10% of all non-fibre petro-plastics [1]. Bio-PE and bio-PET make up 25.6% and 9.5% of the bioplastic market respectively [13]. Despite bio-versions of PE and PET having a smaller carbon footprint and identical mechanical properties to their petroleum counterpart, their production growth has relatively stopped [4, 13]. This can be attributed to the cheaper costs of petroleum production instead of a renewable biomass resource.
Recycling Routes
Once plastic waste has been collected, sorted and cleaned, there are four possible recycling routes (Fig. 1). (A) Primary recycling is a closed loop recycling method that can only be carried out on high quality plastic scrap of known history. It refers to either the reuse of the material or the closed loop mechanical recycling of scrap plastic to produce products with the original structure [16, 17]. (B) Secondary recycling refers to the transformation/downgrading of waste plastic into a less demanding product via mechanical means (screw extrusion, injection moulding, blow moulding, etc.) [16,17,18,19,20]. Mechanical recycling offers the following advantages over chemical recycling: a lower processing cost, lower global warming potential, less non-renewable energy use and a less acidification and eutrophication [21]. (C) Tertiary recycling (chemical recycling) is used to describe chemical processes which depolymerise and degrade plastic waste into monomeric units or directly into other useful materials. Chemical recycling methods are diverse and numerous, and depending on the polymer type each technique will have its advantages and disadvantages. Chemical recycling has the following advantages over mechanical recycling: the opportunity to produce value added materials, the potential for a circular polymer production economy since recovered virgin monomers can be repolymerised for an indefinite amount of recycles [22, 23]. (D) Quaternary recycling refers to the energy recovery via incineration of low grade plastic waste. Plastics are high-yielding energy sources, when burned the resulting heat energy is used to generate steam and electricity [16]. This type of recycling should only be carried out as a last resort since not only is the imbedded energy of the polymers molecular structure lost, but also harmful chemicals and dioxins are released into the atmosphere [16, 24].

Different plastic waste treatment options and associated plastic quality
Biopolymers
By definition a bioplastic is either bio-based, biodegradable or a combination of both [13, 25, 26]. A bio-based polymer has been derived from carbon biomass sources such as vegetable fats, cellulose or corn starch. The biopolymer can be either fully or partially bio-based [11]. Bio-based polymers are more sustainable materials than petroleum-based polymers. The plants which provide the biomass feedstocks for biopolymer synthesis absorb CO2 as they grow, this way the net carbon footprint of bioplastic production is reduced [27]. Furthermore the biopolymers themselves emit smaller quantities of CO2 when disposed in an incinerator compared to conventional petro-plastics [25]. There is an important distinction between first- and second-generation biomass, the first refers to food biomass (e.g. sugar cane) whereas the latter refers to inedible biomass (lignocellulosic material) [28]. Using a second-generation biomass such as forest residues to synthesize bioplastics that could replace petro-plastics in their applications, would significantly mitigate climate change effects due to smaller carbon footprints [29, 30]. In order for bioplastics to replace conventional petro-plastics a number of issues need to be addressed. There needs to be a cost reduction in their production, a need to improve their thermomechanical and barrier properties, improved speed of biodegradability and a wider availability [25]. As the bioplastic market continues its rapid growth, its total production capacity increases and so its production prices will fall [31]. The key issue then is if the properties of bioplastics can be improved to become competitive with petro-plastics.
Degradation Routes
Biodegradation
Biodegradation of a polymer is the combination of abiotic reactions (photodegradation, oxidation, hydrolysis) and the enzymatic cleavage of its polymer chains into metabolic products (H2O, CO2, biomass, etc.) of the microorganism responsible for the enzymes [32, 33]. Microbial characteristics affecting the biodegradation rate include: the type of microorganism present, the microorganisms distribution, the growth conditions of the microbes (pH, temperature, moisture content, oxygen, nutrients) and the types of enzymes used by the microbes (intracellular, extracellular) [32, 34,35,36]. Plastic characteristics that affect the rate include the surface conditions (hydrophilic, hydrophobic), the first order structure (chemical structure, molecular weight) and the high order structure (Tg, Tm, crystallinity) [35, 36]. Biodegradation occurs in two discrete stages. In the first stage polymer chains are degraded and shortened by both abiotic reactions and extracellular enzymatic attack; enzymes preferentially degrade the less-ordered amorphous regions of a plastic. Therefore, the crystallinity of a plastic will rapidly increase and level off near 100% [36]. The second stage occurs when the polymer chains have reached a sufficiently small size, thus allowing for transportation into the cells of the microorganisms where they can bioassimilate and mineralize [33].
There are several international standards which define the biodegradability of bioplastics each under specific conditions. The standard EN 13,432:2000, for instance, states that for bioplastics to be compostable they must decompose by 90% and mineralize within 3 months in an industrial composter between 50 and 60 °C [37]. In principle it is not necessary to collect biodegradable plastics since they can be left to biodegrade in the environment which offers several advantages such as: increased soil fertility, lower accumulation of plastic in landfills and a reduction in the cost of waste management [25, 34, 38]. In reality, however, depending on the type of bioplastic and the conditions of the compose its rate of degradation may be significantly less than desired. Another disadvantage of biodegradation is that any value imbedded in the polymer’s molecular structure is lost. Instead, a circular economy approach should be worked towards, where bioplastics are recycled both mechanically and chemically to recapture the material value [14, 22].
Pyrolysis
(Thermal cracking) is the degradation of a polymer by heating in an inert environment with or without catalysts [17]. The pyrolysis process converts plastic waste through thermal decomposition to organic vapors, char and gases in the absence of oxygen. The produced organic vapors are converted into oil by a condensation process.
Solvolysis
Chemical recycling is normally achieved via solvolytic chain cleavage and heat. Solvolysis is an umbrella term and includes many depolymerisation and partial depolymerisation techniques. A variety of solvents and heat is used to depolymerise a polymer into its monomers or to fractionally depolymerise the polymer into its oligomers or other chemicals [39]. Common solvolysis methods are hydrolysis, alcoholysis, glycolysis, aminolysis and ammonolysis. Hydrolysis should be thought of as two components, water diffusion into polymer mass and the hydrolysis reaction occurring simultaneously. If water diffusion is slow compared to rate of hydrolysis, degradation of the polymer occurs primarily on the surface (heterogeneous erosion); conversely, if water diffusion is fast compared to the rate of hydrolysis, degradation occurs through the whole bulk of the polymer (homogeneous erosion) [40]. Alcoholysis refers to reactions where alcohol is the nucleophile. In terms of chemical recycling of polyesters a transesterification reaction occurs where the alcohol group cleaves the ester bonds. The polymer depolymerises into its monomers or value added products. Glycolysis in terms of chemical recycling of polyesters refers to the insertion of a glycol into the polymeric chains, breaking the ester linkages and replacing them with hydroxyl terminals.
Polylactic Acid
PLA is a polyester thermoplastic; depending on its tacticity it can have semi-crystalline or even completely amorphous characteristics. PLA can be synthesized directly from lactic acid (LA) via a polycondensation reaction; this method suffers from back-biting reactions due to trace amounts of water and impurities which limits the achievable Mw of the polymer [41, 42]. The industrially preferred method for PLA production is catalytic ring-opening polymerisation (ROP) of lactide, via the coordination-insertion of tin(II) octoate [43]. Other ROP pathways are possible such as anionic, cationic and organocatalytic, however coordination-insertion has been accepted as the most effective method in terms of MW (Mw > 100,000 g/mol) and microstructure of PLA [44]. The relief of the ring strain of the lactide during ROP acts as the thermodynamic driving force for the reaction overcoming the unfavorable entropy of polymerisation [44]. The majority of LA in circulation has been produced via fermentation [45]. A fermentation broth contains a complex mixture of impurities such as nutrients and cell debris which makes downstream processing of LA a costly and crucial step in the overall production of PLA [46]. Instead this could be avoided by recovering LA using a chemical recycling method. The mechanical properties of PLA depend on both the Mw and the degree of crystallinity (dictated by its tacticity) (Table 1) [47]. A higher Mw will increase the Tg and Tm as well as the tensile strength and the elastic modulus. PLA is limited by its inferior mechanical/barrier properties and its relative brittleness compared to existing petroleum derived polymers (Table 2). PLA’s brittleness can be improved by blending with tough polymers or via plasticization block copolymerisation, which has been shown to increase the strain at break but reduces the material’s tensile strength [48]. Despite these limitations PLA is still a promising bioplastic. Since PLA mechanical properties are comparable with those of PS, it is potentially a more sustainable replacement material.
Mechanical Recycling
The mechanical recycling of PLA is superior than its chemical recycling in terms of human and ecosystem health and use of resources. On the other hand mechanical recycling causes downgrading so the recycled PLA will be of a lower quality that if it was chemically recycled [53]. When mechanically recycling PLA, it is possible to add a chain extender which helps partially recover the impaired molar mass and other mechanical properties, making the recycled PLA more comparable with virgin PLA [54]. Niaounakis [55] estimated that in order for a specific biopolymer packaging and postconsumer mechanical recycling plant to be profitable, there needs to be a global production of at least 200 kT of biopolymer and the recycling facility should be able to process at least 5–18 kT annually. Once recycled PLA becomes low grade it should be chemically recycled to recover its virgin monomers.
Biodegradation
It is important to define the environmental conditions needed to decompose a biodegradable polymer. PLA biodegradation only occurs in a favorable environment of high temperature and humidity when appropriate microbes are present [56, 57]. While PLA fully decomposes into CO2 and H2O in a large scale composter at 50–60 °C in 90 days, its biodegradation in land littering or seawater is less than desired [35, 43]. PLA degrading microorganisms occur significantly less in the environment compared to other polyester degraders. The percentage of PLA degrading microbes in soil is only 0–0.04%, whereas degrading microbes for another biopolymer P3HB is between 0.2 and 11.4% [58]. After one year in a marine environment at 30 °C PLA only biodegrades by about 8% whereas biopolymer PHBV biodegrades by about 80% (Table 3) [59]. PLA composites can be prepared which increases PLA biodegradability. In soil, the rate of weight loss of pure PLA is ≈0% /year, whereas that of starch/PLA composite is between 0 and 15% /year, and that of starch/poly(hydroxyester-ether)/PLA of around 4–50% /year [60]. A PLA/P3HB blend was shown to biodegrade and mineralize in a composite pile at 52–59 °C after 70 days [61].
Hydrolysis
PLA can be hydrolysed with a 95% conversion to LA within 2 h at 160–180 °C [62]. The hydrolytic degradation of PLA follows third order kinetics since the hydrolysis rate depends on the concentration of polymer bonds, water and the acidic hydrolysis products [62]. It is important to note that the hydrolysis of PLA is autocatalytic, meaning that the carboxyl groups generated with each hydrolytic cleavage will further catalyze the reaction. If the hydrolysis medium is basic, then the mechanism of ester cleavage is random, but if the medium is acidic, then the cleavage mechanism is chain-end unzipping. The parameters governing hydrolytic degradation are: the rate constant, the amount of absorbed water, the diffusion coefficient of chain fragments, the solubility of degradation products, pH, temperature and the crystallinity of the PLA [63]. One clear example of the potential of chemical recycling is in the recovery of LA. It has been estimated that the production of LA from the fermentation of corn is around 55 MJ/kg of LA produced; in contrast, the formation of LA via the hydrolysis of PLA is much less energy intensive at about 14 MJ/kg of LA produced [40]. Not only is the chemical recycling less energy intensive thus saving money on operating costs, it also avoids the costly separation needed to separate LA from a fermentation broth.
