
Abstract
A strategy for complete backbone and side-chain resonance assignment of disordered proteins with highly repetitive sequence is presented. The protocol is based on three resolution-enhanced NMR experiments: 5D HN(CA)CONH provides sequential connectivity, 5D HabCabCONH is utilized to identify amino acid types, and 5D HC(CC-TOCSY)CONH is used to assign the side-chain resonances. The improved resolution was achieved by a combination of high dimensionality and long evolution times, allowed by non-uniform sampling in the indirect dimensions. Random distribution of the data points and Sparse Multidimensional Fourier Transform processing were used. Successful application of the assignment procedure to a particularly difficult protein, δ subunit of RNA polymerase from Bacillus subtilis, is shown to prove the efficiency of the strategy. The studied protein contains a disordered C-terminal region of 81 amino acids with a highly repetitive sequence. While the conventional assignment methods completely failed due to a very small differences in chemical shifts, the presented strategy provided a complete backbone and side-chain assignment.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Unstructured polypeptide sequences attract an increased attention of biochemists and structural biologists. It has been recognized that a large portion of the genome encodes proteins that remain fully or partially disordered in the cell. An automatic analysis of putative amino acid sequences predicted that one third of eukaryotic proteins, including human ones, contains disordered regions longer than 30 amino acids (Ward et al. 2004). These intrinsically disordered proteins have well-defined biological functions. Some of them may adopt a regular structure on binding to their interacting partners, and many of them are related to various human diseases (Bussell and Eliezer 2001; Fink 2005; Dyson and Wright 2005; Dunker et al. 2008). The lack of a unique structure makes the disordered proteins poor targets for crystallographic studies. Even when a highly ordered monocrystal is successfully prepared, the structure calculated from diffraction data does not reflect the distribution of conformations in the native ensemble in solution. Therefore, nuclear magnetic resonance (NMR) plays a crucial role in studies of disordered proteins (Dyson and Wright 2004; Eliezer 2007, 2009). NMR can provide valuable information on residual secondary structure, possible long-range contacts, and internal dynamics of the disordered polypeptide chain. A particularly important step in NMR investigation is the assignment of observed spectral frequencies to individual atoms. Not only the assignment is a necessary prerequisite for interpretation of NMR spectra, but the obtained frequency list itself provides perhaps the most powerful tool for identification of partially ordered segments (Eliezer 2007). The chemical shift of every nucleus is influenced by the molecular environment and a local extended or partially folded conformations can be therefore identified by a deviation from the “random coil” frequency value. Many intrinsically disordered proteins can be assigned using a standard set of triple-resonance NMR experiments applied to 13C,15N-labeled samples (Peti et al. 2001; Yao et al. 2001; Marion. 2006; Mukrasch et al. 2009; Motáčková et al. 2009). However, combination of the structural disorder with a high incidence of sequential repeats often results in spectra with severely overlapped peaks, impossible to assign by the traditional approach. Here we present a strategy that allows to assign frequencies of all backbone and side-chain nuclei even in cases when the standard methods completely fail. The strategy is based on an extreme improvement of spectral resolution and is applicable to standard 13C,15N-labeled protein samples. The power of the presented approach is documented on a case study of a particularly difficult sample, the delta subunit of RNA polymerase unique for gram-positive bacteria (Clark et al. 1974). The 20 kDa protein contains a disordered C-terminal region of 81 amino acids with a highly repetitive sequence (DeSaro et al. 1995). Biochemical studies indicated that the δ subunit may control specificity of RNA polymerase, but molecular basis for its function remains unknown. It represents a strong motivation for NMR investigation of the partially disordered δ subunit.
Materials and methods
Sample preparation
The 13C,15N-uniformly labeled sample of the RNA polymerase δ subunit was prepared from 2 l of cell culture grown in enriched M9 medium using a standard expression protocol, and purified by the ion exchange chromatography as described earlier (DeSaro et al. 1995). Mass spectrometric analysis of unlabeled protein revealed a correct mass of the δ subunit, with the first residue (Met1) cleaved off during the expression. The protein was dialyzed twice against 20 mM phosphate buffer, pH 6.6, containing 10 mM NaCl. The sample was concentrated to yield 0.8 mM δ protein solution.
