
Abstract
Leukocyte entry from the blood into inflamed tissues, exit into the lymphatics, and migration to regional lymph nodes are all crucial processes for mounting an effective adaptive immune response. Leukocytes must cross two endothelial cell layers, the vascular and the lymphatic endothelial cell layers, during the journey from the blood to the lymph node. The proteins and cellular interactions which regulate leukocyte migration across the vascular endothelium are well studied; however, little is known about the factors that regulate leukocyte migration across the lymphatic endothelium. Here, we will summarize evidence for a role for galectins, a family of carbohydrate-binding proteins, in regulating leukocyte migration across the vascular endothelium and propose that galectins are also involved in leukocyte migration across the lymphatic endothelium.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Specific Leukocyte Subsets Enter Inflamed Tissues and Exit into the Lymphatic System
Leukocytes migrate into inflamed tissues from the blood and can exit into the lymphatics after encountering antigen at sites of inflammation. This migration and the subsequent activation of the adaptive immune system in downstream lymph nodes are imperative for mounting an effective immune response. However, surprisingly little is known about how immune cells migrate out of inflamed tissues and traffic towards lymph nodes [1].
During inflammation, neutrophils are the first immune cells to arrive at tissue sites, followed by monocytes and then effector and memory T and B cells [2]. Expression of pro-inflammatory cytokines at sites of inflammation induces vascular endothelial cells to up-regulate the expression of adhesion molecules that facilitate the entry of leukocytes into the inflamed tissue. These adhesion molecules include selectins and integrins, whose roles in leukocyte adhesion to the vascular endothelium and leukocyte entry into tissues have been analyzed in detail [2, 3].
A role for galectins in leukocyte migration across the vascular endothelium has also been described [4]. Galectins are a family of carbohydrate-binding proteins that are expressed by vascular endothelial cells and regulate immune cell migration during inflammation [5–9]. However, in contrast to selectins and integrins, less is known about how galectins influence leukocyte capture, rolling, and adhesion to and migration across the vascular endothelial cell layer and through the extracellular matrix (ECM) to the site of inflammation.
Of the immune cells that enter sites of inflammation, only certain types of leukocytes exit tissues and travel to lymph nodes. Neutrophils, monocyte-derived dendritic cells (DCs) and macrophages, as well as memory CD4 T cells, have all been shown to make the journey through the afferent lymph towards draining lymph nodes [10–15]. However, little is known about the adhesion molecules and signaling intermediates that are expressed by lymphatic endothelial cells and how these can modulate leukocyte exit from sites of inflammation [1]. One of the few reports analyzing this question found that, similar to vascular endothelial cells, lymphatic endothelial cells up-regulated expression of vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) under inflammatory conditions and that this up-regulation facilitated DC migration across a layer of lymphatic endothelial cells [16]. Recently, the same group demonstrated that the chemokine CCL21 was secreted from lymphatic endothelial cells under inflammatory conditions and also promoted DC migration across a layer of lymphatic endothelial cells [17].
As mentioned above, galectin expression by vascular endothelial cells also changes under inflammatory conditions [5, 6]. It is therefore conceivable that galectins also regulate leukocyte migration across the lymphatic endothelium, as described for VCAM-1, ICAM-1, and CCL21. This idea is supported by two recent reports, which demonstrate that galectin-1 and galectin-3 are involved in the exit of tissue-resident DCs into the lymphatics and migration to draining lymph nodes [18, 19]. In this review, we will discuss what is known about the influence of galectins on leukocyte entry from blood into sites of inflammation and propose a role for galectins in leukocyte exit into the lymphatic system.
Galectins Function Through Binding and Clustering Glycan Ligands on Glycoprotein Counterreceptors on Leukocytes
There are 14 galectin family members (galectin-1 to -14) in mammals, all of which contain conserved carbohydrate recognition domains (CRDs). All galectins can oligomerize into higher order carbohydrate-binding proteins. The prototype galectins (galectin-1, -2, -5, -7, -10, -11, -13, and -14) exist as monomers or homodimers of one CRD. The tandem-repeat type galectins (galectin-4, -6, -8, -9, and -12) harbor two distinct CRDs joined by a peptide linker and can oligomerize via CRD–CRD interactions [20]. The chimera-type galectin-3 consists of a CRD attached to a peptide linker that can pentamerize upon binding to glycan ligands on cells or matrix [21].