Alcoholysis
PLA can be depolymerised via alcoholysis to generate value added products. A variety of alcohol groups can be used to attack the ester bonds of PLA via a transesterification reaction to produce lactate esters. A transesterification catalyst is also required for sufficient depolymerisation with relatively mild reaction conditions [64]. Depolymerising PLA waste using methanol, ethanol, propanol etc. will yield methyl lactate, ethyl lactate and propyl lactate respectively which are industrially valuable chemicals [65]. Román-Ramírez et al. [64, 66] depolymerised PLA via methanolysis using Zn(Et)2 and Zn(Pr)2 catalysts (synthesized from ethylenediamine and propylenediamine Schiff bases), for each catalyst they calculated activation energies of Ea1 = 37.89 kJ mol−1 and a barrierless Ea1 = − 5.7 kJ mol−1 respectively. The conversion of PLA waste into lactate esters via alcoholysis offers several advantages such as: value added products derived from waste, high yield of lactate ester, simple purification and retention of stereochemistry [67]. Additionally alkyl lactates can be converted into lactide, which could then be converted into PLA via ROP creating a truly circular economy (Fig. 2) [68, 69]. Furthermore this type of recycling adds value to the PLA supply chain since virgin PLA is valued at £1.69 per kg whereas ethyl lactate sells at £2.54–3.49 per kg [67, 70]. In 2011 the U.S. had a solvent demand of ≈ 4.5 × 105 tons per year with prices ranging from £1.38 to 2.62 per kg [70]. For ethyl lactate to compete directly with petroleum derived solvents, processing advances need to be made in order to lower the price of ethyl lactate to a comparable level. The volatile nature of oil prices makes ethyl lactate a more commercially attractive solvent, and as global environmental consciousness continues to rise more consumers will be willing to pay extra for products that are less detrimental to the environment.

One possible circular route for PLA production
Optimal Recycling Route
All plastics including PLA should be reused before recycling as this has the lowest energy requirement. In most cases, however, this is not possible. The next best scenario is to mechanically recycle the plastic for as long as possible, until its properties deteriorate and the material becomes low grade. The monomers of the low grade plastic should be recovered via a chemical recycling method. For PLA both hydrolysis and alcoholysis routes have smaller activation energies than pyrolysis (Table 4). Although PLA depolymerises via hydrolysis under relatively mild conditions without a catalyst, depolymerisation via methanolysis has a smaller activation energy, furthermore, chemically recycling PLA via an alcoholysis route has the additional benefit of obtaining value added products (alkyl lactates). The lowest PLA depolymerisation activation energy − 5.7 kJ/mol is accomplished via methanolysis using Zn(Pr)2, this barrierless reaction has huge potential for industrial scale up [66]. The next lowest activation energy 20.96 kJ/mol belongs to the ionic liquid 2[Bmim][OAc]:Zn(OAc)2 [71]. Like other ionic liquids it has excellent recovery and reusability, it was reused five times without significant decrease in catalytic activity achieving PLA conversions of 97% in 2 h [71]. The products from both hydrolysis and alcoholysis can be converted into lactide which can then be polymerised into PLA. Both types of recycling can be used to create a circular economy for PLA production. In theory, the biodegradation of PLA is also a circular economy since the generated CO2 is absorbed by plants and converted to glucose, this glucose can then be used to produce LA and dimerized to lactide which can then be repolymerised back to PLA. In practice, however, the very low levels of PLA degraders in soil and the ocean means it has a very slow degradation rate of years. Even in an industrial composter it takes 90 days to fully decompose [43]. Low grade PLA should not be biodegraded instead its monomers should be recovered via the lowest activation energy alcoholysis route.
Polyglycolide
Polyglycolide (polyglycolic acid) (PGA) is a petroleum derived biopolymer and has the simplest polyester molecular structure. PGA is synthesized either by the polycondensation of the monomer glycolic acid to produce low Mw PGA, or via the catalytic ROP of glycolide to produce high Mw PGA (200,00–140,000 g/mol suitable for suture manufacturing) [75, 76]. Glycolide is synthesized from the dimerization of the glycolic acid monomer. PGA is highly crystalline (45–55%) with a high Tm between 220 and 225 °C and a Tg of 35–40 °C [75]. PGA’s strong mechanical properties along with its high biodegradability makes it a very suitable material as absorbable sutures in medicine. In 2010 the biomaterial suture market was valued at £1.1 billion [15]. PGA and its copolymers represent the largest production by volume for medical sutures compared to other commercial biopolymers [77]. Glycolide is often copolymerised with L-lactide to form a polyglycolide-co-lactide (90:10) copolymer (PGLA). PGA and its copolymers are used in a range of medical applications and procedures such as screws, nails, treating bone fractures and injuries of internal organs [78]. PGA represent less than 1% of the biopolymer market, its collection and recycling is not necessary since the only PGA in circulation is then used in medical applications where they biodegrade inside the body.
Biodegradation and Biocompatibility
The ratio between glycolide and L-lactide of the PGLA copolymer will determine both, the crystallinity and the rate of absorption. Sutures fabricated from 100% glycolide have the fastest in vivo absorption, while sutures fabricated from 100% L-lactide have the longest absorption times (longer than 5 years to completely absorb) (Table 5) [79]. For wound closure applications PGA sutures must have enough crystallinity to attain adequate tensile strength required for the application, it must also retain sufficient strength during its degradation and absorption into the body [79, 80]. A completely amorphous biodegradable polymer would degrade too fast to be useful for wound closure. A copolymer of PGA-co-PDLLA has higher amorphous content compared to a copolymer of PGA-co-PLLA; so degrades faster since more amorphous content means easier penetration of water molecules into the polymers chains causing chain scission reactions [80].
Polyhydroxyalkanoates
Polyhydroxyalkanoates (PHA) represents a family of optically active biological polyesters, these polyesters naturally occur in a wide range of Gram-positive and Gram-negative microorganisms [51]. Anaerobic fermentation of abundant lignocellulosic wastes generates volatile fatty acids (VFA), under conditions of nutrient stress native PHA producing organisms will utilize VFA for their growth and will accumulate PHA as intracellular carbon storage and energy reserve, to levels as high as 90% of the cell dry weight [51, 81, 82]. Bacteria such as Alcaligenes latus and recombinant E. Coli do not require nutrient limitation in order to synthesis PHA [83]. Once accumulated the polyesters are then extracted from the bacteria and purified before they can be used in a variety of applications [84, 85]. End-of-life PHA could be composited and biodegraded to CO2 which would assimilate into plants. The resulting plant biomass could then be fermented to generate more PHA ensuring in a circular economy [86]. More than 100 different monomer units have been identified as constituents for PHA. The general structure of the monomers is (R)-hydroxy fatty acids where the side group varies from C1–C14. PHA monomers are classified as either: short chain length (SCL) (C3–C5) which have high crystallinity and are brittle, or medium chain length (MCL) (C6–C14) which are more flexible but have lower tensile strength and Tm [87]. Additionally, the monomer chain can be saturated, unsaturated, branched or have a substituted side chain [51, 81]. By controlling the composition of these monomers as well the polymer composition (homo- or co-polyester) creates the possibility of generating PHA with drastically different properties (Table 6).
PHA only makes up 1.4% of the biopolymer market despite having superior barrier and mechanical properties to PLA, but its production levels are set to quadruple by 2023 [13]. PHA shares the same brittleness limitation as PLA and likewise its brittleness can be improved using tailored blends with plasticizers [89]. Although PHA has vastly superior barrier properties compared to other biopolymers its drawback is a higher production cost [90]. PHA has high biodegradability and good barrier properties meaning it could replace PET in bottle applications (Table 7); its excellent ultimate strain means it could replace LDPE in film applications as a more environmentally sustainable material [91]. Since the monomer of P3HB is a normal metabolic of the human blood, the polymer also has applications in medicine where it can be used as surgical implants [87, 90, 92].
Industrial PHA production is commonly carried out using pure microbial culture (PMC) fermentation. This route has high costs associated with the use of refined sugar substrates, fermentation operation and downstream processing. As much as 50% of the total production costs is due to the refined sugar substrates [93]. In an attempt to reduce the production costs, research has focused on both the optimizion of (PMC) fermentation via genetic engineering, and the use of mixed microbial cultures (MMC) that exploit waste feedstocks. Using MMC for PHA production has both a smaller economic and environment cost compared to PMC fermentation; this is because they can utilize the surplus of waste feedstocks and do not require sterile conditions [94]. Amulya et al. [95] reported the industrial production of copolymer poly(3-hydroxybutyrate-co-3-hydroxyvalerate (PHBV) from food waste using MMC fermentation, achieving a PHA production of 23.7% of the cell dry weight, this route has the additional benefit of generating biohydrogen as a side product.
Poly(3-Hydroxybutyrate)
The most well know member of PHA is Poly(3-hydroxybutyrate) (P3HB) which is produced naturally in the bacterium Alcaligenes eutrophus. There are three well-known biosynthetic pathways for PHA production: a carbon source pathway that generates 3-hydroxybutyrate monomers, a fatty acid degradation pathway and a fatty acid biosynthesis pathway that generate different monomers [88]. P3HB is derived from a three-step pathway of enzymatic reactions, the genes responsible for these enzymes (phbA, phbB and phbC) are conveniently organized on a single operon (phbCAB) (Fig. 3) [81]. The reaction pathway: (A) β-ketoacyl CoA thiolase (encoded by the phbA gene) condenses two Acetyl-CoA molecules into Acetoacetyl-CoA, (B) Acetoacetyl-CoA dehydrogenase (encoded by the phbB gene) reduces Acetoacetyl-CoA to the monomer D-Hydroxybutyryl-CoA, (C) PHB polymerase (encoded by the phbC gene) polymerises the monomer into P3HB [96]. DNA fragments containing the phbCAB operon can be used as a cartridge in other bacteria that express A. eutrophus PHB-biosynthetic genes, thus giving them the ability to synthesize P3HB from aceyl-CoA [96]. P3HB has also be generated in transgenic plants, which provides even greater control over monomer composition and thus final polymer properties [97].

Adapted from Reddy et al. [81]
Simplified pathway of P3HB synthesis. Operon shows genes responsible for the enzymes of the three step pathway.