NMR experiments
All NMR experiments were performed on the 13C,15N-uniformly labeled sample at 301 K. Standard HNCA, HN(CO)CA, HNCACB, CBCA(CO)NH, HNCO, HCCH-TOCSY (Sattler et al. 1999), 13C-edited NOESY spectra (Sattler et al. 1995), and 13C detected CON, CBCACON, and CBCANCO spectra (Bermel et al. 2006a), were acquired on a Bruker Avance 600MHz spectrometer equipped with the cryogenic 1H/13C/15N TCI probehead with the z-axis gradient coil. Experimental details are listed in Supplementary Material. Acquisition of 5D HN(CA)CONH, 5D HabCabCONH, and 5D HC(CC-TOCSY)CONH spectra (Kazimierczuk et al. 2009, 2010) and of 3D HNCO auxiliary spectrum was carried out on a Varian NMR System 700 spectrometer equipped with a Performa xyz-PFG with the 5 mm1H/13C/15N room temperature triple resonance probehead. These experiments were performed with a non-uniform sampling of the indirectly detected dimensions. Poisson disk sampling, introducing distance constraints between the time points, was chosen to generate the time schedule. Further, the constant density of the generated time points was transformed to the decaying density according to a Gaussian distribution (σ = 0.5), with regard to the maximum evolution time. Such constrained random sampling scheme was shown to reduce the level of artifacts in the peak vicinity (Kazimierczuk et al. 2008). All the experiments were measured with four scans per increment. The HNCO experiment was acquired with the interscan delay of 1.2 s, with spectral widths set to 12,000 (aq) × 2,500 (15N) × 3,700 (13C’) Hz, and with 484 points measured in the indirect dimensions. Maximal evolution times were adjusted to 100 ms for the 13C’ dimension and to 120 ms for the 15N dimension. The spectrum was acquired in 3 h, which represents 0.43 % of time needed for a conventional experiment with a similar setting. The 5D HN(CA)CONH experiment was performed with the interscan delay set to 1.2 s and spectral widths of 12,000 (aq) × 2,800 (15N) × 3,000 (13C′) × 2,800 (15N) × 6,000 (1H) Hz were used. The maximal evolution times were adjusted to 20 ms for t 1 (1H), 50 ms for the t 2 and t 4 (15N), and 30 ms for t 3 (13C′). A total of 740 points in the indirect dimensions were measured. Experimental time of 20 h fills 0.00034 % of time needed for a similar experiment with conventional settings. The 5D HabCabCONH experiment was run with the interscan delay of 1.2 s and with spectral widths set to 12,000 (aq) × 2,800 (15N) × 3,000 (13C′) × 14,000 (13Caliph) × 4,000 (1Haliph). The maximal evolution times were set to 10 ms for 1Haliph, 7.1 ms for 13Caliph, 30 ms for 13C’, and 50 ms for 15N indirect dimensions. A total of 880 indirect dimension points were measured which represents 0.0002 % points needed for a similar experiment with conventional settings. The 5D HC(CC-TOCSY)CONH experiment was measured with the interscan delay of 1.6 s and with the spectral widths set to 12,000 (aq) × 2,800 (15N) × 3,000 (13C′) × 18,000 (13Caliph) × 8,000 (1Haliph) Hz. A total of 2260 points were measured in the indirect dimensions. The maximal evolution times were adjusted to 10 ms for 1Haliph and 13Caliph, 30 ms for 13C′, and 50 ms for 15N. Total experimental time of 2 days and 14 h constitutes 0.0012 % of duration of a corresponding conventional experiment. Other experimental details are presented in Supplementary Material and the pulse sequences for Varian spectrometers are available from the authors (W.K.) on request.
Data processing
All uniformly sampled data were processed using spectral processing and analysis system NMRPipe/NMRDraw 3.0 (Delaglio et al. 1995). The 3D HNCO spectrum acquired with non-uniform sampling was processed with the Multidimensional Fourier Transform (MFT) algorithm (Kazimierczuk et al. 2006a, b), while the 5D data were processed with the Sparse Multidimensional Fourier Transform (SMFT) algorithm (Kazimierczuk et al. 2009). The processing software is available from the authors (W.K.) on request. The direct dimension was square cosine weighted and zero-filled to 2048 complex points and processed using the standard FFT algorithm. Prior to calculating 2D cross-sections using SMFT, the number of spectral points in ω1 and ω2 dimensions was set to 432 (ω1, 1H) and 512 (ω2, 15N) in the case of 5D HN(CA)CONH, to 256 (ω1, 1H) and 512 (ω2, 13C) in the case of 5D HabCabCONH, and to 256 (ω1, 1H) and 560 (ω2, 13C) in the case of 5D HC(CC-TOCSY)CONH. The remaining frequencies ω3, ω4, and ω5 were fixed to values identified as 13C′, 15N, and 1HN coordinates of individual peaks in the 3D HNCO spectrum, respectively, and SMFT was performed. The assignment of the NMR spectra was performed in a graphical NMR assignment and integration software Sparky 3.115 (T.D. Goddard and D. G. Kneller, University of California, San Francisco, USA).
