
Abstract
Four novel 5D (HACA(N)CONH, HNCOCACB, (HACA)CON(CA)CONH, (H)NCO(NCA)CONH), and one 6D ((H)NCO(N)CACONH) NMR pulse sequences are proposed. The new experiments employ non-uniform sampling that enables achieving high resolution in indirectly detected dimensions. The experiments facilitate resonance assignment of intrinsically disordered proteins. The novel pulse sequences were successfully tested using δ subunit (20 kDa) of Bacillus subtilis RNA polymerase that has an 81-amino acid disordered part containing various repetitive sequences.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Intrinsically disordered proteins (IDPs) are a group of macromolecules with peculiar structure, dynamics and interactions (Dyson and Wright 2001; Tompa et al. 2006) that allow them to play a number of important biological functions (Ward et al. 2004; Dyson and Wright 2005; Tompa 2010). Solution state NMR spectroscopic techniques appear ideally suited for the studies of these proteins as IDPs’ fast conformational dynamics results in relatively slow transverse relaxation rates. The problem is that the rapid interconversion rate for the various conformations causes averaging of chemical shifts and very poor peak separation, making resonance assignment difficult, even for relatively small disordered protein fragments. However, there is a promise for solving this obstacle in multidimensional NMR methods utilizing non-uniform sampling of indirectly detected dimensions (Felli and Brutscher 2009; Coggins et al. 2010; Kazimierczuk et al. 2010a), as this sampling facilitates acquisition of high-resolution and high-dimensional spectra.
The established approach for backbone resonance assignment of globular proteins consists of a suite of triple-resonance experiments where sequential connectivities are found using carbonyl 13C, aliphatic 13C and/or aliphatic 1H chemical shifts (Sattler et al. 1999). However, this approach often fails in the case of IDPs because of poor dispersion of side-chain chemical shifts (which depend mostly on the residue identity). In Fig. 1, we show HA-CA, HB-CB and N-CO chemical shift correlations for the 81 a.a. unstructured part of the δ subunit of B. subtilis RNA polymerase (Motáčková et al. 2010; Nováček et al. 2011). These graphs clearly show the best dispersion of amide nitrogen and carbonyl carbon chemical shifts. The utility of these nuclei for sequential assignment is additionally supported by their relatively slow (as compared to that of aliphatic protons or aliphatic carbon atoms) transverse relaxation and lack of non-refocused homonuclear couplings, the presence of which may limit resolution even stronger than does the relaxation limit, e.g. in the case of aliphatic carbon atoms.

Correlation of HA–CA (a), HB–CB (b), and N–CO (c) chemical shifts for the 81 a.a. unstructured part of the δ subunit of B. subtilis RNA polymerase. The 1H and 13C chemical shifts depend mostly on particular amino acid residue, whereas 13CO and 15N frequencies are much better resolved and enable identification of backbone connectivity. (BMRB entry 16912)
To date, several strategies have been proposed for effective backbone resonance assignment of IDPs. These strategies include 13C detection (Bermel et al. 2006, 2009), automated projection spectroscopy (APSY; Narayanan et al. 2010), HA detection (Mäntylahti et al. 2011), and the sparsely sampled 4D (Wen et al. 2011) and 5D experiments (Motáčková et al. 2010; Nováček et al. 2011). Below, we propose a set of new pulse sequences that feature high resolution and high dimensionality resulting from the use of sparse random sampling in the indirectly detected dimensions. The novel experiments, which require (because of multiple coherence transfer steps involved) a slow transverse relaxation, were designed for IDPs and are superior in terms of peak resolution and the easiness of resonance assignment of the proteins. The pulse sequences were tested using the 20 kDa δ subunit of B. subtilis RNA polymerase. Having an 81 a.a. unstructured part with various repetitive sequences, this macromolecule is an excellent example of an IDP whose resonance assignment is extremely difficult using conventional methods.