Galectins bind discrete but overlapping sets of glycan ligands that are displayed on cell surface glycoprotein counterreceptors [8, 9, 22–25]. Glycan ligands for specific galectins are presented on distinct N- and O-glycan structures, which are anchored on glycoprotein counterreceptors that can be expressed by different cell types. For example, galectin-1 and galectin-3, but not galectin-9 [23, 24, 26–28], bind to N- and O-glycans attached to the glycoprotein counterreceptor CD45 [29–31]. Interestingly, the availability of discrete galectin-1 and galectin-3 glycan ligands on CD45 in T cells is developmentally regulated, and only certain T cell subpopulations that express the specific glycan ligands are susceptible to galectin-1- and galectin-3-induced cell death [23, 31, 32]. Taken together, these results illustrate that the availability of specific glycan ligands dictates cellular responses to galectin binding.
It has been proposed that multivalent galectins act as scaffolding molecules by simultaneously binding glycan ligands on multiple glycoprotein counterreceptors on the cell surface to either cluster different glycoprotein counterreceptors together or segregate the glycoproteins into discrete domains. For example, galectin-1 can simultaneously bind glycan ligands attached to CD43 and CD45 on DCs and cluster CD43 and CD45 into patches on the cell surface (heterotypic clustering) [18]. On T cells, however, galectin-1 binds to CD43 and CD45 and segregates CD43 and CD45 into different membrane domains (homotypic clustering) [30]. Clustering of surface receptors can significantly modulate their function and thus influence cellular responses. For example, heterotypic clustering of CD43 and CD45 on DCs induces DC activation and migration [18]. In contrast, homotypic clustering of CD43 on T cells inhibits T cell migration through the ECM and across vascular endothelial cells [33] while homotypic clustering of CD45 on T cells inhibits CD45 phosphatase activity [27, 34]. Several studies have shown that these glycoprotein clusters or aggregates, termed galectin–glycoprotein lattices, can regulate many important leukocyte functions, such as T cell receptor signaling [35, 36], CD8 T cell anergy [37], thymocyte selection [38], DC migration [18], and others [39–41].
Galectins Are Expressed by Leukocytes at Sites of Inflammation
Galectins are expressed by all leukocytes that enter into tissues and exit into the lymphatics [42–46]. Expression of galectin-1, galectin-3, and galectin-9 in leukocytes has been analyzed in a variety of inflammatory processes, including atherosclerosis [47], atopic dermatitis [48], acute peritonitis [42, 49], and airway inflammation [50, 51]. These studies revealed that the expression of specific galectins is increased in certain leukocyte subsets found at sites of inflammation. In particular, galectin-3 expression was increased in macrophages and CD4 T cells in tissue samples extracted from atherosclerotic lesions and skin lesions in atopic dermatitis, respectively [47, 48]. In experimental models for acute peritonitis and airway inflammation, galectin-3 expression in macrophages was also increased [42, 49–51]. One study described an increase in galectin-1 expression in macrophages in a model of acute peritonitis [42], but galectin-1 expression was not analyzed in other leukocyte subsets or in different models of acute inflammation. Furthermore, DCs that were matured in vitro by inflammatory cytokines increased expression of galectin-9 messenger RNA (mRNA) and down-regulated expression of galectin-3 mRNA in comparison to immature DCs [44]. While the dynamic expression of isolated galectins has been described in certain leukocyte subsets under specific inflammatory conditions, it will be important to characterize the complete profile of galectin expression in all leukocytes that enter and exit tissues in different models of inflammation.
Galectins Are Expressed by Endothelial Cells
Vascular endothelial cells express galectin-1, galectin-3, galectin-8, and galectin-9 under steady-state conditions, and changes in expression under inflammatory conditions have been described. Specifically, in activated vascular endothelium, galectin-1 expression is increased while galectin-8 and galectin-9 expression are decreased [5, 6]. Galectin-3 expression, however, remains unaltered upon activation of vascular endothelial cells [6] indicating that the expression level of specific galectin family members in vascular endothelial cells is differentially regulated during inflammation.
In contrast, little is known about galectin expression in lymphatic endothelial cells. Only one publication has described the expression of a galectin family member, galectin-8, by lymphatic endothelial cells [52], and this group proposed that galectin-8 is involved in the attachment of lymphatic endothelial cells to the ECM. Given the complex regulation of galectin expression by vascular endothelial cells, other galectin family members are likely expressed by lymphatic endothelial cells and it will be important to further characterize the expression pattern of all galectin family members in lymphatic endothelial cells.
Galectins Regulate Leukocyte Entry into Sites of Inflammation
Galectin-3 and Galectin-9 Promote Leukocyte Entry into Sites of Inflammation
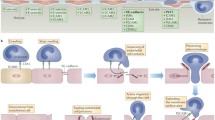
Although the roles played by galectins in lymphatic endothelial cells are still elusive, it is well established that the dynamic expression of galectins in leukocytes and vascular endothelial cells controls inflammatory processes through modulating cell survival and death [23, 53], activation and cytokine secretion [9, 53, 54], and migration [4, 54, 55]. The ability to migrate to and from sites of inflammation is crucial for mounting a proper immune response. Galectins, in particular galectin-1, galectin-3, and galectin-9, have been shown to influence both cell entry into and cell exit from inflamed tissues (Table I and Fig. 1).