Results and discussion
As the first stage of the assignment procedure, standard triple-resonance experiments including HNCA, HN(CO)CA, HNCACB, CBCA(CO)NH, and HNCO were applied to assign frequencies of backbone nuclei 1HN, 15N, 13C′, 13Cα, and 13Cβ. The HCCH-TOCSY spectrum was acquired in order to assign the side-chain frequencies of aliphatic residues. A 13C-edited NOESY spectrum was recorded to assign aromatic residues. The process of assignment resulted in a complete list of individual resonance frequencies of nuclei from the structured part of the protein (G2–Q92), with the exception of the carbonyl frequency of D65 (not determined due to the conformational exchange). The assignment was validated by a comparison with the frequency list of a separated N-terminal δ domain (BMRB accession code 16635), obtained within the frame of a previous structural study (Motáčková et al. 2010). The peaks of nuclei from the ordered part in the spectra of the full-length protein nicely overlaid with the corresponding peaks in spectra of the N-terminal construct.
In contrast to the straightforward and efficient assignment of the well-ordered N-terminal domain, the disordered and highly repetitive C-terminal part of the δ sequence (Fig. 1) was found to be extremely difficult to assign. While the standard assignment approach was sufficient to assign many disordered proteins (Peti et al. 2001; Yao et al. 2001; Marion 2006; Mukrasch et al. 2009; Motáčková et al. 2009), it completely failed in the case of the C-terminal domain of δ. Flexibility of the domain together with a very similar chemical environment of all nuclei in the short sequential repeats caused severe peak overlaps. To demonstrate this problem, Fig. 2 shows the 1H,15N-HSQC spectrum of the δ subunit (with assignment obtained as described below) compared with the same spectrum of another partially disordered protein studied in our laboratory, retroviral protease of the murine intracisternal A-type particle. The dimeric 34 kDa protease was 2H, 13C, 15N-labeled and almost completely assigned including a disordered 50 amino acid C-terminal domain, using standard triple-resonance experiments (Motáčková et al. 2009). The 1HN chemical shift dispersion of the disordered C-terminal domain of the δ subunit is lower than that of the retroviral protease: 0.5 ppm versus 0.7 ppm. The 15N chemical shifts of the unstructured regions cover 6.4 and 11.1 ppm for the δ subunit and retroviral protease, respectively, with the most crowded regions spanning 3.4 ppm and 4.8 ppm, respectively. The aliphatic 13C dimension of the 3D triple resonance experiments did not help to resolve the overlapped peaks. As a consequence, a reliable peak-picking was impossible in the 1H,15N-HSQC spectrum of the δ subunit because only 40 signals out of expected 80 could be distinguish.

Amino-acid sequence of δ protein with the unstructured C-terminal domain highlighted in red

1H,15N-HSQC spectra of RNA polymerase δ subunit (A, C, E, F) and of retroviral protease of the murine intracisternal A-type particle (B, D). Entire spectra are shown in Panels A and B, while the central regions are expanded in Panels C and D. Close views of the most crowded areas of the δ subunit spectrum are displayed in Panels E and F. Both spectra were acquired and processed in the same manner and identical scales are used in Panels A and B and in Panels C and D. All assigned signals of backbone amides are labeled, with the exception of L78 of the δ subunit (1H and 15N chemical shifts 4.077 and 115.926 ppm, respectively) and of I15 (1H and 15N chemical shifts 9.955 and 127.403 ppm, respectively), G77 (1H and 15N chemical shifts 6.504 and 106.764 ppm, respectively), and V88 (1H and 15N chemical shifts 6.852 and 108.601 ppm, respectively) of the retroviral protease. Unassigned peaks of the retroviral protease are marked with asterisks
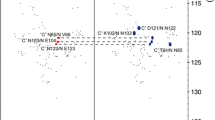
As the next step, a set of 13C-detected CON, CBCACON, and CBCANCO spectra was acquired. The better resolution of carbonyl 13C chemical shifts, exploited in the 13C-detected experiments, is routinely utilized for the assignment of unfolded proteins (Bermel et al. 2006b; Perez et al. 2009; Knoblich et al. 2009). The 13C-detected experiments allowed us to assign carbonyl 13C signals of residues preceding prolines in the well-ordered N-terminal domain, which are not observable in standard 1H-detected spectra. The 2D CON spectrum exhibited higher resolution of the peaks than the 1H,15N-HSQC spectrum (Fig. 3), allowing to distinguish 55 signals out of 80. However, sequential connectivities still could not be established due to the lack of resolution in the aliphatic 13C dimension.