Methods
The uniformly 13C,15N-labeled sample of B. subtilis RNA polymerase δ subunit was prepared as described previously (Motáčková et al. 2010). All spectra were acquired in a 0.7 mM protein solution sample on a Varian NMR System 700 spectrometer equipped with a Performa XYZ PFG unit, using the standard 5 mm 1H-13C-15N triple-resonance probehead. High-power 1H, 13C and 15N π/2 pulses of 5.9, 13.5 and 31.0 μs, respectively, were used. Selective CA and CO pulses were realized as phase-modulated (for off-resonance excitation or inversion) sinc shapes, with B1 field strength adjusted to have a minimal effect on CO and CA, respectively. In all cases, four scans per each data set were acquired with acquisition time of 85 ms and relaxation delay of 1.2 s. For processing of directly detected dimension, cosine square weighting function was used prior to Fourier transform with zero-filling to 2,048 complex points. The experiments were performed using random off-grid Poisson disk sampling with sampling density set according to a Gaussian distribution (σ = 0.5) with regard to maximum evolution time (Kazimierczuk et al. 2008). No apodization was applied in indirect dimensions. The number of complex points Mi in the frequency domain of ith indirectly detected dimension was set as Mi ≥ 3 × swi × t maxi . The sparse multidimensional Fourier transform (SMFT) procedure (Kazimierczuk et al. 2009), with ‘fixed’ frequencies derived from 3D HNCO and 4D HNCOCA peak list, was used to obtain F 1/F 2 cross-sections in 5D and 6D experiments, respectively. The remaining relevant experimental parameters are shown in Table 1.
The pulse sequences were written using own-developed programming library. The resulting spectra were analyzed using the SPARKY software (Goddard and Kneller 2002). The pulse sequence code for Agilent spectrometers as well as the SMFT software used for data processing are available from the authors upon request.
Results and discussion
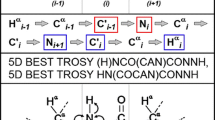
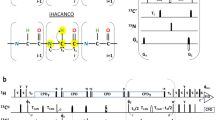
The first two pulse sequences (5D HACA(N)CONH and 5D (HACA)CON(CA)CONH) are depicted schematically in Figs. 2a and 3a, respectively, and the corresponding coherence transfer pathways are given in Figs. 2b and 3b. Both experiments employ equilibrium magnetization of HA protons. This allows identifying certain chemical shifts of the proline residues whose successors’ amide protons are detected. In both experiments, the effective separation of F 1/F 2 cross-sections is obtained due to the good peak separation in CO–N subspectra. In the HACA(N)CONH experiment, the sequential connectivities may be obtained from 1HA and 13CA chemical shifts, which approach usually fails in the case of IDPs due to poor peak separation. On the other hand, the (HACA)CON(CA)CONH spectra allow, at the expense of additional coherence transfer steps, finding of connectivities with the use 13CO and 15N frequencies that are more uniformly distributed over the entire spectral band and therefore are more suitable for the studies of IDPs. This experiment requires extension of the spectral width in the first two dimensions to accommodate correlations of proline residues. In Figs. 2c and 3c we show an example of 2D cross-sections for the I118-L123 fragment of the disordered part of δ subunit of B. subtilis RNA polymerase.