Galectin family members regulate leukocyte migration across vascular and lymphatic endothelium. Schematic representation of leukocyte entry from the blood vasculature into inflamed tissue and exit into the lymphatic system. Neutrophils (N), monocytes (M), monocyte-derived dendritic cells (DC), and lymphocytes (L) are shown. Galectin-3 and galectin-9 support the migration of leukocytes from the blood into tissues under inflammatory conditions while this process is inhibited by galectin-1 (left panels). Leukocyte exit into the lymphatic vasculature is increased in the presence of galectin-1 and galectin-3, but any role for galectins in negatively regulating this process has not yet been examined (right panels)
To promote cell entry into tissues and migration across the vascular endothelial cell layer, leukocytes utilize molecular cues, such as adhesion molecules and chemokine gradients that guide cells to sites of inflammation. Galectin-3 has been shown to be a chemoattractant for neutrophils, monocytes, and macrophages in vitro [56, 57]. In addition, using an in vivo mouse air pouch model for leukocyte migration, Sano et al. showed that injection of recombinant galectin-3 into the air pouch attracted increased numbers of neutrophils, eosinophils, and monocytes compared to animals that were not treated with recombinant galectin-3 [57]. Moreover, galectin-3 has been shown to promote granulocyte, monocyte, and lymphocyte migration to inflamed tissues in several in vivo models of inflammation [48, 49, 51, 58, 59]. For example, in two models of acute peritonitis, the numbers of infiltrating granulocytes, macrophages, and lymphocytes were significantly decreased in galectin-3 knock-out mice compared to wild-type animals [49, 58]. Similarly, the number of mononuclear cells and eosinophils was significantly decreased in skin lesions in an atopic dermatitis model when galectin-3 knock-out mice were compared to wild-type animals [48]. In addition, in models of lung inflammation, galectin-3 knock-out mice had fewer leukocytes in the bronchoalveolar lavage fluid than wild-type animals [51, 59] indicating that, as in the other models described above, the presence of galectin-3 supports leukocyte entry into sites of inflammation.
While most reports describe a role for galectin-3 in promoting granulocyte, monocyte, and lymphocyte entry into inflamed tissues, galectin-9 primarily acts as a chemoattractant for eosinophils in vitro and in vivo [60–63]. However, one report demonstrated that not only eosinophils, but also neutrophils and macrophages were increased upon injection of recombinant galectin-9 into the peritoneum in the absence of any other inflammatory stimulus [62]. This suggests that galectin-9 may be a direct chemoattractant for eosinophils, neutrophils, and macrophages under specific conditions.
Galectin-1 Inhibits Leukocyte Entry into Sites of Inflammation
Galectin-1, in contrast to galectin-3 and galectin-9, inhibits leukocyte entry into inflamed tissues (Table I and Fig. 1). In vitro experiments revealed that recombinant galectin-1 reduced chemotaxis of neutrophils towards IL-8 and transmigration of these cells across a layer of vascular endothelial cells [64]. Similarly, Norling et al. observed a dose-dependent decrease in lymphocyte capture, adhesion, and rolling along a layer of vascular endothelial cells when endothelial cells were treated with increasing concentrations of recombinant galectin-1 [65]. Taken together, these data suggest that addition of exogenous galectin-1 influences several different steps during leukocyte migration, such as leukocyte capture, rolling, and adhesion.
In addition to being expressed by vascular endothelial cells, galectin-1 also can bind to components of the ECM [66]. It has been shown that galectin-1 deposition in the ECM can impede T cell migration through the ECM and across a layer of vascular endothelial cells [33]. This inhibition of T cell migration was accompanied by the re-organization and clustering of the galectin-1 glycoprotein counterreceptor CD43 on the T cell surface [33]. Upon binding of galectin-1, CD43 molecules were clustered into patches at the T cell/ECM interface. Since CD43 must localize to the trailing edge of migrating T cells [67], binding of galectin-1 to CD43 may inhibit T cell migration by preventing CD43 re-distribution to the trailing edge of the cell.
The inhibitory role of galectin-1 in leukocyte entry into inflamed tissues has been substantiated in several in vivo models of inflammation, including acute peritonitis, contact hypersensitivity and PLA2-induced paw edema [42, 64, 65, 68]. For example, the role of galectin-1 has been analyzed in a model of acute peritonitis, which showed a decrease in the number of neutrophils in the peritoneum when recombinant galectin-1 was injected at the site of inflammation [64]. Analysis of galectin-1 knock-out mice in a contact hypersensitivity model revealed an increase in the number of lymphocytes found at the site of inflammation [65] implying that endogenous galectin-1 inhibits leukocyte migration. Remarkably, endogenous galectin-1 promotes DC migration through the ECM, but not across a layer of vascular endothelial cells [69]; this observation indicates that galectin-1 affects discrete steps during DC entry into tissues.