13C-detected CON spectrum of the RNA polymerase δ subunit (A), with the central region enlarged in Panel B
The results listed above clearly showed that a completely different strategy is needed to assign disordered proteins exhibiting such degree of peak overlaps as observed for the δ subunit. In particular, NMR techniques providing extreme resolution are necessary to distinguish residues with very small chemical shift differences in several dimensions. A methodology promising such resolution enhancement has been recently developed in one of our laboratories (Kazimierczuk et al. 2009) bud so far tested only on well-structured proteins (human ubiquitin, 76 amino acids long, CsPin interacting with NIMA-kinase from Cenarcheaum symbiosum, 96 amino acids long, and maltose binding protein, 341 amino acids long). Quality of the spectra obtained for the ordered proteins encouraged us to employ this approach in the novel assignment strategy reported in this paper.
The proposed and successfully applied assignment strategy is based on a set of three 5D experiments utilizing non-uniform sampling in indirectly detected dimensions: HN(CA)CONH, HabCabCONH, and HC(CC-TOCSY)CONH. As the technical details has been published elsewhere (Kazimierczuk et al. 2009, 2010), only the basic principles will be reviewed below.
Ultra-high resolution can be achieved by increasing the number of dimensions and maximal evolution times in each dimension. Two potential drawbacks of such an approach must be considered. First, the long polarization transfer pathways of high-dimensional experiments make the transverse relaxation more pronounced. Fortunately, favorable relaxation properties of the disordered proteins usually keep the resulting signal loss in acceptable ranges. Second, the required overall experimental time would largely exceed any practical limit if all indirect time dimensions were uniformly sampled in ranges given by the desired resolution on one side and by the Nyquist theorem on the other side. Therefore, only a sparse data set can be recorded in order to keep the measurement time reasonable. In our case, Poisson disk sampling, weighted to fulfill the condition of time-decaying density of points (Kazimierczuk et al. 2008), was used. Such sampling scheme is optimal with respect to the artifacts related to the incomplete size of the data set (Kazimierczuk et al. 2008; Dippé and Wold 1985) and can be easily implemented on any NMR spectrometer. The non-uniform sampling dramatically shortened the time of data collection to 0.0002–0.0012% of the time needed to acquire standard spectra of the same resolution.
Various sophisticated methods have been proposed in the literature to process NMR data sets acquired in a non-conventional manner (Kupče and Freeman, 2008; Coggins et al. 2005; Ding and Gronenborn 2002; Kim and Szyperski 2003; Koźmiński and Zhukov 2003; Malmodin and Billeter 2004; Coggins and Zhou 2006; Kazimierczuk et al. 2006a; Marsh et al. 2006; Coggins and Zhou 2007; Rovnyak et al. 2004; Kupče and Freeman 2003; Tugarinov et al. 2005; Atreya et al. 2005; Hiller et al. 2007). The sampling scheme employed in this study allows to follow a procedure closely resembling the conventional NMR data processing, based on a slightly modified Fourier transform algorithm, Multidimensional Fourier Transform (MFT) (Kazimierczuk et al. 2006a, b). In principle, complete 5D spectra can be obtained, but the size of the full 5D matrix would require terabytes of disk space. Therefore, the amount of data was reduced by extracting 2D cross-sections from the 5D spectra during the processing (Kazimierczuk et al. 2009; Zawadzka-Kazimierczuk et al. 2010). The procedure started with acquisition of an auxiliary 3D HNCO spectrum. The spectrum was recorded with non-uniform sampling in order to achieve a very high resolution. Each 5D experiment used in this work can be considered as an expansion of the HNCO correlation (squares in Fig. 4). While the HNCO correlation does not bring any new structural information, it identifies a residue whose side-chain or sequential neighbour is searched for. Therefore, the three coordinates of the HNCO peaks can be used to fix three frequencies when performing MFT on the 5D data. Equation 1 shows the procedure of calculation of k-th 2D cross-section from the 5D data, revealing correlations of frequencies ω1 and ω2 with the fixed frequencies ω k3 , ω k4 , and ω k5 (13C′, 15N, and 1HN resonances, respectively) (Kazimierczuk et al. 2010).
where M denotes the overall number of indirectly detected points, j 1-5 are the imaginary units for commutative Clifford algebra of the 5-th order, and t m i is the i-th dimension coordinate of the m-th time point.