5D HACA(N)CONH technique. (a) Pulse sequence, 1H, 13CA, and 15N evolution is in semi-constant-time mode: ai = (t i + Δ)/2, b i = t i(1−Δ/t maxi )/2, c i = Δ(1−t i/t maxi )/2 (where Δ stands for coherence transfer delays listed below, t i is the evolution time in ith dimension, and t maxi is maximal length of the evolution time delay). Delays were set as follows: Δ’H-C = 2.6 ms, ΔCA-N = 28.0 ms, ΔCA-N-CO = 28.0 ms, ΔN-CO = 28.0 ms, and ΔN-H = 5.4 ms. The four-step phase cycle was used: ϕ1 = x, −x, ϕ2 = 2x, 2(−x) and Rec = ϕ1 + ϕ2. Simultaneous inversion of CA and CO spins was achieved using 6-element composite pulse (Shaka 1985). The coherence selection gradients (marked xyz) were applied at the magic angle. The phase ψ was inverted simultaneously with the last gradient pulse. (b) Coherence transfer in the peptide chain. HN, N, and CO frequencies (filled rectangles) are ‘fixed’ for Fourier transform. Frames for HA and CA indicate the dimensions of 2D cross-sections obtained by SMFT procedure. (c) 2D spectral planes for the δ subunit of B. subtilis RNA polymerase, which were obtained by SMFT procedure performed on the 5D randomly sampled signal (Poisson disk sampling) with ‘fixed’ frequencies obtained from 3D HNCO peak list. Each cross-section contains two cross-peaks: for’fixed’ H Ni , Ni and COi−1, the peaks correspond to HAi–CAi and HAi−1–CAi−1 correlations

5D (HACA)CON(CA)CONH technique. (a) Pulse sequence, 15N (in t 2 and t 4) and 13CO (in t 1 and t 3) evolution is in semi-constant-time mode: ai = (t i + Δ)/2, b i = t i(1−Δ/t maxi )/2, c i = Δ(1−t i/t maxi )/2 (where Δ stands for coherence transfer delays listed below, t i is the evolution time in ith dimension, and t maxi is maximal length of the evolution time delay). Delays were set as follows: ΔH-C = 3.7 ms, Δ’H-C = 2.6 ms, ΔCA-CO = 6.8 ms, ΔCA-N = 28.0 ms, ΔCA-N-CO = 28.0 ms, ΔN-CO = 28.0 ms, ΔN-H = 5.4 ms. The four-step phase cycle was used: ϕ1 = x, −x, ϕ2 = 2x, 2(−x) and Rec = ϕ1 + ϕ2. Simultaneous inversion of CA and CO spins was achieved using 6-element composite pulse (Shaka 1985). The coherence selection gradients (marked by xyz) were applied at the magic angle. The phase ψ was inverted simultaneously with the last gradient pulse. (b) Coherence transfer in the peptide chain. HN, N, and CO frequencies (filled rectangles) are ‘fixed’ for Fourier transform. Frames for N and CO indicate the dimensions of 2D cross-sections obtained by SMFT procedure. (c) 2D spectral planes for the δ subunit of B. subtilis RNA polymerase, which were obtained by SMFT procedure performed on the 5D randomly sampled signal (Poisson disk sampling) with ‘fixed’ frequencies obtained from 3D HNCO peak list. Each cross-section contains two cross-peaks: for ‘fixed’ H Ni , Ni and COi−1 the peaks correspond to Ni-COi−1 and Ni−1-COi−2 correlations
The out-and-back 5D HNCOCACB experiment shown in Fig. 4 correlates 1H Ni , 15Ni, 13COi−1 with 13CA i−1 and 13CB i−1 chemical shifts. Contrary to the established CBCANH, CBCA(CO)NH and HNCACB experiments (Grzesiek and Bax 1992a, 1992b, 1993; Wittekind and Müller 1993), the CA and CB evolutions are performed in separate dimensions. This allows to increase the CA-CB coupling evolution delay to 0.5/JCACB and therefore, to double (providing one ignores the relaxation) the sensitivity, which in the case of IDPs compensates for the extended pulse sequence. Although this sequence does not provide sequential connectivities, it allows to assign CA and CB chemical shifts and to identify a.a. residues by comparing the respective chemical shifts with typical values for each amino acid.