Galectins Regulate Leukocyte Exit into the Lymphatic System
As mentioned above, there are two recent reports that suggest a role for galectins in DC migration across the lymphatic endothelium. Both groups utilized fluorescein-isothiocyanate (FITC) skin painting models to analyze the migration of tissue-resident, dermal DCs to lymph nodes in vivo in the presence or absence of galectin-1 or galectin-3, respectively [18, 19]. Fulcher et al. showed that injection of recombinant galectin-1 prior to FITC skin painting increased DC migration to draining lymph nodes in MRL-fas mice [18]. Similarly, after FITC skin painting, there was a reduction of dermal DC migration to draining lymph nodes in galectin-3 knock-out mice compared to wild-type mice [19]. Taken together, these results strongly imply a role for galectin-1 and galectin-3 in promoting DC exit from tissues into the lymphatics. This would be independent of any role for galectins in DC migration across the vascular endothelium since the DC populations that were analyzed were already tissue-resident and only had to migrate across the lymphatic endothelium to traffic to draining lymph nodes. Interestingly, these data indicate that both galectin-1 and galectin-3 promote DC exit into the lymphatic vasculature; this is different from their effect on migration across the vascular endothelium, where galectin-1 generally inhibits leukocyte entry into tissues [42, 64, 65, 68] while galectin-3 promotes leukocyte entry into tissues [48, 49, 51, 58, 59].
While galectin-1 inhibits leukocyte entry into tissues from the blood stream, it is unknown if any galectin family member(s) can inhibit leukocyte exit from tissues into the lymphatics (Fig. 1). Thus, as mentioned above, it will be critical to fully characterize the expression pattern of galectin family members in lymphatic endothelial cells. In addition, it will be important to determine how galectin expression in these cells is dynamically regulated during inflammation and whether this affects leukocyte exit into the afferent lymphatics.
Mechanisms by Which Galectins Regulate Leukocyte Migration Across Endothelial Cells
An important yet unanswered question is how do galectins regulate leukocyte migration during inflammation? As yet, there is little known about the molecular interactions among galectins, immune cells, and endothelial cells that modulate leukocyte entry into tissues and exit into the lymphatic system. Several possible mechanisms, some of which have been explored in leukocyte migration across vascular endothelium, may also regulate leukocyte migration across the lymphatic endothelium.
Galectins can indirectly regulate cellular interactions by inducing molecular changes in a specific cell type that then contribute to the outcome of the interaction. For example, binding of galectins to specific counterreceptors on leukocytes or endothelial cells can induce expression of other molecules that are involved in migration, such as adhesion molecules or matrix metalloproteinases [65, 69]. In addition, it has been shown that galectin binding to cell surface glycoproteins can re-organize the cytoskeleton in T cells [66] and thus may modulate the migratory behavior of leukocytes.
Direct regulation of cellular interactions is another way by which galectins can modulate leukocyte entry into and exit from tissues. For example, galectins expressed by endothelial cells can act as chemoattractants for specific immune cells [57, 60]. Once the immune cell is in close proximity to the endothelial cell layer, galectins might bind to glycoprotein counterreceptors on both cell types and thus facilitate or inhibit migration. This idea is supported by the observation that increased expression of galectin-1 by vascular endothelial cells decreases capture, rolling, and adhesion of lymphocytes and inhibits migration of T cells [33, 65]. Another possible role for galectins in directly modulating cellular interactions would be the re-organization of surface molecules, such as CD43 re-organization by galectin-1 on T cells, that can contribute to the efficiency by which the cell can cross the endothelial cell layer and/or migrate through the ECM [33].
As discussed in the previous paragraphs, galectins can regulate cellular interactions directly by binding to glycoprotein ligands on the cell surface, such as galectin-1 binding to CD43 and CD45 on T cells and DCs [18, 29–31, 33]. This interaction is a highly complex process that can be modulated in each cell type at several different levels. First, the expression of different galectins is dynamic and changes during development and upon activation of leukocytes and endothelial cells [5, 6, 42–46, 52]. Second, the expression of glycan ligands on glycoprotein counterreceptors is dynamic since expression or activity of many glycosyltransferases changes during leukocyte development, maturation, and activation [23, 32, 70, 71]. Third, the availability of glycan ligands on specific glycoproteins, such as CD45, can be modulated by the expression of different protein isoforms that bear different glycan ligands [34, 72–74]. Finally, the expression of glycoprotein counterreceptors that can bear glycan ligands is dynamic since expression can vary from one leukocyte subset to another or change during development [32, 75]. Thus, there are a number of factors that contribute to the effect of galectin interactions with specific glycoprotein counterreceptors on specific cell types and that modulate context-dependent functions, such as cell migration.