Mechanical Recycling
There are only a few studies on the mechanical and chemical recycling of PHA due to both its high production costs and low amounts in circulation. If a P3HB homopolymer is mechanically recycled by the third extrusion cycle there is a 50% reduction in its tensile strength [98]. The mechanical recycling of PHA copolymers has more potential. Zaverl et al. [99] investigated copolymers of PHBV which only suffers an 8% loss in tensile strength after five extrusion cycles. PLA can be added to the copolymer which has a stabilizing effect on its recyclability. PHBV/PLA blends have been reported to maintain their mechanical properties even after six extrusion cycles (Table 8) [100]. Once mechanical options have been exhausted low grade PHA could either be recycled via pyrolysis to obtain valuable chemicals, or biodegraded into CO2 and assimilated by plants which could then be used in fermentation.
Pyrolysis
Pyrolysis of PHA can also be undertaken at relatively mild temperatures and activation energies to yield several products (Table 9). The thermal degradation mechanism of P3HB occurs exclusively via a random chain scission reaction (cis-elimination) of the ester groups, forming the major products trans/cis-crotonic acid (CA) and its oligomers [101]. Through simple esterification reactions CA can be converted into crotonate esters [102]. From here there is promising potential to polymerase these stereo-specific crotonate esters into optically active polymers, which would be obtaining a value added product from PHA waste [103]. Catalytic pyrolysis improves the product distribution giving a higher proportion of valuable products [104].
Biodegradation
PHA will readily biodegrade in compost, soil and marine environments [92, 105]. In the absence of microorganisms and ambient conditions PHA remains stable. The copolymer poly(3-hydroxybutyrate-co-3-hydroxyhexanoate) (P3HB-co-3HH) has similar biodegradation rates to cellulose powder. Under anaerobic conditions and room temperature P3HB-co-3HH experiences a 62.9% conversion of its carbon to gas in just 85 days [105]. Under aerobic seawater conditions at room temperate the same copolymer has a gaseous carbon loss of 83% in 195 days [105]. There are numerous microorganisms capable of degrading PHA, each microbe will have a preferential environment where it has optimal growth. Although some microorganisms are capable of degrading extracellular SCL-PHA and MCL-PHA, the majority of PHA degraders are for SCL-PHA and can be found in soil, sewage sludge, compost and marine water. Microbes in the soil environment for SCL-PHA is between 0.2 and 11.4% [58, 106].
Short chain fatty acids (SCFA) have been shown to exhibit bacteriostatic activity, interestingly SCL-PHA can be degraded in the gastrointestinal tract of some animals resulting in biocontrol effects similar to SCFA [83]. Freier et al. [107] showed that a PHB patch could be used to repair bowel defects of rats, in one week it degraded by 10%. It has also been reported that copolymer PHBV pretreated with NaOH is digested in pigs by 37% [108]. The same pretreated copolymer has a digestibility of 85% in sheep [109]. Defoirdt et al. [110] reported that monomer 3HB completely inhibits growth of pathogenic Vibrio campbelli in brine shrimp, 3HB could be used as a more ecologically and economically sustainable alternative to fight infections in aquacultures instead of antibiotics. Additionally, 3HB have been shown to have antimicrobial activity against Vibrio, E. Coli and Salmonella in the gastro intestinal tract of higher organisms [111]. End-of-life PHA should be used as an energy providing feed ingredient as it has no adverse effects on animal health and provides bacteriostatic effects.
Optimal Recycling Route
The mechanical and chemical recycling routes for PHA have not been thoroughly explored due to its high production costs and low amounts in circulation. Similarly to the other biopolymers, PHA should first be reused then mechanically recycled until its properties deteriorate. Low grade PHA then has two recycling options, pyrolysis or biodegradation. Although pyrolysis leads to the generation of CA which itself is a value added product, end-of-life PHA should instead be biodegraded. This is because biodegraded PHA allows for a circular economy of PHA production. The degradation products CO2 and water are assimilated into plants which could be used in fermentation for PHA production [86]. In terms of biodegradation, PHA degrades in composite, soil and marine environments, and it also degrades in some animals. The ideal route would utilize end-of-life PHA as an additional feed ingredient for animals that host the correct microorganisms. 3HB the most common monomer of PHA, has been shown to have antimicrobial activity against harmful pathogens. Biological recycling reduces the need for antibiotics and decreases the cost of animal feed since proteins, lipids and minerals of the bacterial PHA degraders will be used as nutrition in the animals [112]. Undigested PHA is excreted in fecal matter this is then an ideal medium for MMC fermentation to generate new PHA ensuring a circular economy.
Bio-Polyethylene Terephthalate
Another important commercial polyester is polyethylene terephthalate (PET), it has excellent material properties such as high mechanical strength, good barrier properties and strong solvent resistance [113]. PET has many applications in construction, transport and packaging, but is predominantly used in the textile and drinking bottle industries [113, 114]. Production of PET consists of four stages, first, bis(hydroxyethyl)terephthalate (BHET) is produced from ethylene glycol (EG) via either a transesterification reaction with dimethyl terephthalate (DMT) or an esterification reaction of terephthalic acid (TA). The second stage is a pre-polymerisation of BHET; the third stage is a melt polycondensation to form low Mw PET (suitable for fibres), and the fourth stage is used to generate high Mw PET (suitable for bottles) via solid state polymerisation [115]. The esterification of EG with TA is the industrially preferred method over transesterification with DMT since: it has a higher reaction rate, it uses water instead of methanol as the condensation agent, it requires no transesterification catalyst and generates higher molecular weight PET [116, 117].
Bio-PET—Although PET is non-biodegradable and its precursors TA and EG are fossil fuel derived, it is entirely possible to obtain the monomers from renewable resources and therefore synthesis bio-based PET [118]. The majority of bio-based PET currently in circulation is only 30% partially bio-based where only one of its monomers, EG, is produced from biomass. Technical constraints have limited TA production to fossil fuel based [119]. A few beverage companies such as Dasani, Coca-cola, Pepsi and Nestle already sell some of their drinks in 30% partially bio-based plant bottles [120]. One possible solution to generate 100% bio-based PET is to use abundant lignocellulosic biomass from forest residues to generate bio-TA [29, 121]. Yeast microorganisms can be used to convert lignocellulosic biomass into isobutanol (IBA), from here IBA can be further processed into paraxylene (PX) which is a precursor to TA (Fig. 4) [122, 123]. While a variety of biomass sources can be used to generate bio-versions of TA and EG, different sources will have different environmental impacts, with the smallest impact belonging to second-generation sources. Bio-PET bottles with precursor TA obtained from forest residues have a CO2 emission of 4.14–4.92 kg CO2 equivalent per kg PET bottle. This is 27% lower than Bio-PET with precursor TA derived from corn stover, and 21% lower than Bio-PET with fossil derived TA [121]. This highlights that just because a precursor is obtained from a renewable resource does not necessarily mean, it will have a lower environmental impact compared to its petroleum counterpart. In the TA from corn stover example, production is very energy intensive as it includes farming and harvesting, once the corn stover is processed into its sugars it then has to be converted to IBA which is further processed into PX and then TA, which overall produces more CO2 emissions than deriving TA straight from petroleum based PX [121, 124]. Similarly, in the TA obtained from forest residue example, the biggest CO2 impact is from the initial IBA production. It takes a considerable amount of energy to destruct lignocellulose in woody biomass into fermentable sugars that can be processed to IBA [121, 125]. Bio-PET has identical properties as petroleum derived PET and can be processed using the same equipment and same polymerisation techniques and conditions. Furthermore bio-PET can be recycled in the same recycling stream.

Adapted from Chen et al. [121]
PET production from biomass.
Mechanical Recycling
The mechanical recycling of PET bottles is well established in part because of the large amount of PET already in circulation but also because the recycled PET is utilized in a variety of applications. Around 72% of recycled PET is used in fibre applications [21, 126]. Mechanically recycled PET suffers from yellowing discoloration but this has been shown to be nontoxic [127]. Recycled PET has a reduction in rheological, mechanical and thermal properties; the reduction significantly increases with the number of recycles (Table 10) [128]. This can be explained by a buildup of impurities causing chain scission reactions and cleavage of the ester bonds [129]. Another downside is that PET waste streams are easily contaminated by both PVC and PLA. PVC content as low as 100 ppm causes significant degradation of the recycled PET during the high temperatures required for PET melt reprocessing (160 °C). PVC generates acids that act as catalysts causing chain scission reactions lowering the recycled PET Mw and its commercial value [129,130,131]. PLA content as low as 1,000 ppm causes noticeable hazing and degradation of the recycled PET. The low Tm of PLA (155 °C) results in serious disruption during melt reprocessing of PET [130]. Once recycled PET becomes low grade it can no longer be mechanically recycled, at this point it should be chemically recycled to recover its monomers.
Hydrolysis
PET can be hydrolysed into its monomers TA and EG. Hydrolysis conditions are either acidic, alkaline or neutral. The disadvantage of hydrolysis is its high temperature requirements (200–250 °C) and pressures (1.4–2 Mpa), along with long reaction times. Commercially, hydrolysis of PET is limited as there are additional costs associated with purifying the recycled TA [132, 133]. Alkaline hydrolysis of PET is typically carried out using an aqueous alkaline solution of NaOH at a concentration of 4–20 wt.%. The reaction conditions are 3–5 h at 210–250 °C and 1.4–2 Mpa and generates products EG and a salt TA-Na2 [132]. To recover the products the mixture is heated to 340 °C to recover EG then the remaining TA salt has to be neutralized with a strong acid [133]. Acid hydrolysis of PET is carried out using a concentrated acid, industry frequently use concentrated sulfuric acid (H2SO4 > 14.5 M). A number of patented processes use H2SO4 (minimum 87 wt.%) which allows for a reaction at atmospheric pressure and low temperature [134]. Major drawbacks to this method are: the high corrosion of the system, the generation of large amounts of waste inorganic salts, the need to purify EG from H2SO4 [133]. Neutral hydrolysis of PET is carried out in water therefore it has the advantage of high ecological purity. Its drawbacks are, however, that any mechanical impurities from the polymer will be left in the recovered TA, it requires a higher temperature (300 °C) and pressure (4 MPa) than other types of hydrolysis [133, 134]. Recovered TA and EG can be repolymerised to form virgin PET.
Alcoholysis
PET can be depolymerised via methanolysis which results in the stoichiometric formation of its monomers DMT and EG. Methanolysis of PET flakes is typically carried out at high temperatures (180–280 °C) and high pressures (2–4 Mpa), and divalent metal catalysts are usually employed to enhance depolymerisation rates [114, 135]. The methanolysis of waste PET also generates several byproducts such as BHET, 2-hydroxyethyl methyl terephthalate (MHET), oligomers, and dimers of DMT and BHET (Fig. 5). The separation and refinement of these byproducts make PET methanolysis a costly process. Another major problem with this method is that the product is DMT, and nowadays the majority of PET is based synthesis from TA. Therefore, additional conversion of DMT to TA is required which adds considerable costs to the whole process [136]. In comparison to glycolysis of PET, methanolysis has a simpler product purification and can accept lower quality feed of PET wastes [136]. The methanolysis of waste PET can also be achieved without the use of a catalyst, however, in order to get reasonable yields supercritical methanol (239 °C, 8.09 MPa) must be used (Fig. 5) [135, 137, 138]. The optimal conditions for supercritical methanolysis of PET are a temperature range of 250–270 °C at 9–22 MPa and weight ratio methanol to PET from 6 to 8; 98% conversion are achieved in as little as 40 min [137]. An increase in the ratio of methanol to PET and an increase in reaction temperature, both increased the degree of depolymerisation and DMT selectivity [137]. PET can also be depolymerised via supercritical ethanolysis (241 °C, 6.14 MPa), were PET waste completely depolymerises at 255 °C and 7.6–11.6 MPa in 5 h with a DMT yield of 98.5% [139].