5D HNCOCACB technique. (a) Pulse sequence, 13CO and 15N evolution is in semi-constant-time mode: ai = (t i + Δ)/2, b i = t i(1−Δ/t maxi )/2, c i = Δ(1−t i/t maxi )/2 (where Δ stands for listed below coherence transfer delays, t i is the evolution time in ith dimension and t maxi is maximal length of the evolution time delay). 13CA chemical shift evolution is in constant-time mode. Delays were set as follows: ΔN-H = 5.4 ms, ΔN-CO = 28.0 ms, ΔCA-CO = 9.1 ms, and ΔCACB = 14.3 ms. The four-step phase cycle was used: ϕ1 = x, −x, ϕ2 = 2x, 2(−x) and Rec = ϕ1 + ϕ2. The coherence selection gradients (marked by xyz) were applied at the magic angle. The phase ψ was inverted simultaneously with the last gradient pulse. (b) Coherence transfer in the peptide chain. HN, N, and CO frequencies (filled rectangles) are ‘fixed’ for Fourier transform. Frames for CA and CB indicate the dimensions of 2D cross-sections obtained by SMFT procedure. (c) 2D spectral planes for the δ subunit of B. subtilis RNA polymerase, which were obtained by SMFT procedure performed on the 5D randomly sampled signal (Poisson disk sampling) with ‘fixed’ frequencies obtained from 3D HNCO peak list. Each cross-section contains one cross-peak: for ‘fixed’ H Ni , Ni and COi−1 the peak corresponds to CAi−1-CBi−1 correlation
The 5D (H)NCO(NCA)CONH experiment is schematically depicted in Fig. 5a together with the scheme of coherence transfer pathway in protein backbone 5b. In this case the magnetization of amide proton’s origin is transferred through amide nitrogen and carbonyl carbon nuclei back to nitrogen and to two different CA nuclei, then via the respective CO nuclei to the corresponding coupled NH pairs. In this case, again, 13CO and 15N chemical shifts enable peak resolution in the ‘fixed’ dimensions F 3 and F 4, and establishing sequential connectivities from 13CO and 15N in dimensions F 1 and F 2.

5D (H)NCO(NCA)CONH technique. (a) Pulse sequence, 15N (in t 1 and t 4) and 13CO (in t 3) evolution is in semi-constant-time mode: ai = (t i + Δ)/2, b i = t i(1−Δ/t maxi )/2, c i = Δ(1−t i/t maxi )/2 (where Δ stands for listed below coherence transfer delays, t i is the evolution time in ith dimension and t maxi is maximal length of the evolution time delay). Delays were set as follows: ΔN-H = 5.4 ms, ΔN-CO = 28.0 ms, ΔCO–N-CA = 28.0 ms, ΔN-CA = 28.6 ms, and ΔCA-CO = 9.1 ms. The four-step phase cycle was used: ϕ1 = x, −x, ϕ2 = 2x, 2(−x) and Rec = ϕ1 + ϕ2. Simultaneous inversion of CA and CO spins was achieved using 6-element composite pulse (Shaka 1985). The coherence selection gradients (marked by xyz) were applied at the magic angle. The phase ψ was inverted simultaneously with the last gradient pulse. (b) Coherence transfer in the peptide chain. HN, N, and CO frequencies (filled rectangles) are ‘fixed’ for Fourier transform. Frames for N and CO indicate the dimensions of 2D cross-sections obtained by SMFT procedure. (c) 2D spectral planes for the δ subunit of B. subtilis RNA polymerase, which were obtained by SMFT procedure performed on the 5D randomly sampled signal (Poisson disk sampling) with ‘fixed’ frequencies obtained from 3D HNCO peak list. Each cross-section contains two cross-peaks: for ‘fixed’ H Ni , Ni and COi−1 the peaks correspond to Ni-COi−1 and Ni−1-COi−2 correlations
The 6D (H)NCO(N)CACONH pulse sequence is shown in Fig. 6. This sequence was obtained from the aforementioned 5D variant by introducing constant time evolution of CA frequencies, i.e. with no increase in overall sequence duration. The extra resolution gain resulting from the increased dimensionality may be crucial for IDPs’ spectra that feature high chemical shift degeneracy. Such an example is given in Fig. 7, where the additional dimension enables to resolve peaks that still overlap in 5D spectra. In this case, however, application of SMFT procedure requires the knowledge of CAi−1 chemical shifts. These shifts can be obtained using 5D HNCOCACB, 5D HabCabCONH (Kazimierczuk et al. 2010), or 4D HNCOCA (Zawadzka-Kazimierczuk et al. 2010) experiments.