Conclusions
Appropriate regulation of leukocyte migration into tissues and subsequently to lymph nodes is critical for generating innate and adaptive immune responses. Leukocyte migration defects in humans cause significant morbidity and can affect either leukocyte entry into tissues or leukocyte exit into the lymphatic vasculature. The diseases leukocyte adhesion deficiency (LAD) I and II result in reduced interactions between leukocytes and vascular endothelial cells. Patients suffer from life-threatening infections due to the inability of immune cells to enter infected tissues [76]. While LAD I is caused by mutations in the β2 integrin gene, LAD II is caused by a defect in fucosylation of glycan ligands for selectins which reduces the tethering and rolling of leukocytes on activated vascular endothelial cells [77].
In addition, if leukocytes cannot exit from tissues into the lymphatic vasculature and traffic to lymph nodes, patients develop lymphedema, a condition in which immune cells remain in tissues and secrete inflammatory cytokines that exacerbate inflammation [1, 78]. These patients also cannot mount appropriate adaptive immune responses to pathogens present in the lymphedematous tissue, which in turn can become severely and chronically inflamed [79].
It is crucially important to improve our understanding about the factors that regulate leukocyte exit from tissues into the lymphatic system. Members of the galectin family of glycan-binding proteins are dynamically expressed in vascular endothelial cells under steady-state and inflammatory conditions and regulate leukocyte entry into sites of inflammation. We propose a similar role for galectins in modulating leukocyte exit from tissues into the lymphatic vasculature. This model is supported by recent in vivo evidence, indicating that galectin-1 and galectin-3 support migration of tissue-resident DCs into draining lymph nodes [18, 19]. Galectin-mediated leukocyte migration into and out of inflamed tissues will likely be a complex process regulated by appropriate expression of the galectins, the glycan ligands, and the glycoprotein counterreceptors by the cells.
References
Johnson LA, Jackson DG. Cell traffic and the lymphatic endothelium. Ann NY Acad Sci. 2008;1131:119–33.
Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol. 2005;6:1182–90.
Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–89.
Elola MT, Wolfenstein-Todel C, Troncoso MF, Vasta GR, Rabinovich GA. Galectins: matricellular glycan-binding proteins linking cell adhesion, migration, and survival. Cell Mol Life Sci. 2007;64:1679–700.
Baum LG, Seilhamer JJ, Pang M, Levine WB, Beynon D, Berliner JA. Synthesis of an endogeneous lectin, galectin-1, by human endothelial cells is up-regulated by endothelial cell activation. Glycoconj J. 1995;12:63–8.
Thijssen VL, Hulsmans S, Griffioen AW. The galectin profile of the endothelium: altered expression and localization in activated and tumor endothelial cells. Am J Pathol. 2008;172:545–53.
Vasta GR. Roles of galectins in infection. Nat Rev Microbiol. 2008;7:424–38.
Almkvist J, Karlsson A. Galectins as inflammatory mediators. Glycoconj J. 2004;19:575–81.
Rabinovich GA, Liu FT, Hirashima M, Anderson A. An emerging role for galectins in tuning the immune response: lessons from experimental models of inflammatory disease, autoimmunity and cancer. Scand J Immunol. 2007;66:143–58.
Maletto BA, Ropolo AS, Alignani DO, Liscovsky MV, Ranocchia RP, Moron VG, et al. Presence of neutrophil-bearing antigen in lymphoid organs of immune mice. Blood. 2006;108:3094–102.
Abadie V, Badell E, Douillard P, Ensergueix D, Leenen PJ, Tanguy M, et al. Neutrophils rapidly migrate via lymphatics after Mycobacterium bovis BCG intradermal vaccination and shuttle live bacilli to the draining lymph nodes. Blood. 2005;106:1843–50.
Randolph GJ, Angeli V, Swartz MA. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat Rev Immunol. 2005;5:617–28.
Alvarez D, Vollmann EH, von Andrian UH. Mechanisms and consequences of dendritic cell migration. Immunity. 2008;29:325–42.
Harmsen AG, Muggenburg BA, Snipes MB, Bice DE. The role of macrophages in particle translocation from lungs to lymph nodes. Science. 1985;230:1277–80.
Mackay CR, Marston WL, Dudler L. Naive and memory T cells show distinct pathways of lymphocyte recirculation. J Exp Med. 1990;171:801–17.
Johnson LA, Clasper S, Holt AP, Lalor PF, Baban D, Jackson DG. An inflammation-induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. J Exp Med. 2006;203:2763–77.
Johnson LA, Jackson DG. Inflammation-induced secretion of CCL21 in lymphatic endothelium is a key regulator of integrin-mediated dendritic cell transmigration. Int Immunol. 2010 doi:10.1093/intimm/dxq435.