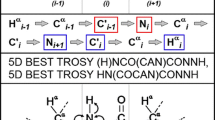
Correlations utilized in the 5D experiments. Correlations of the nuclei in circles are observed in 2D cross-sections extracted from the 5D spectrum at chemical shifts of the nuclei in square boxes. Blue, green, and red circles indicate the correlations observed in the HN(CA)CONH, HabCabCONH, and HC(CC-TOCSY)CONH spectra, respectively
Correlations observed in the 5D experiments used in this study are shown in Fig. 4. The 5D HN(CA)CONH experiment is designed for the sequential assignment of backbone amide resonances based on correlations between amide 1HN and 15N nuclei of adjacent residues. As a result, two peaks are visible in each 2D cross-section extracted from the 5D spectrum at the 1HN, 15N, and 13C’ frequencies of HNCO peaks. One of them corresponds to the cross-peak at 1HN, 15N frequencies of the given residue while the second one shows the 1HN, 15N frequencies of the preceding residue (Fig. 5). In this manner, we were able to easily assign several fragments of the sequence consisting of 5–20 amino acids. The assigned fragments could not be connected to each other due to ambiguities in the cross-sections. In several cases, two residues appeared to have almost identical chemical shifts of their 1HN, 15N, and 13C’ nuclei and therefore three peaks appeared in the cross-sections: an overlapped peak of two intraresidual correlations and two resolved peaks of interresidual correlations, representing two possible sequential connectivities.

Central region of uniformly sampled 1H,15N-HSQC spectrum (left) and the corresponding region of a 2D cross-section extracted from the 5D HN(CA)CONH spectrum (right). The 2D cross-section was obtained by fixing frequencies in ω3, ω4, and ω5 dimensions to the values of chemical shifts of 13C’ of I157, 15N of I158, and 1HN of I158, respectively. Peaks corresponding to the sequential and intraresidual correlations are displayed in black and red, respectively. The contour threshold was set to 50 % of the height of the I158 H-N peak in both spectra
In order to identify the fragments, we used the 5D HabCabCONH experiment which reveals chemical shifts of 1Hα, 1Hβ, 13Cα, and 13Cβ nuclei of the given residue in each cross-section (Fig. 6A) and thus allows to partially classify the type of the amino acid based on the typical 13C chemical shift values. Utilizing this supporting knowledge, all ambiguities in the cross-sections were resolved and the sequential assignment of the δ subunit was completed.

Examples of 2D cross-sections extracted from the 5D HabCabCONH (Panel A) and HC(CC-TOCSY)CONH (Panel B) spectra at chemical shifts of I1581HN, I15815N, and I15713C’
Side-chains of amino acid residues were assigned using the 5D HC(CC-TOCSY)CONH experiment. Each 2D cross-section displays all one-bond correlations between 13C and 1H atoms from the side-chain spin-system of the given residue (Fig. 6B). The cross-peaks in the cross-sections were assigned employing typical chemical shift values obtained from the BMRB database (http://www.bmrb.wisc.edu). Such assignment is facilitated by the fact that disordered polypeptide chains exhibit much smaller deviations from the average chemical shift values than well-structured proteins. The frequencies of 1Hα, 1Hβ, 13Cα, and 13Cβ were cross-validated by the comparison with the results of the 5D HabCabCONH experiment.
In summary, the presented strategy provided assignment of 100% backbone and side-chain resonances of the disordered C-terminal domain of the δ subunit of RNA polymerase from B. subtilis. This is the first report demonstrating that the employed 5D non-uniformly sampled experiments allow efficient and complete assignment of disordered proteins with particularly high signal overlap. It should be noted that the overall experimental time was comparable to the time needed to record conventional 3D triple resonance experiments and that the assignment process was easier and faster than assigning a well-folded protein of a similar size using the standard triple resonance spectra.