6D (H)NCO(N)CACONH technique. (a) Pulse sequence, 15N (in t 1 and t 5) and 13CO (in t 4) evolution is in semi-constant-time mode: ai = (t i + Δ)/2, b i = t i(1−Δ/t maxi )/2, c i = Δ(1−t i/t maxi )/2 (where Δ stands for listed below coherence transfer delays, t i is the evolution time in ith dimension and t maxi is maximal length of the evolution time delay). CA evolution (in t 3) is in constant-time mode. Delays were set as follows: ΔN-H = 5.4 ms, ΔN-CO = 28.0 ms, ΔCO-N-CA = 28.0 ms, ΔN-CA = 28.6 ms, and ΔCA-CO = 9.1 ms. The four-step phase cycle was used: ϕ1 = x, −x, ϕ2 = 2x, 2(−x) and Rec = ϕ1 + ϕ2. Simultaneous inversion of CA and CO spins was achieved using 6-element composite pulse (Shaka 1985). The coherence selection gradients (marked by xyz) were applied at the magic angle. The phase ψ was inverted simultaneously with the last gradient pulse. (b) Coherence transfer in the peptide chain. HN, N, CO and CA frequencies (filled rectangles) are ‘fixed’ for Fourier transform. Frames for N and CO indicate the dimensions of 2D cross-sections obtained by SMFT procedure. (c) 2D spectral planes for the δ subunit of B. subtilis RNA polymerase, which were obtained by SMFT procedure performed on the 6D randomly sampled signal (Poisson disk sampling) with ‘fixed’ frequencies obtained from 4D HNCOCA peak list. Each plane contains two cross-peaks: for ‘fixed’ H Ni , Ni, COi−1 and CAi−1 the peaks correspond to Ni-COi−1 and Ni−1-COi−2 correlations

Comparison of 2D N-CO cross-sections from 5D (H)NCO(NCA)CONH (a) and 6D (H)NCO(N)CACONH (b, c) experiments. In 5D experiment (a) the two pairs of correlation peaks overlap (E130/E131 with E170/E171, and E131/L132 with E171/I172) due to similarity of chemical shifts for all ‘fixed’ dimensions: H NL132 and H NI172 , NL132 and NI172, COE131 and COE171. The additional CA dimension in 6D experiment (b, c) enabled to differentiate and assign all peaks
The experiments shown in Figs. 5 and 6 are conceptually similar to the (HACA)CON(CA)CONH experiment shown in Fig. 3, but the (HACA) fragment is replaced by (H)N. This modification enables application of band-selective excitation short-transient (BEST) approach that is aimed at the acceleration of acquisition (Schanda et al. 2006; Lescop et al. 2007), but does not allow to find the resonances of proline residues. Notably, the (H)NCO(NCA)CONH sequence has a sensitivity advantage over the (HACA)CON(CA)CONH sequence as the CA → CO coherence transfer in the latter is attenuated due to the presence of a concurrent 1JCACB coupling. The respective amplitude transfer functions at this point (i.e. before the first CO evolution period) are shown in (1) and (2) below:
where n is the number of HA protons (for Gly: n = 2).
We have set ΔCACO = 6.8 ms to compromise between J-couplings and relaxation. The same choice was made by Mäntylahti et al. (2011) for other HA-excited experiments and is usually employed in HSQC type HN(CA)CO experiments (for references see Sattler et al. 1999). Assuming T2HN = 50 ms, T2N = 50 ms, T2HA, T2CA = 20 ms, n = 1, and delay times as given in figure captions, we obtain 0.50 and 0.36 for I(HN) and I(HACA), respectively. Setting ΔCACO at 28.5 ms, which is close to 1/JCACB, with evolution of JCACO extended to 9.1 ms, further reduces I(HACA) to 0.18. Using the relaxation times T2HN = 80 ms, T2N = 100 ms, T2HA = 40 ms and T2CA = 50 ms, which seem likely for IDPs (based on our experience), and ΔCACO = 6.8 ms, one obtains I(HN) = 0.68 and I(HACA) = 0.49, whereas for ΔCACO of 28.5 ms I(HACA) = 0.47. Therefore, the latter option seems impractical, especially that the relaxation rates and coupling constants may not be uniform in the entire molecule.