Overall reactions for PET alcoholysis using supercritical methanol
The same alcoholysis conditions that would completely depolymerise PLA leaves PET unconverted. The nucleophilic attack of an alcohol group with a PET ester bond is steric hindered by benzene rings, which explains its lower reactivity towards alcohols in comparison to PLA [140]. It is well known that depolymerisation process are initiated more easily in amorphous regions than crystalline regions. Despite both polymers being semi-crystalline, PLA plastics tend to have more amorphous content in comparison to PET plastics, which also explains PET lower reactivity [141].
Glycolysis
Glycolysis is the most widely used chemical recycling method for PET. Glycolysis is the insertion of a glycol into the PET chains, breaking the ester linkages and replacing them with hydroxyl terminals to produce BHET, oligomers and dimers [114, 132]. The optimum parameters for PET glycolysis are: a temperature range of 180–240 °C, pressures of 0.1–0.6 MPa, a transesterification catalyst, a high EG/PET ratio and 0.5–8 h for completion, product formation is mostly BHET with very little oligomers [132, 136, 142]. The reaction follows first order kinetics with respect to PET, and second order with respect to EG concentration [143]. A higher ratio of EG:PET (≥ 5:1) favors the formation of BHET instead of oligomers [143]. The addition of cyclohexylamine with NaOH in EG significantly enhances the rate of glycolysis and results in the product formation of DMT and EG instead of BHET [144].
In the absence of a catalyst the reaction is slow unless there is very high temperature and pressure [145]. Glycolysis can be carried out using a variety of glycols such as EG, diethylene glycol (DEG), propylene glycol (PG) and dipropylene glycol, however most attention in literature has been devoted to glycolysis by EG [39]. Several catalysts can be used for glycolysis, industry often used a metal acetate and Zinc acetate is considered the best among them (Table 11). These catalysts have several drawbacks such as: difficulty separating the catalyst from the products, side reactions, product impurities [145]. Instead ionic liquid catalysts can be used which have similar activities and selectivity for BHET, while also being more recyclable and have a simpler purification process [143, 146, 147]. Sangalang et al. [148] used a generalized kinetic analysis for PET glycolysis and calculated an activation energy of 29 kJ/mol. This value is somewhat lower than other activation energy’s reported in literature, but it is closer to theoretical values for ester bond degradation and transesterification [148]. Glycolysis can be used to achieve a circular economy since BHET can be repolymerised to virgin PET.
Additionally, value added products can be obtained from glycolysis; BHET, dimers and oligomers can serve as starting materials for the production of unsaturated polyester resins (UPR) [143]. Vaidya and Nadkarni [149] were among the first to study the formation of UPR [149]. They found that if PG was used for glycolysis of PET then the product oligomers could be further reacted with maleic anhydride and mixed with styrene monomer to produce UPR. It has also been reported that the type of glycol used in glycolysis has a significant effect on the characteristics of the cured UPR resins [150]. Both cured EG and DEG UPR exhibited hard and brittle characteristics whereas cured PG UPR exhibits characteristics compatible with PS [149, 150]. UPR are used in applications such as varnishes, lacquers, artificial pearls and ornaments [151].
Optimal Recycling Route
PET materials should be reused where possible, after which PET should be mechanical recycled for as long as possible. Lower grade PET can be used in fibre applications. Low grade PET should be chemically recycled to recover its monomers. A variety of chemical recycling routes can be used to depolymerise PET to its monomers, which once recovered can then be repolymerised to PET achieving a circular economy. Among the different possible chemical routes, glycolysis offers the lowest activation energy (Table 12). Moreover, if glycolysis is carried out using an ionic liquid then product purification is relatively easy. While hydrolysis and alcoholysis generate the monomers TA, DMT and EG, glycolysis generates BHET which requires less polymerisation steps to generate PET. Glycolysis also offers the opportunity to obtain UPR which is a value added product.
Bio-Polyethylene
Polyethylene (PE) is a crystalline thermoplastic and is arguably one of the most important commodity plastics in circulation accounting for 36% of all non-fibre plastic, it has good mechanical resistance and is highly resistant to hydrolysis, solvolysis and oxidation [1, 47]. PE can be separated into two main categories depending on both its density and the degree of branching, high-density PE (HDPE), and low-density PE (LDPE) [159]. LDPE has a combination of desirable properties such as a very low Tg (≈ –120 °C) and moderate Tm (105–115 °C), these properties allow the material to have a good strength, flexibility, impact resistance and melt flow behavior, creating utility for LDPE over a wide temperature range [47]. On the other hand, HDPE which is synthesized via catalytic coordination with a Ziegler–Natta catalyst has little to no branching, this allows for more efficient packing of polymer chains causing stronger intermolecular forces and a higher crystallinity and tensile strength [17, 47].
Bio-PE—Conventionally PE is a fossil fuel derived plastic however it is entirely possible to obtain PE’s monomer ethylene from a renewable resource and thus generating bio-based PE. Bio-ethylene is normally synthesized from the catalytic dehydration of bio-ethanol, which itself is generated from first- or second-generation fermentation of biomass [160, 161]. In theory, any of the existing petrochemical based PE plants could be converted to a bio-PE plant just by using bio-based ethylene instead of petro-based [159]. The same polymerisation techniques and conditions can be used and bio-PE can be processed using the same equipment. Furthermore, bio-PE can be recycled in the same recycling stream, no additional investment into equipment changes or waste sorting is needed to implement bio-PE. Bio-PE offers a reduced carbon footprint (especially when utilizing second generation bio-ethylene) and matches the thermomechanical ability of PE. If bio-PE replaced conventional PE in all of its huge variety of applications and was collected and recycled at end-of-life, it would have a significantly positive impact on the environment [159, 161].
Mechanical Recycling
PE can be mechanical recycled via melt extrusion up to 40 cycles without a significant change in mechanical properties or processability [162]. After this, there is a drop in Tm due to reduced crystallinity of the recycled polymer. The decreased crystallinity is attributed to less effective packing from structural irregularities formed because of thermo-mechanical and thermo-oxidative degradation [162, 163]. PE has a highly stable molecular structure meaning it is not susceptible to any type of solvolysis.
Pyrolysis
Pyrolysis of PE is carried out at high temperatures (≤ 800 °C) or low temperatures (400–500 °C) in an inert environment. Its thermal decomposition leads to the formation of a solid char and a volatile fraction. The fraction can be separated into a condensable hydrocarbon oil and a non-condensable gas that has a very high gross calorific value (GCV) [17, 164, 165]. The hydrocarbon oil produced from the pyrolysis of mixed plastic waste streams (predominantly PE, PS, PET, metals, paper, etc.) consists of a complex mixture of organic compounds such as styrene, toluene, ethyl-benzene etc.[166]. These valuable chemicals can be used as raw materials for many other applications, e.g. styrene could be used in UPR production [150]. The non-condensable gas is a mixture of high GCV hydrocarbons along with CO2 and CO [166]. The chemical energy of pyrolysis gas increases substantially with increasing process temperature. The gas produced from PE has a GCV of 46.06 MJ kg−1 [167]. High PE content in mixed waste generates higher viscosity liquids and high metal content yields more aromatics in the liquid fraction [166, 168]. The oil is the desired product of pyrolysis while char and gases are by-products [169]. The theoretical energy requirement to pyrolyze 1 kg of PE is 1.047 MJ and can generate around 72–84 wt.% liquid oil [170]. The energy value of the oil is around 43 MJ/kg so the energy profit is very high for this process [169, 171]. The high heating value of char means it has the potential to be used as an energy source, it can be crushed into powder and made into briquettes. Char derived from HDPE pyrolysis has a calorific value of 18.82 MJ kg−1 [172]. Chars derived from 100% PE have higher combustion rates and heat release in comparison to a mixed waste char [173].
The distribution of products depends on several factors including temperature, retention time, moisture content, heating rate, particle size of feedstock, the type and operation mode of the reactor [165, 169]. With an increasing temperature the liquid fraction and char amount decreases and the gas fraction increases [166, 174]. Increasing the pyrolysis temperature results in shorter chain products because of increased C–C bond cracking [169]. PE is only suitable for pyrolysis if a catalyst is used, without it PE is mostly converted into wax instead of liquid oil [168].
Catalytic pyrolysis has a narrower distribution of hydrocarbon products, through careful selection and experiment design products of higher market value could preferentially be generated (Table 13) [17, 175, 176]. Solids such as silica alumina, ZSM-5, and zeolites have been reported to effectively convert PE into liquid fuel, giving lighter fractions as compared to standard thermal cracking [16, 177]. ZSM-5 zeolite is commonly used for catalytic pyrolysis of PE achieving higher conversions of valuable aromatics in the oil compared to thermal degradation only; furthermore, the catalyst has been shown to decrease pyrolysis energy consumption [178]. A downside is that ZSM-5 loses its activity after use however, the deactivation can be reversed upon heating at 550 °C with oxygen [178]. ZSM-5 has a high micropore area and volume meaning it has high internal porosity, this along with its acidic nature make it an effect catalyst [169]. The acid nature of the catalysts enhance conversion by protonating defective sites along the polymer chain which then undergo β-scission, acid strength and textural properties of the catalyst are the main parameters dictating performance [179]. Porosity, surface area characteristics and particle size, determine to a large extent the accessibility of bulky polymeric molecules to the internal catalytic acid sites of the catalyst [175].
Optimal Recycling Route
Bio-PE/PE materials should be reused where possible, after which should be mechanical recycled for as long as possible. Low grade PE should then be chemically recycled to recover its value. Since PE is highly resistance to solvolysis the only chemical recycling route capable of degrading it is pyrolysis. Catalytic pyrolysis should be carried out as it has a lower energy requirement than just thermal decomposition and generates a greater proportion of valuable products (Table 14). ZSM-5 is robust catalyst that is well suited for the industrial pyrolysis of PE achieved very high conversions of oil fraction with a large proportion of aromatics in the liquid. There is a strong synergistic interaction between cellulose and PE, suggesting that PE should be mixed with lignocellulosic waste before pyrolysis [183]. The oil consists of very valuable aromatics that can be used as feedstocks in a variety of applications. Additionally, the byproducts gas and char are also valuable as they can be used as fuel substitutes.