Fulcher JA, Chang MH, Wang S, Almazan T, Hashimi ST, Eriksson AU, et al. Galectin-1 co-clusters CD43/CD45 on dendritic cells and induces cell activation and migration through Syk and protein kinase C signaling. J Biol Chem. 2009;284:26860–70.
Hsu DK, Chernyavsky AI, Chen HY, Yu L, Grando SA, Liu FT. Endogenous galectin-3 is localized in membrane lipid rafts and regulates migration of dendritic cells. J Invest Dermatol. 2009;129:573–83.
Nagae M, Nishi N, Murata T, Usui T, Nakamura T, Wakatsuki S, et al. Crystal structure of the galectin-9 N-terminal carbohydrate recognition domain from Mus musculus reveals the basic mechanism of carbohydrate recognition. J Biol Chem. 2006;281:35884–93.
Ahmad N, Gabius HJ, Andre S, Kaltner H, Sabesan S, Roy R, et al. Galectin-3 precipitates as a pentamer with synthetic multivalent carbohydrates and forms heterogeneous cross-linked complexes. J Biol Chem. 2004;279:10841–7.
Stowell SR, Arthur CM, Mehta P, Slanina KA, Blixt O, Leffler H, et al. Galectin-1, -2, and -3 exhibit differential recognition of sialylated glycans and blood group antigens. J Biol Chem. 2008;283:10109–23.
Earl LA, Baum LG. CD45 glycosylation controls T-cell life and death. Immunol Cell Biol. 2008;86:608–15.
Bi S, Earl LA, Jacobs L, Baum LG. Structural features of galectin-9 and galectin-1 that determine distinct T cell death pathways. J Biol Chem. 2008;283:12248–58.
Song X, Xia B, Stowell SR, Lasanajak Y, Smith DF, Cummings RD. Novel fluorescent glycan microarray strategy reveals ligands for galectins. Chem Biol. 2009;16:36–47.
Nguyen JT, Evans DP, Galvan M, Pace KE, Leitenberg D, Bui TN, et al. CD45 modulates galectin-1-induced T cell death: regulation by expression of core two O-glycans. J Immunol. 2001;167:5697–707.
Amano M, Galvan M, He J, Baum LG. The ST6Gal I sialyltransferase selectively modifies N-glycans on CD45 to negatively regulate galectin-1-induced CD45 clustering, phosphatase modulation, and T cell death. J Biol Chem. 2003;278:7469–75.
Hernandez JD, Klein J, Van Dyken SJ, Marth JD, Baum LG. T-cell activation results in microheterogeneous changes in glycosylation of CD45. Int Immunol. 2007;19:847–56.
Perillo NL, Pace KE, Seilhamer JJ, Baum LG. Apoptosis of T cells mediated by galectin-1. Nature. 1995;378:736–9.
Pace KE, Lee C, Stewart PL, Baum LG. Restricted receptor segregation into membrane microdomains occurs on human T cells during apoptosis induced by galectin-1. J Immunol. 1999;163:3801–11.
Stillman BN, Hsu DK, Pang M, Brewer CF, Johnson P, Liu FT, et al. Galectin-3 and galectin-1 bind distinct cell surface glycoprotein receptors to induce T cell death. J Immunol. 2006;176:778–89.
Daniels MA, Hogquist KA, Jameson SC. Sweet ‘n’ sour: the impact of differential glycosylation on T cell responses. Nat Immunol. 2002;3:903–10.
He J, Baum LG. Endothelial cell expression of galectin-1 induced by prostate cancer cells inhibits T-cell transendothelial migration. Lab Invest. 2006;86:578–90.
Earl LA, Bi S, Baum LG. N- and O-glycans modulate galectin-1 binding, CD45 signaling, and T cell death. J Biol Chem. 2010;285:2232–44.
Demetriou M, Granovsky M, Quaggin S, Dennis JW. Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature. 2001;409:733–9.
Chen IJ, Chen HL, Demetriou M. Lateral compartmentalization of T cell receptor versus CD45 by galectin–N-glycan binding and microfilaments coordinate basal and activation signaling. J Biol Chem. 2007;282:35361–72.
Demotte N, Stroobant V, Courtoy PJ, Van Der Smissen P, Colau D, Luescher IF, et al. Restoring the association of the T cell receptor with CD8 reverses anergy in human tumor-infiltrating lymphocytes. Immunity. 2008;28:414–24.
Liu SD, Whiting CC, Tomassian T, Pang M, Bissel SJ, Baum LG, et al. Endogenous galectin-1 enforces class I-restricted TCR functional fate decisions in thymocytes. Blood. 2008;112:120–30.