Analysis of the obtained chemical shifts revealed why both high dimensionality and resolution enhancement, achieved with the help of non-uniform sampling in the indirect dimensions, were crucial for the successful assignment. Variations in the 13Cα and 13Cβ chemical shifts of amino acids of the same type were particularly low (Figs. S1–S2 in Supplementary Material). We observed up to 17 residues within a 13Cα chemical shift range of ± 0.2 ppm, which is the second highest number among 27 proteins described as disordered, unfolded, or unstructured in their BMRB entry titles. It explains why the 5D HN(CA)CONH experiment was needed for the sequential assignment. Differences in 1HN and 15N chemical shifts of adjacent residues, obtained in a similar manner as in the 6D APSY HNCACONH experiment (Fiorito et al. 2006), allowed to distinguish residues with identical 13Cα and 13Cβ frequencies. 3D triple resonance experiments, relying on the 13Cα and 13Cβ chemical shifts, completely failed even in versions with the resolution enhanced by non-uniform sampling (data not shown), similar to those proposed by Pannetier et al. (Pannetier et al. 2007).
It should be emphasized that dispersion of the 1HN and 15N chemical shifts of the δ subunit was also very poor, even when compared to other disordered proteins (Fig. 2 and Figs. S3–S12 in Supplementary Material), and required a very high spectra resolution. Especially, resonances of amino acids of the same type in the same sequential repeats were almost identical. For example, resonances of four leucines L110, L112, L123, L151 almost completely coincide in the 1HN and 15N dimensions, as demonstrated in Fig. 2F and Fig. S1C in Supplementary Material. Acidic residues (Fig. 2E and Figs. S1A,B in Supplementary Material) may serve as another example of complete peak overlap. However, our approach of analysing cross-sections of full-dimensional 5D HN(CA)CONH spectra allowed us to overcome the clustering of resonance frequencies.
It should be noted that approaches eliminating the problem of degeneracy of aliphatic 13C resonances, not requiring high dimensionality and non-uniform sampling, were reported in the literature. For example, Mukrasch et al. ( 2009) assigned a 441-residue Tau protein using 3D (HA)CANNH and HNN spectra recorded at 900 MHz on the full-length protein and three shorter polypeptides with partially overlapping sequences. However, the degeneration of the 13Cα connectivities was caused by a large number of amino acids of the Tau protein, while the dispersion of 1H and 15N resonances was better than in our case.
The methodology utilized in this study does not represent the only way how to acquire high-dimensional spectra. Several groups have reported successful implementation of high-dimensional experiments (5D–7D) in combination with sophisticated methods of data processing to perform a sequential assignment of disordered proteins (Atreya et al. 2005; Hiller et al. 2007). These approaches are based either on the reduced dimensionality spectroscopy exploiting the aliphatic carbon frequencies (13Cα, 13Cβ) for sequential assignment (Atreya et al. 2005) or on the projection spectroscopy utilizing the 15N and 1HN chemical shifts for sequential assignment (Hiller et al. 2007). Such techniques are very fast and elegant. The reason why we propose a more traditional approach for our application (assignment of disordered proteins with severe peak overlaps) is its transparency. As true 5D spectra are obtained, it is always possible to visually inspect problematic regions of the spectra and correct the peak picking if it was affected by signal overlaps. At the same time, the analyzed cross-sections of the 5D spectra are extremely simple (Figs. 5, 6) and offer direct interpretation, while the experimental time is maintained on the similar time scale as for approaches mentioned above.
Conclusions
We present a novel strategy which allows a complete assignment of unstructured portions of proteins, especially in the cases where this task is strongly complicated by frequent repetitions in the sequence. The obtained frequency assignment is the most important source of information on residual structural features of disordered proteins. The complete list of chemical shifts of the full-length δ protein was deposited in the BMRB database under the accession code 16912.