Conclusion
Rising CO2 levels and climate change means there is an increasing pressure to shift from non-renewable fossil-fuel based plastics to renewable bioplastics. Bioplastics should be synthesized from second generation biomass instead of first generation and should then replace petro-plastics in as many applications as possible due to their inherit lower carbon footprint. In applications that require the superior properties of petro-plastics, instead of replacing them with a mechanically inferior bioplastic the petro-plastic itself should be derived from a renewable resource. This review mentioned two such cases, the commodity plastics PE and PET are conventionally derived from fossil fuels even though bio-versions of both can be synthesized that match the mechanical ability just at a fraction of the carbon footprint.
The optimal recycling route for each of the mentioned polymers should first be reuse, after which the polymers should be mechanically recycled for as long as possible until they become low grade. Bio-PET and bio-PE both maintain their mechanical properties for a decent number of recycles. Once the polymers become low grade their monomers should be recovered via a chemical route, the monomers can then be repolymerised resulting in a circular production economy. Each biopolymer has an optimum route with the lowest activation energy. PLA should be recycled via alcoholysis since it generates a value added product. Similarly, bio-PET should be recycled via glycolysis since it also generates a value added product. Bio-PE has strong solvent resistance so it can only be recycled via pyrolysis. PGA has a small amount in circulation and is only used in medical applications where it fully biodegrades. PHA should be biologically recycled as it reduces the need for antibiotics and decreases the cost of animal feed. Undigested PHA in fecal matter is an ideal medium for MMC fermentation to generate new PHA.
In terms of challenges that remain, biopolymer production costs need to be reduced and the biopolymer mechanical ability needs to be improved. The technology and literature for bio-versions of commodity plastics and chemical recycling routes is already well established. All that remains then is for the chemical recycling infrastructure to develop and better plastic waste collection schemes to be put into place.
References
Geyer R, Jambeck JR, Law KL (2017) Production, use, and fate of all plastics ever made. Sci Adv 3:1–5. https://doi.org/10.1126/sciadv.1700782
World Economic Forum (2016) The New Plastics Economy Rethinking the future of plastics. World Economic Forum, Cologny, pp 1–36. https://doi.org/10.1103/Physrevb.74.035409
United Nations Enviroment Programme (2014) UNEP Year Book 2014 emerging issues update. United Nations Environment Programme, Nairobi, pp 1–68
Dilkes-Hoffman LS, Pratt S, Lant PA, Laycock B (2019) The role of biodegradable plastic in solving plastic solid waste accumulation. Plastics to energy. Elsevier, Amsterdam, pp 469–505
Jambeck J, Geyer R, Wilcox C et al (2015) Plastic waste inputs from land into the ocean. Publ Am Assoc Adv Sci 347:768–771. https://doi.org/10.1126/science.1260352
Cole M, Lindeque P, Fileman E et al (2013) Microplastic ingestion by zooplankton. Environ Sci Technol 47:6646–6655. https://doi.org/10.1021/es400663f
Zhang C, Chen X, Wang J, Tan L (2017) Toxic effects of microplastic on marine microalgae Skeletonema costatum: interactions between microplastic and algae. Environ Pollut 220:1282–1288. https://doi.org/10.1016/j.envpol.2016.11.005
Igamberdiev AU, Lea PJ (2006) Land plants equilibrate O2 and CO2 concentrations in the atmosphere. Photosynth Res 87:177–194. https://doi.org/10.1007/s11120-005-8388-2
Wang F, Wong CS, Chen D et al (2018) Interaction of toxic chemicals with microplastics: a critical review. Water Res 139:208–219. https://doi.org/10.1016/j.watres.2018.04.003
Cox KD, Covernton GA, Davies HL et al (2019) Human consumption of microplastics. Environ Sci Technol 53:7068–7074. https://doi.org/10.1021/acs.est.9b01517
Niaounakis M (2015) Chapter 1—introduction. Biopolymers: processing and products. Elsevier, Amsterdam, pp 1–77
Zhu Y, Romain C, Williams CK (2016) Sustainable polymers from renewable resources. Nature 540:354–362. https://doi.org/10.1038/nature21001
European Bioplastics Association (2019) Bioplastics facts and figures. European Bioplastics Association, Berlin. https://doi.org/10.1136/bmj.1.5165.62
Niaounakis M (2019) Recycling of biopolymers—the patent perspective. Eur Polym J 114:464–475. https://doi.org/10.1016/j.eurpolymj.2019.02.027
Pillai CKS, Sharma CP (2010) Review paper: absorbable polymeric surgical sutures: chemistry, production, properties, biodegradability, and performance. J Biomater Appl 25:291–366. https://doi.org/10.1177/0885328210384890
Al-Salem SM, Lettieri P, Baeyens J (2009) Recycling and recovery routes of plastic solid waste (PSW): a review. Waste Manag 29:2625–2643. https://doi.org/10.1016/j.wasman.2009.06.004
Kumar S, Panda AK, Singh RK (2011) A review on tertiary recycling of high-density polyethylene to fuel. Resour Conserv Recycl 55:893–910. https://doi.org/10.1016/j.resconrec.2011.05.005
Grigore M (2017) Methods of recycling, properties and applications of recycled thermoplastic polymers. Recycling 2:24. https://doi.org/10.3390/recycling2040024
Siddiqui J, Pandey G (2013) A review of plastic waste management strategies. Int Res J Environ Sci 2:84–88
Singh N, Hui D, Singh R et al (2017) Recycling of plastic solid waste: a state of art review and future applications. Compos Part B Eng 115:409–422. https://doi.org/10.1016/j.compositesb.2016.09.013
Shen L, Worrell E, Patel MK (2010) Open-loop recycling: a LCA case study of PET bottle-to-fibre recycling. Resour Conserv Recycl 55:34–52. https://doi.org/10.1016/j.resconrec.2010.06.014
Payne J, McKeown P, Jones MD (2019) A circular economy approach to plastic waste. Polym Degrad Stab 165:170–181. https://doi.org/10.1016/j.polymdegradstab.2019.05.014
Hopewell J, Dvorak R, Kosior E (2009) Plastics recycling: challenges and opportunities. Philos Trans R Soc B Biol Sci 364:2115–2126. https://doi.org/10.1098/rstb.2008.0311
Hites RA (2011) Dioxins: an overview and history. Environ Sci Technol 45:16–20. https://doi.org/10.1021/es1013664
Niaounakis M (2013) Introduction to biopolymers. Biopolymers reuse, recycling, and disposal. Elsevier, Amsterdam, pp 1–75
De Clercq R, Dusselier M, Sels BF (2017) Heterogeneous catalysis for bio-based polyester monomers from cellulosic biomass: advances, challenges and prospects. Green Chem 19:5012–5040. https://doi.org/10.1039/c7gc02040f
Piemonte V (2011) Bioplastic wastes: the best final disposition for energy saving. J Polym Environ 19:988–994. https://doi.org/10.1007/s10924-011-0343-z
Stephen JD, Mabee WE, Saddler JN (2011) Will second-generation ethanol be able to compete with first-generation ethanol? Opportunities for cost reduction. Biofuels, Bioprod Biorefining 6:159–176. https://doi.org/10.1002/bbb.331
Repo A, Känkänen R, Tuovinen JP et al (2012) Forest bioenergy climate impact can be improved by allocating forest residue removal. GCB Bioenergy 4:202–212. https://doi.org/10.1111/j.1757-1707.2011.01124.x
de Paula FC, de Paula CB, Contiero J (2018) Prospective biodegradable plastics from biomass conversion processes. Intech, London, pp 246–271. https://doi.org/10.5772/intechopen.75111
Schmidt J, Jia J (2015) Time and cost to commercialize an oil shale surface retorting technology time and cost to commercialize an oil shale surface retorting technology. Technology 15:17
Mohan SK, Srivastava T (2010) Microbial deterioration and degradation of polymeric materials. J Biochem Technol 2:210–215
Luckachan GE, Pillai CKS (2011) Biodegradable polymers—a review on recent trends and emerging perspectives. J Polym Environ 19:637–676. https://doi.org/10.1007/s10924-011-0317-1
Tokiwa Y, Calabia BP, Ugwu CU, Aiba S (2009) Biodegradability of plastics. Int J Mol Sci 10:3722–3742. https://doi.org/10.3390/ijms10093722
Shah AA, Hasan F, Hameed A, Ahmed S (2008) Biological degradation of plastics: a comprehensive review. Biotechnol Adv 26:246–265. https://doi.org/10.1016/j.biotechadv.2007.12.005
Artham T, Doble M (2008) Biodegradation of aliphatic and aromatic polycarbonates. Macromol Biosci 8:14–24. https://doi.org/10.1002/mabi.200700106
European Bioplastics Association (2015) EN 13432 certified bioplastics performance in industrial composting. European Bioplastics Association, Berlin
Song JH, Murphy RJ, Narayan R, Davies GBH (2009) Biodegradable and compostable alternatives to conventional plastics. Philos Trans R Soc B Biol Sci 364:2127–2139. https://doi.org/10.1098/rstb.2008.0289
Malik N, Kumar P, Shrivastava S, Ghosh SB (2017) An overview on PET waste recycling for application in packaging. Int J Plast Technol 21:1–24. https://doi.org/10.1007/s12588-016-9164-1
Piemonte V, Sabatini S, Gironi F (2013) Chemical recycling of PLA: a great opportunity towards the sustainable development? J Polym Environ 21:640–647. https://doi.org/10.1007/s10924-013-0608-9
Henton DE, Gruber P, Lunt J, Randall J (2005) Chapter 16. Polylactic acid technology. Natural fibers, biopolymers, and biocomposites. CRC Press LLC, Boca Raton, pp 527–578
Rabnawaz M, Wyman I, Auras R, Cheng S (2017) A roadmap towards green packaging: the current status and future outlook for polyesters in the packaging industry. Green Chem 19:4737–4753. https://doi.org/10.1039/c7gc02521a
Madhavan Nampoothiri K, Nair NR, John RP (2010) An overview of the recent developments in polylactide (PLA) research. Bioresour Technol 101:8493–8501. https://doi.org/10.1016/j.biortech.2010.05.092
