
Abstract
A new computer program, called SHIFTX2, is described which is capable of rapidly and accurately calculating diamagnetic 1H, 13C and 15N chemical shifts from protein coordinate data. Compared to its predecessor (SHIFTX) and to other existing protein chemical shift prediction programs, SHIFTX2 is substantially more accurate (up to 26% better by correlation coefficient with an RMS error that is up to 3.3× smaller) than the next best performing program. It also provides significantly more coverage (up to 10% more), is significantly faster (up to 8.5×) and capable of calculating a wider variety of backbone and side chain chemical shifts (up to 6×) than many other shift predictors. In particular, SHIFTX2 is able to attain correlation coefficients between experimentally observed and predicted backbone chemical shifts of 0.9800 (15N), 0.9959 (13Cα), 0.9992 (13Cβ), 0.9676 (13C′), 0.9714 (1HN), 0.9744 (1Hα) and RMS errors of 1.1169, 0.4412, 0.5163, 0.5330, 0.1711, and 0.1231 ppm, respectively. The correlation between SHIFTX2’s predicted and observed side chain chemical shifts is 0.9787 (13C) and 0.9482 (1H) with RMS errors of 0.9754 and 0.1723 ppm, respectively. SHIFTX2 is able to achieve such a high level of accuracy by using a large, high quality database of training proteins (>190), by utilizing advanced machine learning techniques, by incorporating many more features (χ2 and χ3 angles, solvent accessibility, H-bond geometry, pH, temperature), and by combining sequence-based with structure-based chemical shift prediction techniques. With this substantial improvement in accuracy we believe that SHIFTX2 will open the door to many long-anticipated applications of chemical shift prediction to protein structure determination, refinement and validation. SHIFTX2 is available both as a standalone program and as a web server (http://www.shiftx2.ca).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chemical shifts are often called the mileposts of NMR spectroscopy. They are easily measured, highly reproducible spectroscopic parameters that can be readily used to identify, annotate or locate individual atoms. Chemical shifts also contain a considerable amount of information pertaining to a molecule’s covalent and non-covalent structure. Indeed, their sensitivity to the type and character of neighbouring atoms has long made chemical shifts a favourite tool of organic synthetic chemists to help decipher the structure of small molecules. Likewise, their sensitivity to a variety of important protein structural features has made chemical shifts equally valuable to protein chemists and biomolecular NMR spectroscopists. In fact, protein chemical shifts can be used to identify secondary structures (Pastore and Saudek 1990; Williamson 1990; Wishart et al. 1991), estimate backbone torsion angles (Spera and Bax 1991; Wishart and Nip 1998), determine the location of aromatic rings (Perkins and Dwek 1980; Osapay and Case 1991), assess cysteine oxidation states (Sharma and Rajarathnam 2000), estimate solvent exposure (Vranken and Rieping 2009) or measure backbone flexibility (Berjanskii and Wishart 2005).
While the extraction of approximate structural features from protein chemical shifts has become almost routine, the extraction of precise structural features is not. In fact, the inherently complex geometric, dynamic and electronic dependencies of protein chemical shifts has made the calculation of precise chemical shifts from protein structures or the calculation of precise structures from chemical shifts a significant challenge for more than 40 years (Sternlicht and Wilson 1967). For the specific task of calculating chemical shifts from structure (i.e. protein chemical shift prediction), at least two different routes have emerged. One is based on using sequence/structure alignment against chemical shift databases (i.e. sequence-based methods) and the other is based on directly calculating chemical shifts from atomic coordinates (i.e. structure-based methods).
Sequence-based methods take advantage of the continuous growth of today’s protein chemical shift databases. The idea behind predicting shifts via sequence homology lies in the simple observation that similar protein sequences share similar structures, which in turn, share similar chemical shifts (Gronwald et al. 1998; Potts and Chazin 1998; Wishart et al. 1997). The first implementation of this concept appeared in 1997 in a program called SHIFTY (Wishart et al. 1997). This relatively simple program takes an input sequence and uses sequence alignment against the BRMB (Seavey et al. 1991) or other chemical shift databases (Zhang et al. 2003) to identify a matching homologue. Once found, the complete set of homologous shifts of the matching protein is “assigned” to the query protein using a set of empirically defined rules. Chemical shifts predicted via sequence homology can be very accurate if a good homologue is found (Wishart and Nip 1998; Wishart et al. 1997). A key advantage to sequence-based methods is that as the chemical shift database (e.g. BMRB) expands, the predictions tend to improve as the odds of finding a suitable sequence homologue tends to increase. A key disadvantage of sequence-based approaches is that no predictions will be performed if no sequence homologue can be found.
A more recent extension to standard sequence-based shift prediction methods is SPARTA (Shen and Bax 2007). Rather than looking for global similarity, as is done with SHIFTY, SPARTA assesses similarity over a much smaller sequence range (just three residues). To predict chemical shifts for a given query protein, each tripeptide in the query structure is searched against the SPARTA tripeptide database and scored on the basis of its sequence and torsion angle (ϕ, ψ, and χ1) similarity. This information is combined with additional structural information (H-bond effects and ring current effects) to calculate a final set of chemical shifts. SPARTA and its successor SPARTA+ (Shen and Bax 2010), have proven to be remarkably accurate, especially for predicting 13C and 15N backbone shifts.