Despite long duration of the proposed pulse sequences and high sparsity of the sampling schedules employed we have found all expected peaks for the disordered fragment of B. subtilis RNA polymerase δ subunit. We have not found any false peaks, i.e. all resonances found were unambiguously assigned in a sequential manner.
In Fig. 8, non-specificity of aliphatic 1H and 13C chemical shifts is demonstrated using E168-E171 correlations for δ subunit of RNA polymerase from B. subtilis. It is shown that in the repeated glutamic acid fragment the aliphatic chemical shifts do not differ sufficiently for the sequential assignment, while amide nitrogen and carbonyl carbon chemical shifts enable unambiguous assignment.

F 1–F 2 2D cross-sections of HACA(N)CONH (a), HNCOCACB (b), and (H)NCO(NCA)CONH (c) 5D experiments, acquired for the δ subunit of B. subtilis RNA polymerase, show E168-E171correlations. The two intra- and inter-residual HA-CA correlation peaks are not resolved in (a); CA-CB correlation peaks in (b) have identical coordinates, however, the resolved pairs of N-CO peaks shown in (c) enable unambiguous sequential assignment
Conclusions
Random sampling and SMFT processing allow developing novel NMR experiments of high dimensionality and high resolution that would not be feasible using conventional sampling. The new experiments enable simple and unambiguous backbone assignment of IDPs. Importantly, not all of the presented techniques must be used to obtain complete sequential assignment. One can combine various experiments (also from among those published before) to construct an optimal set for a given protein. The use of the techniques we present in this paper provides sequential connectivities via 13CO and 15N chemical shifts and enables more straightforward sequential assignment than the 5D experiments published previously. Moreover, the separation of individual spin systems on 2D cross-sections could be very useful for a possible automatic assignment algorithm, which would allow fast and simple resonance assignment, also for large IDPs.
References
Bermel W, Bertini I, Felli IC, Piccioli M, Pierattelli R (2006) 13C detected protonless NMR spectroscopy of proteins in solution. Prog Nucl Magn Reson Spectrosc 48:25–45
Bermel W, Bertini I, Csizmok V, Felli IC, Pierattelli R, Tompa P (2009) H-start for exclusively heteronuclear NMR spectroscopy: the case of intrinsically disordered protein. J Magn Reson 198:275–281
Coggins BE, Venters RA, Zhou P (2010) Radial sampling for fast NMR: concepts and practices over three decades. Prog Nucl Magn Reson Spectrosc 57:381–419
Dyson HJ, Wright PE (2001) Nuclear magnetic resonance methods for elucidation of structure and dynamics in disordered states. Methods Enzymol 339:258–270
Dyson HJ, Wright PE (2005) Intrinsically unstructured proteins and their functions. Natl Rev Mol Cell Biol 6:197–208
Felli IC, Brutscher B (2009) Recent advancements in solution NMR: fast methods and heteronuclear direct detection. ChemPhysChem 10:1356–1368
Goddard TD, Kneller DG (2002) SPARKY 3. University of California, San Francisco. http://www.cgl.ucsf.edu/home/sparky
Grzesiek S, Bax A (1992a) Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR. J Am Chem Soc 114:6291–6293
Grzesiek S, Bax A (1992b) An efficient experiment for sequential backbone assignment of medium sized isotopically enriched proteins. J Magn Reson 99:201–207
Grzesiek S, Bax A (1993) Amino acid type determination in the sequential assignment procedure of uniformly 13C/15 N enriched proteins. J Biomol NMR 3:185–204
Kazimierczuk K, Zawadzka A, Koźmiński W (2008) Optimization of random time domain sampling in multidimensional NMR. J Magn Reson 192:123–130
Kazimierczuk K, Zawadzka A, Koźmiński W (2009) Narrow peaks and high dimensionalities: exploiting the advantages of random sampling. J Magn Reson 197:219–228