Cha SK, Ortega B, Kurosu H, Rosenblatt KP, Kuro OM, Huang CL. Removal of sialic acid involving Klotho causes cell-surface retention of TRPV5 channel via binding to galectin-1. Proc Natl Acad Sci USA. 2008;105:9805–10.
Garner OB, Baum LG. Galectin-glycan lattices regulate cell-surface glycoprotein organization and signalling. Biochem Soc Trans. 2008;36:1472–7.
Brewer CF, Miceli MC, Baum LG. Clusters, bundles, arrays and lattices: novel mechanisms for lectin–saccharide-mediated cellular interactions. Curr Opin Struct Biol. 2002;12:616–23.
Gil CD, Cooper D, Rosignoli G, Perretti M, Oliani SM. Inflammation-induced modulation of cellular galectin-1 and -3 expression in a model of rat peritonitis. Inflamm Res. 2006;55:99–107.
Gil CD, La M, Perretti M, Oliani SM. Interaction of human neutrophils with endothelial cells regulates the expression of endogenous proteins annexin 1, galectin-1 and galectin-3. Cell Biol Int. 2006;30:338–44.
Dietz AB, Bulur PA, Knutson GJ, Matasic R, Vuk-Pavlovic S. Maturation of human monocyte-derived dendritic cells studied by microarray hybridization. Biochem Biophys Res Commun. 2000;275:731–8.
Huang Q, Liu D, Majewski P, Schulte LC, Korn JM, Young RA, et al. The plasticity of dendritic cell responses to pathogens and their components. Science. 2001;294:870–5.
Haining WN, Angelosanto J, Brosnahan K, Ross K, Hahn C, Russell K, et al. High-throughput gene expression profiling of memory differentiation in primary human T cells. BMC Immunol. 2008;9:44.
Nachtigal M, Al-Assaad Z, Mayer EP, Kim K, Monsigny M. Galectin-3 expression in human atherosclerotic lesions. Am J Pathol. 1998;152:1199–208.
Saegusa J, Hsu DK, Chen HY, Yu L, Fermin A, Fung MA, et al. Galectin-3 is critical for the development of the allergic inflammatory response in a mouse model of atopic dermatitis. Am J Pathol. 2009;174:922–31.
Colnot C, Ripoche MA, Milon G, Montagutelli X, Crocker PR, Poirier F. Maintenance of granulocyte numbers during acute peritonitis is defective in galectin-3-null mutant mice. Immunology. 1998;94:290–6.
Liu FT, Hsu DK, Zuberi RI, Kuwabara I, Chi EY, Henderson WR. Expression and function of galectin-3, a beta-galactoside-binding lectin, in human monocytes and macrophages. Am J Pathol. 1995;147:1016–28.
Zuberi RI, Hsu DK, Kalayci O, Chen HY, Sheldon HK, Yu L, et al. Critical role for galectin-3 in airway inflammation and bronchial hyperresponsiveness in a murine model of asthma. Am J Pathol. 2004;165:2045–53.
Cueni LN, Detmar M. Galectin-8 interacts with podoplanin and modulates lymphatic endothelial cell functions. Exp Cell Res. 2009;315:1715–23.
Stowell SR, Qian Y, Karmakar S, Koyama NS, Dias-Baruffi M, Leffler H, et al. Differential roles of galectin-1 and galectin-3 in regulating leukocyte viability and cytokine secretion. J Immunol. 2008;180:3091–102.
Rabinovich GA, Toscano MA. Turning ‘sweet’ on immunity: galectin–glycan interactions in immune tolerance and inflammation. Nat Rev Immunol. 2009;9:338–52.
Rubinstein N, Ilarregui JM, Toscano MA, Rabinovich GA. The role of galectins in the initiation, amplification and resolution of the inflammatory response. Tissue Antigens. 2004;64:1–12.
Sato S, Ouellet N, Pelletier I, Simard M, Rancourt A, Bergeron MG. Role of galectin-3 as an adhesion molecule for neutrophil extravasation during streptococcal pneumonia. J Immunol. 2002;168:1813–22.
Sano H, Hsu DK, Yu L, Apgar JR, Kuwabara I, Yamanaka T, et al. Human galectin-3 is a novel chemoattractant for monocytes and macrophages. J Immunol. 2000;165:2156–64.
Hsu DK, Yang RY, Pan Z, Yu L, Salomon DR, Fung-Leung WP, et al. Targeted disruption of the galectin-3 gene results in attenuated peritoneal inflammatory responses. Am J Pathol. 2000;156:1073–83.
Nieminen J, St-Pierre C, Bhaumik P, Poirier F, Sato S. Role of galectin-3 in leukocyte recruitment in a murine model of lung infection by Streptococcus pneumoniae. J Immunol. 2008;180:2466–73.