References
Atreya H, Eletsky A, Szyperski T (2005) Resonance assignment of proteins with high shift degeneracy based on 5D spectral information encoded in G(2)FT NMR experiments. J Am Chem Soc 127(13):4554–4555
Bermel W, Bertini I, Felli I, Piccioli M, Pierattelli R (2006a) C-13-detected protonless NMR spectroscopy of proteins in solution. Prog Nucl Mag Res Sp 48(1):25–45
Bermel W, Bertini I, Felli I, Lee Y, Luchinat C, Pierattelli R (2006b) Protonless NMR experiments for sequence-specific assignment of backbone nuclei in unfolded proteins. J Am Chem Soc 128(12):3918–3919
Bussell R, Eliezer D (2001) Residual structure and dynamics in Parkinson’s disease-associated mutants of alpha-synuclein. J Biol Chem 276(49):45,996–46,003
Clark S, Losick R, Pero J (1974) New RNA-polymerase from Bacillus-subtilis infected with phage PBS2. Nature 252(5478):21–24
Coggins B, Zhou P (2006) Polar Fourier transforms of radially sampled NMR data. J Magn Reson 182(1):84–95
Coggins B, Zhou P (2007) Sampling of the NMR time domain along concentric rings. J Magn Reson 184(2):207–221
Coggins B, Venters R, Zhou P (2005) Filtered backprojection for the reconstruction of a high-resolution (4,2)D CH3-NHNOESY spectrum on a 29 kDa protein. J Am Chem Soc 127(33):11,562–11,563
Delaglio F, Grzesiek S, Vuister G, Zhu G, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6(3):277–293
DeSaro F, Woody A, Helmann J (1995) Structural-analysis of the Bacillus-subtilis delta-factor—a polyanion which displaces RNA from RNA-polymerase. J Mol Biol 252(2):189–202
Ding K, Gronenborn A (2002) Novel 2D triple-resonance NMR experiments for sequential resonance assignments of proteins. J Magn Reson 156(2):262–268
Dippé M, Wold E (1985) Antialiasing through stochastic sampling, ACM SIGGRAPH. Comput Graphics 19:69–78
Dunker AK, Silman I, Uversky VN, Sussman JL (2008) Function and structure of inherently disordered proteins. Curr Opin Struct Biol 18(6):756–764
Dyson H, Wright P (2004) Unfolded proteins and protein folding studied by NMR. Chem Rev 104(8):3607–3622
Dyson H, Wright P (2005) Intrinsically unstructured proteins and their functions. Nature Rev Mol Cell Biol 6(3):197–208
Eliezer D (2007) Characterizing residual structure in disordered protein states using nuclear magnetic resonance. Methods Mol Biol 350:49–67
Eliezer D (2009) Biophysical characterization of intrinsically disordered proteins. Curr Opin Struct Biol 19(1):23–30
Fink A (2005) Natively unfolded proteins. Curr Opin Struct Biol 15(1):35–41
Fiorito F, Hiller S, Wider G, Wüthrich K (2006) Automated resonance assignment of proteins: 6D APSY-NMR. J Biomol NMR 35(1):27–37
Hiller S, Wasmer C, Wider G, Wüthrich K (2007) Sequence-specific resonance assignment of soluble nonglobular proteins by 7D APSY-NMR spectroscopy. J Am Chem Soc 129(35): 10,823–10,828
Kazimierczuk K, Koźmiński W, Zhukov I (2006a) Two-dimensional Fourier transform of arbitrarily sampled NMR data sets. J Magn Reson 179(2):323–328
Kazimierczuk K, Zawadzka A, Koźmiński W, Zhukov I (2006b) Random sampling of evolution time space and Fourier transform processing. J Biomol NMR 36(3):157–168
Kazimierczuk K, Zawadzka A, Koźmiński W (2008) Optimization of random time domain sampling in multidimensional NMR. J Magn Reson 192(1):123–130
Kazimierczuk K, Zawadzka A, Koźmiński W (2009) Narrow peaks and high dimensionalities: exploiting the advantages of random sampling. J Magn Reson 197(2):219–228
Kazimierczuk K, Zawadzka-Kazimierczuk A, Koźmiński W (2010) Non-uniform frequency domain for optimal exploitation of non-uniform sampling. J Magn Reson 205(2):286–292
Kim S, Szyperski T (2003) GFT NMR, a new approach to rapidly obtain precise high-dimensional NMR spectral information. J Am Chem Soc 125(5):1385–1393
Knoblich K, Whittaker S, Ludwig C, Michiels P, Jiang T, Schaffhausen B, Guenther U (2009) Backbone assignment of the N-terminal polyomavirus large T antigen. Biomol NMR Assignments 3(1):119–123
Koźmiński W, Zhukov I (2003) Multiple quadrature detection in reduced dimensionality experiments. J Biomol NMR 26(2):157–166
Kupče E, Freeman R (2003) Projection-reconstruction of three-dimensional NMR spectra. J Am Chem Soc 125(46):13,958–13,959
Kupče E, Freeman R (2008) Hyperdimensional NMR spectroscopy. Prog Nucl Mag Res Sp 52(1):22–30