Thomas CM (2010) Stereocontrolled ring-opening polymerization of cyclic esters: synthesis of new polyester microstructures. Chem Soc Rev 39:165–173. https://doi.org/10.1039/b810065a
Auras R, Harte B, Selke S (2004) An overview of polylactides as packaging materials. Macromol Biosci 4:835–864. https://doi.org/10.1002/mabi.200400043
Inkinen S, Hakkarainen M, Albertsson AC, Södergård A (2011) From lactic acid to poly(lactic acid) (PLA): characterization and analysis of PLA and Its precursors. Biomacromol 12:523–532. https://doi.org/10.1021/bm101302t
Odian G (2004) Principles of polymerization, 4th edn. Wiley, New York
Lemmouchi Y, Murariu M, Dos SAM et al (2009) Plasticization of poly(lactide) with blends of tributyl citrate and low molecular weight poly(d, l-lactide)-b-poly(ethylene glycol) copolymers. Eur Polym J 45:2839–2848. https://doi.org/10.1016/j.eurpolymj.2009.07.006
Farah S, Anderson DG, Langer R (2016) Physical and mechanical properties of PLA, and their functions in widespread applications—a comprehensive review. Adv Drug Deliv Rev 107:367–392. https://doi.org/10.1016/j.addr.2016.06.012
Fabra MJ, Lopez-Rubio A, Lagaron JM (2014) Nanostructured interlayers of zein to improve the barrier properties of high barrier polyhydroxyalkanoates and other polyesters. J Food Eng 127:1–9. https://doi.org/10.1016/j.jfoodeng.2013.11.022
Lu J, Tappel RC, Nomura CT (2009) Mini-review: Biosynthesis of poly(hydroxyalkanoates). Polym Rev 49:226–248. https://doi.org/10.1080/15583720903048243
Hamad K, Kaseem M, Yang HW et al (2015) Properties and medical applications of polylactic acid: a review. Express Polym Lett 9:435–455. https://doi.org/10.3144/expresspolymlett.2015.42
Cosate de Andrade MF, Souza PMS, Cavalett O, Morales AR (2016) life cycle assessment of poly(lactic acid) (PLA): comparison between chemical recycling, mechanical recycling and composting. J Polym Environ 24:372–384. https://doi.org/10.1007/s10924-016-0787-2
Cosate de Andrade MF, Fonseca G, Morales AR, Mei LHI (2018) Mechanical recycling simulation of polylactide using a chain extender. Adv Polym Technol 37:2053–2060. https://doi.org/10.1002/adv.21863
Niaounakis M (2015) Chapter 16—recycling. Biopolymers: processing and products. Elsevier, Amsterdam, pp 481–530
Qi X, Ren Y, Wang X (2017) New advances in the biodegradation of poly ( lactic ) acid. Int Biodeterior Biodegrad 117:215–223. https://doi.org/10.1016/j.ibiod.2017.01.010
Haider TP, Vçlker C, Kramm J et al (2019) Plastics of the future? The impact of biodegradable polymers on the environment and on society. Angew Chemie Int Ed 58:50–62. https://doi.org/10.1002/anie.201805766
Tokiwa Y, Calabia BP (2007) Biodegradability and biodegradation of polyesters. J Polym Environ 15:259–267. https://doi.org/10.1007/s10924-007-0066-3
Greene J (2012) PLA and PHA biodegradation in the marine environment. California Department of Resources Recycling and Recovery, Sacramento
Shogren RL, Doane WM, Garlotta D et al (2003) Biodegradation of starch/polylactic acid/poly(hydroxyester-ether) composite bars in soil. Polym Degrad Stab 79:405–411
Sikorska W, Musiol M, Nowak B et al (2015) Degradability of polylactide and its blend with poly[(R, S)-3-hydroxybutyrate] in industrial composting and compost extract. Int Biodeterior Biodegrad 101:32–41. https://doi.org/10.1016/j.ibiod.2015.03.021
Piemonte V, Gironi F (2013) Kinetics of hydrolytic degradation of PLA. J Polym Environ 21:313–318. https://doi.org/10.1007/s10924-012-0547-x
Schliecker G, Schmidt C, Fuchs S, Kissel T (2003) Characterization of a homologous series of D, L-lactic acid oligomers; a mechanistic study on the degradation kinetics in vitro. Biomaterials 24:3835–3844. https://doi.org/10.1016/S0142-9612(03)00243-6
Román-Ramírez LA, Mckeown P, Jones MD, Wood J (2018) Poly(lactic acid) degradation into methyl lactate catalyzed by a well-defined Zn(II) complex. ACS Catal Acscatal 9:409–416. https://doi.org/10.1021/acscatal.8b04863
Bowmer CT, Hooftman RN, Hanstveit AO et al (1998) The ecotoxicity and the biodegradability of lactic acid, alkyl lactate esters and lactate salts. Chemosphere 37:1317–1333. https://doi.org/10.1016/S0045-6535(98)00116-7
Román-Ramírez LA, McKeown P, Jones MD, Wood J (2020) Kinetics of methyl lactate formation from the transesterification of polylactic acid catalyzed by Zn(II) complexes. ACS Omega 5:5556–5564. https://doi.org/10.1021/acsomega.0c00291
Leibfarth FA, Moreno N, Hawker AP, Shand JD (2012) Transforming polylactide into value-added materials. J Polym Sci Part A Polym Chem 50:4814–4822. https://doi.org/10.1002/pola.26303
De Clercq R, Dusselier M, Poleunis C et al (2018) Titania-silica catalysts for lactide production from renewable alkyl lactates: structure-activity relations. ACS Catal 8:8130–8139. https://doi.org/10.1021/acscatal.8b02216
Upare PP, Hwang YK, Chang JS, Hwang DW (2012) Synthesis of lactide from alkyl lactate via a prepolymer route. Ind Eng Chem Res 51:4837–4842. https://doi.org/10.1021/ie202714n
Pereira CSM, Silva VMTM, Rodrigues AE (2011) Ethyl lactate as a solvent: properties, applications and production processes—a review. Green Chem 13:2658–2671. https://doi.org/10.1039/c1gc15523g
Song X, Bian Z, Hui Y et al (2019) Zn-acetate-containing ionic liquid as highly active catalyst for fast and mild methanolysis of poly(lactic acid). Polym Degrad Stab 168:108937. https://doi.org/10.1016/j.polymdegradstab.2019.108937
Tsuji H, Daimon H, Fujie K (2003) A new strategy for recycling and preparation of poly(L-lactic acid): hydrolysis in the melt. Biomacromol 4:835–840. https://doi.org/10.1021/bm034060j
Song X, Zhang X, Wang H et al (2013) Methanolysis of poly(lactic acid) (PLA) catalyzed by ionic liquids. Polym Degrad Stab 98:2760–2764. https://doi.org/10.1016/j.polymdegradstab.2013.10.012
Zou H, Yi C, Wang L et al (2009) Thermal degradation of poly(lactic acid) measured by thermogravimetry coupled to Fourier transform infrared spectroscopy. J Therm Anal Calorim 97:929–935. https://doi.org/10.1007/s10973-009-0121-5
Nair LS, Laurencin CT (2007) Biodegradable polymers as biomaterials. Prog Polym Sci 32:762–798. https://doi.org/10.1016/j.progpolymsci.2007.05.017
Pina S, Ferreira JMF (2012) Bioresorbable plates and screws for clinical applications : a review. J Healthc Eng 3:243–260
Ikada Y, Tsuji H (2000) Biodegradable polyesters for medical and ecological applications. Macromol Rapid Commun 21:117–132. https://doi.org/10.1002/(sici)1521-3927(20000201)21:3<117:aid-marc117>3.3.co;2-o
Sinisaari I (2004) Infections and bioabsorbable implants in orthopadic and trauma surgery—with special reference to the treatment of ankle fractures 1
Chu CC (2013) Materials for absorbable and nonabsorbable surgical sutures. Biotextiles as medical implants. Woodhead Publishing Limited, Sawston, pp 275–334
Zong XH, Wang ZG, Hsiao BS et al (1999) Structure and morphology changes in absorbable poly(glycolide) and poly(glycolide-co-lactide) during in vitro degradation. Macromolecules 32:8107–8114. https://doi.org/10.1021/ma990630p
Reddy CSK, Ghai R, Rashmi KVC (2003) Polyhydroxyalkanoates: an overview. Bioresour Technol 87:137–146. https://doi.org/10.1016/S0960-8524(02)00212-2
Albuquerque MGE, Martino V, Pollet E et al (2011) Mixed culture polyhydroxyalkanoate (PHA) production from volatile fatty acid (VFA)-rich streams: effect of substrate composition and feeding regime on PHA productivity, composition and properties. J Biotechnol 151:66–76. https://doi.org/10.1016/j.jbiotec.2010.10.070
Defoirdt T, Boon N, Sorgeloos P et al (2009) Short-chain fatty acids and poly- β-hydroxyalkanoates: (new) biocontrol agents for a sustainable animal production. Biotechnol Adv 27:680–685. https://doi.org/10.1016/j.biotechadv.2009.04.026
Jacquel N, Lo C, Wei Y et al (2008) Isolation and purification of bacterial poly(3-hydroxyalkanoates). Biochem Eng J 39:15–27. https://doi.org/10.1016/j.bej.2007.11.029
Samorì C, Abbondanzi F, Galletti P et al (2015) Extraction of polyhydroxyalkanoates from mixed microbial cultures: impact on polymer quality and recovery. Bioresour Technol 189:195–202. https://doi.org/10.1016/j.biortech.2015.03.062
Shogren R, Wood D, Orts W, Glenn G (2019) Plant-based materials and transitioning to a circular economy. Sustain Prod Consum 19:194–215. https://doi.org/10.1016/j.spc.2019.04.007
Rai R, Keshavarz T, Roether JA et al (2011) Medium chain length polyhydroxyalkanoates, promising new biomedical materials for the future. Mater Sci Eng R Rep 72:29–47. https://doi.org/10.1016/j.mser.2010.11.002
Philip S, Keshavarz T, Roy I (2007) Polyhydroxyalkanoates: biodegradable polymers with a range of applications. J Chem Technol Biotechnol 82:233–247. https://doi.org/10.1002/jctb
Scaffaro R, Dintcheva NT, Marino R, La Mantia FP (2012) Processing and properties of biopolymer/polyhydroxyalkanoates blends. J Polym Environ 20:267–272. https://doi.org/10.1007/s10924-011-0385-2
Lueft KP, Stelzer F, Wiesbrock F (2015) Poly(hydroxy alkanoate)s in medical applications. Chem Biochem Eng Q 29:287–297. https://doi.org/10.15255/CABEQ.2014.2261
Bugnicourt E, Cinelli P, Lazzeri A, Alvarez V (2014) Polyhydroxyalkanoate (PHA): review of synthesis, characteristics, processing and potential applications in packaging. Express Polym Lett 8:791–808. https://doi.org/10.3144/expresspolymlett.2014.82
Roohi ZMR, Kuddus M (2018) PHB (poly-β-hydroxybutyrate) and its enzymatic degradation. Polym Adv Technol 29:30–40. https://doi.org/10.1002/pat.4126
Shen L, Haufe J, Patel MK (2009) Product overview and market projection of emerging bio-based plastics. Rep PRO-BIP, Berlin