Matsushita N, Nishi N, Seki M, Matsumoto R, Kuwabara I, Liu FT, et al. Requirement of divalent galactoside-binding activity of ecalectin/galectin-9 for eosinophil chemoattraction. J Biol Chem. 2000;275:8355–60.
Yamamoto H, Kashio Y, Shoji H, Shinonaga R, Yoshimura T, Nishi N, et al. Involvement of galectin-9 in guinea pig allergic airway inflammation. Int Arch Allergy Immunol. 2007;143 Suppl 1:95–105.
Tsuboi Y, Abe H, Nakagawa R, Oomizu S, Watanabe K, Nishi N, et al. Galectin-9 protects mice from the Shwartzman reaction by attracting prostaglandin E2-producing polymorphonuclear leukocytes. Clin Immunol. 2007;124:221–33.
Hirashima M, Kashio Y, Nishi N, Yamauchi A, Imaizumi TA, Kageshita T, et al. Galectin-9 in physiological and pathological conditions. Glycoconj J. 2004;19:593–600.
La M, Cao TV, Cerchiaro G, Chilton K, Hirabayashi J, Kasai K, et al. A novel biological activity for galectin-1: inhibition of leukocyte–endothelial cell interactions in experimental inflammation. Am J Pathol. 2003;163:1505–15.
Norling LV, Sampaio AL, Cooper D, Perretti M. Inhibitory control of endothelial galectin-1 on in vitro and in vivo lymphocyte trafficking. FASEB J. 2008;22:682–90.
Rabinovich GA, Ariel A, Hershkoviz R, Hirabayashi J, Kasai KI, Lider O. Specific inhibition of T-cell adhesion to extracellular matrix and proinflammatory cytokine secretion by human recombinant galectin-1. Immunology. 1999;97:100–6.
del Pozo MA, Nieto M, Serrador JM, Sancho D, Vicente-Manzanares M, Martinez C, et al. The two poles of the lymphocyte: specialized cell compartments for migration and recruitment. Cell Adhes Commun. 1998;6:125–33.
Rabinovich GA, Sotomayor CE, Riera CM, Bianco I, Correa SG. Evidence of a role for galectin-1 in acute inflammation. Eur J Immunol. 2000;30:1331–9.
Fulcher JA, Hashimi ST, Levroney EL, Pang M, Gurney KB, Baum LG, et al. Galectin-1-matured human monocyte-derived dendritic cells have enhanced migration through extracellular matrix. J Immunol. 2006;177:216–26.
Bi S, Baum LG. Sialic acids in T cell development and function. Biochim Biophys Acta. 2009;1790:1599–610.
Rifat S, Kang TJ, Mann D, Zhang L, Puche AC, Stamatos NM, et al. Expression of sialyltransferase activity on intact human neutrophils. J Leukoc Biol. 2008;84:1075–81.
Thomas ML. The leukocyte common antigen family. Annu Rev Immunol. 1998;7:339–69.
Hermiston ML, Xu Z, Weiss A. CD45: a critical regulator of signaling thresholds in immune cells. Annu Rev Immunol. 2003;21:107–37.
Furukawa K, Funakoshi Y, Autero M, Horejsi V, Kobata A, Gahmberg CG. Structural study of the O-linked sugar chains of human leukocyte tyrosine phosphatase CD45. Eur J Biochem. 1998;251:288–94.
Daniels MA, Devine L, Miller JD, Moser JM, Lukacher AE, Altman JD, et al. CD8 binding to MHC class I molecules is influenced by T cell maturation and glycosylation. Immunity. 2001;15:1051–61.
Anderson DC, Schmalsteig FC, Finegold MJ, Hughes BJ, Rothlein R, Miller LJ, et al. The severe and moderate phenotypes of heritable Mac-1, LFA-1 deficiency: their quantitative definition and relation to leukocyte dysfunction and clinical features. J Infect Dis. 1985;152:668–89.
Wild MK, Luhn K, Marquardt T, Vestweber D. Leukocyte adhesion deficiency II: therapy and genetic defect. Cells Tissues Organ. 2002;172:161–73.
Cueni LN. Detmar: the lymphatic system in health and disease. Lymphat Res Biol. 2008;6:109–22.
Erickson VS, Pearson ML, Ganz PA, Adams J, Kahn KL. Arm edema in breast cancer patients. J Natl Cancer Inst. 2001;93:96–111.
Acknowledgments
The authors would like to thank Robert Yuan for contributing to the figure and Mary Clark for critically reading the manuscript. This work was supported by NIH grant R21HL102989 (to LGB).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Thiemann, S., Baum, L.G. The Road Less Traveled: Regulation of Leukocyte Migration Across Vascular and Lymphatic Endothelium by Galectins. J Clin Immunol 31, 2–9 (2011). https://doi.org/10.1007/s10875-010-9460-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-010-9460-z