Malmodin D, Billeter M (2004) Robust and versatile interpretation of spectra with coupled evolution periods using multi-way desomposition. Magn Reson Chem 44(S1):185–195
Marion D (2006) Processing of ND NMR spectra sampled in polar coordinates: a simple Fourier transform instead of a reconstruction. J Biomol NMR 36(1):45–54
Marsh J, Singh V, Jia Z, Forman-Kay J (2006) Sensitivity of secondary structure propensities to sequence differences between alpha- and gamma-synuclein: implications for fibrillation. Prot Sci 15(12):2795–2804
Motáčková V, Kubíčková M, Kožíšek M, Grantz-Šašková K, Švec M, Žídek L, Sklenář V (2009) Backbone H-1, C-13, and N-15 NMR assignment for the inactive form of the retroviral protease of the murine intracisternal A-type particle, inMIA-14 PR. Biomol NMR Assign 3(2):261–264
Motáčková V, Šanderová H, Žídek L, Nováček J, Padrta P, Švenková A, Korelusová J, Jonák J, Krásný L, Sklenář V (2010) Solution structure of the N-terminal domain of Bacillus subtilis delta subunit of RNA polymerase and its classification based on structural homologs. Proteins Struct Funct Bioinf 78(7):1807–1810
Mukrasch M, Bibow S, Korukottu J, Jeganathan S, Biernat J, Griesinger C, Mandelkow E, Zweckstetter M (2009) Structural polymorphism of 441-residue Tau at single residue resolution. PLoS Biol 7(2):399–414
Pannetier N, Houben K, Blanchard L, Marion D (2007) Optimized 3D-NMR sampling for resonance assignment of partially unfolded proteins. J Magn Reson 186(1):142–149
Perez Y, Gairi M, Pons M, Bernado P (2009) Structural characterization of the natively unfolded N-terminal domain of human c-Src kinase: insights into the role of phosphorylation of the unique domain. J Mol Biol 391(1):136–148
Peti W, Smith L, Redfield C, Schwalbe H (2001) Chemical shifts in denatured proteins: resonance assignments for denatured ubiquitin and comparisons with other denatured proteins. J Biomol NMR 19(2):153–165
Rovnyak D, Frueh D, Sastry M, Sun Z, Stern A, Hoch J, Wagner G (2004) Accelerated acquisition of high resolution triple-resonance spectra using non-uniform sampling and maximum entropy reconstruction. J Magn Reson 170(1):15–21
Sattler M, Schmidt P, Schleucher J, Schedletzky O, Glaser S, Griesinger C (1995) Novel pulse sequence with sensitivity enhancement fir in-phase coherence transfer employing pulsed-field gradients. J Magn Reson B 108(3):235–242
Sattler M, Schleucher J, Griesinger C (1999) Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog Nucl Mag Res Sp 34(2):93–158
Tugarinov V, Kay L, Ibraghimov I, Orekhov V (2005) High-resolution four-dimensional H-1-C-13 NOE spectroscopy using methyl-TROSY, sparse data acquisition, and multidimensional decomposition. J Am Chem Soc 127(8):2767–2775
Ward J, Sodhi J, McGuffin L, Buxton B, Jones D (2004) Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol 337(3):635–645
Yao J, Chung J, Eliezer D, Wright P, Dyson H (2001) NMR structural and dynamic characterization of the acid-unfolded state of apomyoglobin provides insights into the early events in protein folding. Biochemistry 40(12):3561–3571
Zawadzka-Kazimierczuk A, Kazimierczuk K, Koźmiński W (2010) A set of 4D NMR experiments of enhanced resolution for easy resonance assignment in proteins. J Magn Reson 202(1):109–116
Acknowledgments
This work was supported by the Grants of the Ministry of Education, Youth and Physical Culture of the Czech Republic MSM0021622413 (LŽ), LC06030 (VS), and 2B06065 (LK, HŠ), by the Grants 204/09/0583 (VM, JN, LŽ, LK) and 301/09/H004 (VM) from Czech Science Foundation. Anna Zawadzka-Kazimierczuk thanks the Foundation for Polish Science for supporting her with the MPD Programme that was cofinanced by the EU European Regional Development Fund. Financial support by the Access to Research Infrastructures activity in the 7th Framework Programme of the EC (Contract 228461, EAST-NMR) for conducting the research is gratefully acknowledged by Libor Krásný.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Motáčková, V., Nováček, J., Zawadzka-Kazimierczuk, A. et al. Strategy for complete NMR assignment of disordered proteins with highly repetitive sequences based on resolution-enhanced 5D experiments. J Biomol NMR 48, 169–177 (2010). https://doi.org/10.1007/s10858-010-9447-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-010-9447-3