Gurieff N, Lant P (2007) Comparative life cycle assessment and financial analysis of mixed culture polyhydroxyalkanoate production. Bioresour Technol 98:3393–3403. https://doi.org/10.1016/j.biortech.2006.10.046
Amulya K, Jukuri S, Mohan SV (2015) Sustainable multistage process for enhanced productivity of bioplastics from waste remediation through aerobic dynamic feeding strategy : process integration for up-scaling. Bioresour Technol 188:231–239. https://doi.org/10.1016/j.biortech.2015.01.070
Jari M, Khatami SR, Galehdari H, Shafiei M (2015) Cloning and expression of poly 3-hydroxybutyrate operon into Escherichia coli. Jundishapur J Microbiol 8:3–6. https://doi.org/10.5812/jjm.16318
Poirier Y, Dennis DE, Klomparens K, Somerville C (2020) Polyhydroxybutyrate, a biodegradable thermoplastic, produced in transgenic plants. Publ Am Assoc Adv Sci 256:520–523. https://doi.org/10.1007/s11738-018-2742-4
Rivas LF, Casarin SA, Nepomuceno NC et al (2017) Reprocessability of PHB in extrusion: ATR-FTIR, tensile tests and thermal studies. Polimeros 27:122–128. https://doi.org/10.1590/0104-1428.2406
Zaverl M, Seydibeyoglu MO, Misra M, Mohanty A (2012) Studies on recyclability of polyhydroxybutyrate-co- valerate bioplastic: multiple melt processing and performance evaluations. J Appl Polym Sci 125:E324–E331. https://doi.org/10.1002/app
Zembouai I, Bruzaud S, Kaci M et al (2014) Mechanical recycling of poly(3-hydroxybutyrate-co-3-hydroxyvalerate)/polylactide based blends. J Polym Environ 22:449–459. https://doi.org/10.1007/s10924-014-0684-5
Aoyagi Y, Yamashita K, Doi Y (2002) Thermal degradation of poly[(R)-3-hydroxybutyrate], poly[e-caprolactone], and poly[(S)-lactide]. Polym Degrad Stab 76:53–59. https://doi.org/10.1016/S0141-3910(01)00265-8
Kulawska M, Grzesik M, Skrzypek J (2003) Kinetics of the esterification of crotonic acid with octyl, decyl, or dodecyl alcohol. Chem Pap 57:424–426
Ute K, Tarao T, Nakao S, Kitayama T (2003) Preparation and properties of disyndiotactic poly(alkyl crotonate)s. Polym 44:7869–7874. https://doi.org/10.1016/j.polymer.2003.10.005
Ariffin H, Nishida H, Shirai Y, Hassan MA (2010) Highly selective transformation of poly[(R)-3-hydroxybutyric acid] into trans-crotonic acid by catalytic thermal degradation. Polym Degrad Stab 95:1375–1381. https://doi.org/10.1016/j.polymdegradstab.2010.01.018
Wang S, Lydon KA, White EM et al (2018) Biodegradation of poly(3-hydroxybutyrate-co-3-hydroxyhexanoate) plastic under anaerobic sludge and aerobic seawater conditions: gas evolution and microbial diversity. Environ Sci Technol 52:5700–5709. https://doi.org/10.1021/acs.est.7b06688
Kim DY, Kim HW, Chung MG, Rhee YH (2007) Biosynthesis, modification, and biodegradation of bacterial medium-chain-length polyhydroxyalkanoates. J Microbiol 45:87–97
Freier T, Kunze C, Nischan C et al (2002) In vitro and in vivo degradation studies for development of a biodegradable patch based on poly (3-hydroxybutyrate). Biomaterials 23:2649–2657
Forni D, Wenk C, Bee G et al (1999) Digestive utilization of novel biodegradable plastic in growing pigs. Ann Zootech 48:163–171
Forni D, Bee G, Kreuzer M, Wenk C (1999) Novel biodegradable plastics in sheep nutrition 2. Effects of NaOH pretreatment of poly (3-hydroxybutyrate-co-3-hydroxyvalerate) on in vivo digestibility and on in vitro disappearance (Rusitec). J Anim Physiol Anim Nutr 81:41–50
Defoirdt T, Halet D, Vervaeren H et al (2007) The bacterial storage compound poly b-hydroxybutyrate protects Artemia franciscana from pathogenic Vibrio campbellii. Environ Microbiol 9:445–452. https://doi.org/10.1111/j.1462-2920.2006.01161.x
Boon N, Defoirdt T, De Windt W et al (2013) Hydroxybutyrate and poly-hydroxybutyrate as components of animal feed or feed additives. US 8603518 B2
Ong SY, Zainab-l I, Pyary S, Sudesh K (2018) A novel biological recovery approach for PHA employing selective digestion of bacterial biomass in animals. Appl Microbiol Biotechnol 102:2117–2127. https://doi.org/10.1007/s00253-018-8788-9
Welle F (2011) Twenty years of PET bottle to bottle recycling—an overview. Resour Conserv Recycl 55:865–875. https://doi.org/10.1016/j.resconrec.2011.04.009
Karayannidis GP, Achilias DS (2007) Chemical recycling of poly(ethylene terephthalate). Macromol Mater Eng 292:128–146. https://doi.org/10.1002/mame.200600341
Awaja F, Pavel D (2005) Recycling of PET. Eur Polym J 41:1453–1477. https://doi.org/10.1016/j.eurpolymj.2005.02.005
Kopnick H, Schmidt M, Brugging W et al (2012) Polyesters, vol 29. Ullmann’s Encycl Ind Chem, Weinheim, pp 6.5–621. https://doi.org/10.1002/14356007.a21
PE INTERNATIONAL AG (2014) Process data set: biopolyethylene terephthalate granulate (PET) via terepht. acid + EG (corn); partially biobased via terephthalic acid and ethylene glycol from bioethylene based on corn; single route, at plant; PET bottle grade, Ethylene glycol biobased. https://gabi-documentation-2014.gabi-software.com/xml-data/processes/b8c7e89d-5f0c-4bc1-90d8-84c12b6c4d2e.xml. Accessed 14 Dec 2019
Shen L, Worrell E, Patel MK (2012) Comparing life cycle energy and GHG emissions of bio- based PET, recycled PET, PLA, and man-made cellulosics. Biofuels, Bioprod Biorefining 6:625–639. https://doi.org/10.1002/bbb
Tabone MD, Cregg JJ, Beckman EJ, Landis AE (2010) Sustainability metrics: life cycle assessment and green design in polymers. Environ Sci Technol 44:8264–8269. https://doi.org/10.1021/es103760e
Barrett A (2018) PepsiCo Goes for bioplastic bottles. In: Bioplasticnews. https://bioplasticsnews.com/2018/09/10/pepsico-goes-for-bioplastic-bottles/. Accessed 14 Dec 2019
Chen L, Pelton REO, Smith TM (2016) Comparative life cycle assessment of fossil and bio-based polyethylene terephthalate (PET) bottles. J Clean Prod 137:667–676. https://doi.org/10.1016/j.jclepro.2016.07.094
Feldman RMR, Gunawardena U, Urano J et al (2011) Yeast organism producing isobutanol at a high yield. US 2011/020889 A1
Peters M, Taylor JD, Jenni M et al (2010) Integrated process to selectively convert renewable isobutanol to p-xylene. WO 2011/044243 A1
Luo L, van der Voet E, Huppes G (2009) An energy analysis of ethanol from cellulosic feedstock-Corn stover. Renew Sustain Energy Rev 13:2003–2011. https://doi.org/10.1016/j.rser.2009.01.016
Hendriks ATWM, Zeeman G (2009) Pretreatments to enhance the digestibility of lignocellulosic biomass. Bioresour Technol 100:10–18. https://doi.org/10.1016/j.biortech.2008.05.027
Barman NK, Bhattacharya SS, Mandot A (2015) Research article mechanical properties of melt-spun monofilaments produced feom virgin and recycyled poly (ethylene terephthalate ) blends. Int J Recent Sci Res 6:4517–4525
Masmoudi F, Fenouillot F, Mehri A et al (2018) Characterization and quality assessment of recycled post-consumption poly(ethylene terephthalate) (PET). Environ Sci Pollut Res 25:23307–23314. https://doi.org/10.1007/s11356-018-2390-7
López DMC, Ares Pernas AI, Abad López MJ et al (2014) Assessing changes on poly(ethylene terephthalate) properties after recycling: mechanical recycling in laboratory versus postconsumer recycled material. Mater Chem Phys 147:884–894. https://doi.org/10.1016/j.matchemphys.2014.06.034
Elamri A, Zdiri K, Harzallah O, Lallam A (2017) Progress in polyethylene terephthalate recycling. Polyethylene terephthalate uses, properties and degradtion. Nova Science Publishers, New York, pp 155–185
Cornell DD (2007) Biopolymers in the existing postconsumer plastics recycling stream. J Polym Environ 15:295–299. https://doi.org/10.1007/s10924-007-0077-0
Burat F, Güney A, Olgaç Kangal M (2009) Selective separation of virgin and post-consumer polymers (PET and PVC) by flotation method. Waste Manag 29:1807–1813. https://doi.org/10.1016/j.wasman.2008.12.018
Sinha V, Patel MR, Patel JV (2010) Pet waste management by chemical recycling: a review. J Polym Environ 18:8–25. https://doi.org/10.1007/s10924-008-0106-7
Karayannidis GP, Chatziavgoustis AP, Achilias DS (2002) Poly(ethylene terephthalate) recycling and recovery of pure terephthalic acid by alkaline hydrolysis. Adv Polym Technol 21:250–259. https://doi.org/10.1002/adv.10029
Paszun D, Spychaj T (1997) Chemical recycling of poly(ethylene terephthalate). Ind Eng Chem Res 36:1373–1383. https://doi.org/10.1021/ie960563c
Al-Sabagh AM, Yehia FZ, Eshaq G et al (2016) Greener routes for recycling of polyethylene terephthalate. Egypt J Pet 25:53–64. https://doi.org/10.1016/j.ejpe.2015.03.001
Scheirs J (1998) Polymer recycling: science, technologoy, and applications. Wiley, Hoboken
Yang Y, Lu Y, Xiang H et al (2002) Study on methanolytic depolymerization of PET with supercritical methanol for chemical recycling. Polym Degrad Stab 75:185–191. https://doi.org/10.1016/S0141-3910(01)00217-8
Kurokawa H, Ohshima MA, Sugiyama K, Miura H (2003) Methanolysis of polyethylene terephthalate (PET) in the presence of aluminium tiisopropoxide catalyst to form dimethyl terephthalate and ethylene glycol. Polym Degrad Stab 79:529–533. https://doi.org/10.1016/S0141-3910(02)00370-1
De Castro REN, Vidotti GJ, Rubira AF, Muniz EC (2006) Depolymerization of poly(ethylene terephthalate) wastes using ethanol and ethanol/water in supercritical conditions. J Appl Polym Sci 101:2009–2016. https://doi.org/10.1002/app.23748
Carné Sánchez A, Collinson SR (2011) The selective recycling of mixed plastic waste of polylactic acid and polyethylene terephthalate by control of process conditions. Eur Polym J 47:1970–1976. https://doi.org/10.1016/j.eurpolymj.2011.07.013