Kazimierczuk K, Stanek J, Zawadzka-Kazimierczuk A, Koźmiński W (2010a) Random sampling in multidimensional NMR spectroscopy. Prog Nucl Magn Reson Spectrosc 57:420–434
Kazimierczuk K, Zawadzka-Kazimierczuk A, Koźmiński W (2010b) Non-uniform frequency domain for optimal exploitation of nonuniform sampling. J Magn Reson 205:286–292
Lescop E, Schanda P, Brutscher B (2007) A set of BEST triple-resonance experiments for time-optimized protein resonance assignment. J Magn Reson 187:163–169
Mäntylahti S, Hellman M, Permi P (2011) Extension of the HA-detection based approach: (HCA)CON(CA)H and (HCA)NCO(CA)H experiments for the main-chain assignment of intrinsically disordered proteins. J Biomol NMR 49:99–109
Motáčková V, Nováček J, Zawadzka-Kazimierczuk A, Kazimierczuk K, Žídek L, Šanderová H, Krásný L, Koźmiński W, Sklenář V (2010) Strategy for complete NMR assignment of disordered proteins with highly repetitive sequences based on resolution-enhanced 5D experiments. J Biomol NMR 48:169–177
Narayanan RL, Durr UHN, Bibow S, Biernat J, Mandelkow E, Zweckstetter M (2010) Automatic assignment of the intrinsically disordered protein tau with 441-residues. J Am Chem Soc 132(34):11906–11907
Nováček J, Zawadzka-Kazimierczuk A, Papoušková V, Žídek L, Šanderová H, Krásný L, Koźmiński W, Sklenář V (2011) 5D 13C detected experiments for backbone assignment of unstructured proteins with a very low signal dispersion. J Biomol NMR 50:1–11
Sattler M, Schleucher J, Griesinger C (1999) Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog Nucl Magn Reson Spectrosc 34:93–158
Schanda P, Van Melckebeke H, Brutscher B (2006) Speeding up three-dimensional protein NMR experiments to a few minutes. J Am Chem Soc 128:9042–9043
Shaka AJ (1985) Composite pulses for ultra-broadband spin inversion. Chem Phys Lett 120:201–205
Tompa P (2010) Structure and function of intrinsically disordered proteins. CRC Press, Boca Raton
Tompa P, Dosztanyi Z, Simon I (2006) Prevalent structural disorder in E. coli and S. cerevisiae proteomes. J Proteome Res 5:1996–2000
Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT (2004) Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol 337:635–645
Wen J, Wu J, Zhou P (2011) Sparsely sampled high-resolution 4-D experiments for efficient backbone resonance assignment of disordered proteins. J Magn Reson 209:94–100
Wittekind M, Müller I (1993) HNCACB, a high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the alpha- and beta-carbon resonances in proteins. J Magn Reson B 101:201–205
Zawadzka-Kazimierczuk A, Kazimierczuk K, Koźmiński W (2010) A set of 4D NMR experiments of enhanced resolution for easy resonance assignment in proteins. J Magn Reson 202:109–116
Acknowledgments
This work has been supported by the Bio-NMR project under the 7th framework programme of the EC grant agreement 261863 for conducting the research. The authors thank Dr. Stanisław Chrapusta of the Mossakowski Medical Research Center, Polish Academy of Sciences, for his assistance in language correction. A.Z.-K. thanks the Foundation for Polish Science for supporting her with the MPD Programme that was co-financed by the EU European Regional Development Fund. H.S. and L.K. acknowledge support by Grant No. 204/09/0583 from the Czech Science Foundation and Institutional Research Concept No. AV0Z50200510.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zawadzka-Kazimierczuk, A., Koźmiński, W., Šanderová, H. et al. High dimensional and high resolution pulse sequences for backbone resonance assignment of intrinsically disordered proteins. J Biomol NMR 52, 329–337 (2012). https://doi.org/10.1007/s10858-012-9613-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-012-9613-x