Arnoult M, Dargent E, Mano JF (2007) Mobile amorphous phase fragility in semi-crystalline polymers: comparison of PET and PLLA. Polymer (Guildf) 48:1012–1019. https://doi.org/10.1016/j.polymer.2006.12.053
Chaudhary S, Surekha P, Kumar D et al (2013) Microwave assisted glycolysis of poly (ethylene terepthalate) for preparation of polyester polyols. J Appl Polym Sci 129:2779–2788. https://doi.org/10.1002/app.38970
Raheem AB, Noor ZZ, Hassan A et al (2019) Current developments in chemical recycling of post-consumer polyethylene terephthalate wastes for new materials production: a review. J Clean Prod 225:1052–1064. https://doi.org/10.1016/j.jclepro.2019.04.019
Goje AS, Mishra S (2003) Chemical kinetics, simulation, and thermodynamics of glycolytic depolymerization of poly(ethylene terephthalate) waste with catalyst optimization for recycling of value added monomeric products. Macromol Mater Eng 288:326–336. https://doi.org/10.1002/mame.200390034
Al-Sabagh AM, Yehia FZ, Eissa AMMF et al (2014) Glycolysis of poly(ethylene terephthalate) catalyzed by the Lewis base ionic liquid [Bmim][OAc]. Ind Eng Chem Res 53:18443–18451. https://doi.org/10.1021/ie503677w
Wang Q, Geng Y, Lu X, Zhang S (2015) First-row transition metal-containing ionic liquids as highly active catalysts for the glycolysis of poly(ethylene terephthalate) (PET). ACS Sustain Chem Eng 3:340–348. https://doi.org/10.1021/sc5007522
Wang H, Liu Y, Li Z et al (2009) Glycolysis of poly(ethylene terephthalate) catalyzed by ionic liquids. Eur Polym J 45:1535–1544. https://doi.org/10.1016/j.eurpolymj.2009.01.025
Sangalang A, Bartolome L, Kim DH (2015) Generalized kinetic analysis of heterogeneous PET glycolysis: nucleation-controlled depolymerization. Polym Degrad Stab 115:45–53. https://doi.org/10.1016/j.polymdegradstab.2015.02.012
Vaidya UR, Nadkarni VM (1987) Unsaturated polyester resins from poly(ethylene terephthalate) waste. 1. Synthesis and characterization. Ind Eng Chem Res 26:194–198. https://doi.org/10.1021/ie00062a003
Pimpan V, Sirisook R, Chuayjuljit S (2003) Synthesis of unsaturated polyester resin from postconsumer PET bottles: effect of type of glycol on characteristics of unsaturated polyester resin. J Appl Polym Sci 88:788–792. https://doi.org/10.1002/app.11567
Ul Haq MI (2007) Applications of unsaturated polyester resins. Russ J Appl Chem 80:1256–1269. https://doi.org/10.1134/S1070427207070464
Alzuhairi MAH, Khalil BI, Hadi RS (2017) Nano ZnO catalyst for chemical recycling of polyethylene terephthalate (PET). Eng Technol J 35:831–837
Yue QF, Xiao LF, Zhang ML, Bai XF (2013) The glycolysis of poly(ethylene terephthalate) waste: Lewis acidic ionic liquids as high efficient catalysts. Polymers (Basel) 5:1258–1271. https://doi.org/10.3390/polym5041258
Yoshioka T, Motoki T, Okuwaki A (2001) Kinetics of hydrolysis of poly(ethylene terephthalate) powder in sulfuric acid by a modified shrinking-core model. Ind Eng Chem Res 40:75–79. https://doi.org/10.1021/ie000592u
Yoshioka T, Okayama N, Okuwaki A (1998) Kinetics of hydrolysis of PET powder in nitric acid by a modified shrinking-core model. Ind Eng Chem Res 37:336–340. https://doi.org/10.1021/ie970459a
Ravens DAS, Ward IM (1961) Chemical reactivity of polyethylene terephthalate. Hydrolysis and esterification reactions in the solid phase. Trans Faraday Soc 57:150–159. https://doi.org/10.1039/TF9615700150
Mishra S, Goje AS (2003) Kinetic and thermodynamic study of methanolysis of poly(ethylene terephthalate) waste powder. Polym Int 52:337–342. https://doi.org/10.1002/pi.1147
Diaz Silvarrey LS, Phan AN (2016) Kinetic study of municipal plastic waste. Int J Hydrogen Energy 41:16352–16364. https://doi.org/10.1016/j.ijhydene.2016.05.202
Morschbacker A, Eduardo C, Campos S, Cassiano LC (2010) Bio-polyethylene. Handbook of green materials. World Scientific Publishing, Singapore, pp 89–104
Morschbacker A (2009) Bio-ethanol based ethylene. Polym Rev 49:79–84. https://doi.org/10.1080/15583720902834791
Mendieta CM, Vallejos ME, Felissia FE et al (2019) Review: bio-polyethylene from wood wastes. J Polym Environ 28:1–16. https://doi.org/10.1007/s10924-019-01582-0
Jin H, Gonzalez-Gutierrez J, Oblak P et al (2012) The effect of extensive mechanical recycling on the properties of low density polyethylene. Polym Degrad Stab 97:2262–2272. https://doi.org/10.1016/j.polymdegradstab.2012.07.039
Hamad K, Kaseem M, Deri F (2013) Recycling of waste from polymer materials: an overview of the recent works. Polym Degrad Stab 98:2801–2812. https://doi.org/10.1016/j.polymdegradstab.2013.09.025
Achilias DS, Roupakias C, Megalokonomos P et al (2007) Chemical recycling of plastic wastes made from polyethylene (LDPE and HDPE) and polypropylene (PP). J Hazard Mater 149:536–542. https://doi.org/10.1016/j.jhazmat.2007.06.076
Vasudeo RA, Abitha VK, Vinayak K et al (2016) Sustainable development through feedstock recycling of plastic wastes. Macromol Symp 362:39–51. https://doi.org/10.1002/masy.201500107
López A, De MI, Caballero BM et al (2010) Pyrolysis of municipal plastic wastes : influence of raw material composition. Waste Manag 30:620–627. https://doi.org/10.1016/j.wasman.2009.10.014
Honus S, Kumagai S, Fedorko G et al (2018) Pyrolysis gases produced from individual and mixed PE, PP, PS, PVC, and PET—part I: production and physical properties. Fuel 221:346–360. https://doi.org/10.1016/j.fuel.2018.02.074
Lee K (2012) Effects of the types of zeolites on catalytic upgrading of pyrolysis wax oil. J Anal Appl Pyrolysis 94:209–214. https://doi.org/10.1016/j.jaap.2011.12.015
Miandad R, Rehan M, Nizami A-S et al (2016) The energy and value-added products from pyrolysis of waste plastics. Recycling of solid waste for biofuels and bio-chemicals. Springer, Singapore, pp 333–355
Lee K (2009) Thermal and catalytic degradation of pyrolytic oil from pyrolysis of municipal plastic wastes. J Anal Appl Pyrolysis 85:372–379. https://doi.org/10.1016/j.jaap.2008.11.032
Gao F (2010) Pyrolysis of waste plastics into fuels. PhD Thesis. University of Canterbury, Christchurch
Jamradloedluk J, Lertsatitthanakorn C (2014) Characterization and utilization of char derived from fast pyrolysis of plastic wastes. Procedia Eng 69:1437–1442. https://doi.org/10.1016/j.proeng.2014.03.139
Saptoadi H, Rohmat TA (2017) Combustion of char from plastic wastes pyrolysis. AIP Conference Proceedings, vol 1737. AIP Publishing LLC, Melville, p 030006. https://doi.org/10.1063/1.4949286
Zhao D, Wang X, Miller JB, Huber GW (2020) The chemistry and kinetics of polyethylene pyrolysis: a process to produce fuels and chemicals. Chemsuschem 13:1–12. https://doi.org/10.1002/cssc.201903434
Achilias DS, Andriotis L, Koutsidis IA et al (2012) Recent advances in the chemical recycling of polymers (PP, PS, LDPE, HDPE, PVC, PC, Nylon, PMMA). Material recycling: trends and perspectives. InTech, Rijeka, p 422
Kunwar B, Cheng HN, Chandrashekaran SR, Sharma BK (2016) Plastics to fuel: a review. Renew Sustain Energy Rev 54:421–428. https://doi.org/10.1016/j.rser.2015.10.015
Wang J, Wang L (2011) Catalytic pyrolysis of municipal plastic waste to fuel with nickel-loaded silica-alumina catalysts. Energy Sour Part A Recover Util Environ Eff 33:1940–1948. https://doi.org/10.1080/15567030903436814
López A, De MI, Caballero BM et al (2011) Deactivation and regeneration of ZSM-5 zeolite in catalytic pyrolysis of plastic wastes. Waste Manag 31:1852–1858. https://doi.org/10.1016/j.wasman.2011.04.004
Buekens AG, Huang H (1998) Catalytic plastics cracking for recovery of gasoline-range hydrocarbons from municipal plastic wastes. Resour Conserv Recycl 23:163–181. https://doi.org/10.1016/S0921-3449(98)00025-1
Seo YH, Lee KH, Shin DH (2003) Investigation of catalytic degradation of high-density polyethylene by hydrocarbon group type analysis. J Anal Appl Pyrolysis 70:383–398. https://doi.org/10.1016/S0165-2370(02)00186-9
Lee KH (2008) Composition of aromatic products in the catalytic degradation of the mixture of waste polystyrene and high-density polyethylene using spent FCC catalyst. Polym Degrad Stab 93:1284–1289. https://doi.org/10.1016/j.polymdegradstab.2008.04.009
Al-Salem SM, Lettieri P (2010) Kinetic study of high density polyethylene (HDPE) pyrolysis. Chem Eng Res Des 88:1599–1606. https://doi.org/10.1016/j.cherd.2010.03.012
Wang Z, Shen D, Wu C, Gu S (2018) Thermal behavior and kinetics of co-pyrolysis of cellulose and polyethylene with the addition of transition metals. Energy Convers Manag 172:32–38. https://doi.org/10.1016/j.enconman.2018.07.010
Alsewailem FD, Almutabaqani LA (2014) Activation energy for the pyrolysis of polymer wastes. Eur Chem Bull 3:93–97
Straka P, Bičáková O, Šupová M (2017) Thermal conversion of polyolefins/polystyrene ternary mixtures: kinetics and pyrolysis on a laboratory and commercial scales. J Anal Appl Pyrolysis 128:196–207. https://doi.org/10.1016/j.jaap.2017.10.010
Miskolczi N, Juzsakova T, Sója J (2019) Preparation and application of metal loaded ZSM-5 and y-zeolite catalysts for thermo-catalytic pyrolysis of real end of life vehicle plastics waste. J Energy Inst 92:118–127. https://doi.org/10.1016/j.joei.2017.10.017
Acknowledgements
Fabio M. Lamberti is grateful to the School of Chemical Engineering at University of Birmingham for a studentship and Joseph Wood and Luis Román Ramírez are grateful to the EPSRC for funding via project (Grant No. EP/P016405/1).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lamberti, F.M., Román-Ramírez, L.A. & Wood, J. Recycling of Bioplastics: Routes and Benefits. J Polym Environ 28, 2551–2571 (2020). https://doi.org/10.1007/s10924-020-01795-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10924-020-01795-8