In addition to these sequence-based methods, a substantial number of structure-based methods have emerged over the past 10 years. These include SHIFTCALC (Iwadate et al. 1999), SHIFTS (Moon and Case 2007; Xu and Case 2001), CheShift (Vila et al. 2009), SHIFTX (Neal et al. 2003), PROSHIFT (Meiler 2003) and CamShift (Kohlhoff et al. 2009). All of these programs calculate chemical shifts using only protein coordinates as input. Some methods, such as SHIFTCALC and SHIFTX use empirically derived chemical shift hypersurfaces or related structure/shift tables to translate coordinate data into chemical shifts. Others, such as CheShift and SHIFTS use quantum mechanical models to generate their atom-specific chemical shift hypersurfaces. Still others, such as PROSHIFT, use neural network methods (i.e. machine learning) to predict protein chemical shifts from coordinate data. CamShift employs an ingenious approach to calculate chemical shifts using a set of parameterized distance equations. This makes CamShift’s chemical shift functions both rapid to calculate and easily differentiable. Having a differentiable function is particularly useful for chemical shift refinement via conjugate gradient minimization or molecular dynamics.
All the aforementioned methods are capable of predicting protein chemical shifts with reasonably high accuracy. As a rule, SHIFTX, SHIFTY, CamShift and SPARTA generally perform better than PROSHIFT, SHIFTS, SHIFTCALC and CheSHIFT. Nevertheless, it appears that sequence-based approaches, under certain circumstances, perform better than structure-based approaches, and vice versa. This suggests that by combining the strengths of both approaches, it may be possible to produce a hybrid method that exceeds the performance of any single sequence-based or structure-based method. Here we describe just such a hybrid method, called SHIFTX2. In particular, SHIFTX2 combines many of the structure-based concepts originally introduced in SHIFTX (Neal et al. 2003) with the sequence-based concepts introduced with SHIFTY (Wishart et al. 1997). By making use of a much larger and higher quality training set in combination with a number of other enhancements (using advanced machine learning techniques, employing more structural parameters) the performance of the structure-based component (now called SHIFTX+) was substantially improved. Likewise by using an improved sequence/shift database and by making use of local, instead of global, sequence alignment techniques we were also able to make substantial improvements to the performance of the sequence-based component (now called SHIFTY+). By carefully combining the algorithms for SHIFTX+ and SHIFTY+ we were able to create the hybrid program called SHIFTX2.
As shown below, SHIFTX2 is substantially more accurate (up to 26% better by correlation coefficient and an RMS error that is up to 3.3× smaller) than the next best performing program. It also provides significantly more coverage (up to 10% more), is significantly faster (up to 8.5×) and capable of calculating a wider variety of backbone and side chain chemical shifts (up to 6×) than many other shift predictors. In particular, SHIFTX2 is able to attain correlation coefficients between experimentally observed and predicted backbone chemical shifts of 0.9800 (15N), 0.9959 (13Cα), 0.9992 (13Cβ), 0.9676 (13C′), 0.9714 (1HN), 0.9744 (1Hα) and RMS errors of 1.1169, 0.4412, 0.5163, 0.5330, 0.1711, and 0.1231 ppm, respectively. The correlation coefficients between SHIFTX2’s predicted and observed side chain chemical shifts are 0.9787 (13C) and 0.9482 (1H) with RMS errors of 0.9754 and 0.1723 ppm, respectively. Additional details about SHIFTX2’s algorithms, its training process, its testing protocols and its potential applications is provided in the following pages.
Methods
Key to the development of accurate chemical shift predictors is the creation of high quality chemical shift databases. For sequence-based methods it is necessary to develop a large and accurate database of protein sequences and properly referenced protein assignments. For structure-based methods it is critical to develop a large and accurate database of protein structures with correspondingly accurate and comprehensive chemical shift assignments. In developing the database for our sequence-based method (SHIFTY+) we used the chemical shift assignments from RefDB (Zhang et al. 2003). RefDB, which is updated weekly, currently contains 1903 re-referenced protein assignments that are automatically extracted and processed from the BioMagResBank (Seavey et al. 1991).
In constructing the database for our structure-based method (SHIFTX+) we compiled a preliminary collection of ~300 candidate proteins from a number of sources, including RefDB (Zhang et al. 2003), the SPARTA training set17 and the SHIFTX training set (Neal et al. 2003). This dataset was filtered by selecting only those proteins that had X-ray structures with a resolution <2.1 Å, that were largely monomeric, that were free of bound DNA, RNA or large cofactors and that had mostly (>90%) sequentially complete 1H, 13C and/or 15N assignments. Note that in compiling this database, X-ray structures were given preference over NMR structures. This is because it is widely acknowledged that most NMR structures do not achieve the coordinate accuracy or precision of high quality X-ray structures (Andrec et al. 2007; Berjanskii et al. 2010; Laskowski et al. 1996; Shen and Bax 2007). This collection of ~250 high resolution X-ray structures was then analyzed for structural defects using a number of structure validation programs including VADAR (Willard et al. 2003), PROSA (Wiederstein and Sippl 2007), and WHAT_CHECK (Hooft et al. 1996). A separate program called RefDens (Ginzinger et al. 2010) was used to assess the quality of the protein side chains in each model. Several dozen structures were subsequently excluded due to their poor coordinate geometry or obvious structural defects.
For the remaining structures, we manually matched each structure with their observed chemical shift record from the BioMagResBank (Seavey et al. 1991). SHIFTCOR (Zhang et al. 2003) was used to identify potential chemical shift referencing problems and to re-reference all observed chemical shifts to the IUPAC standard—DSS (2,2-dimethyl-2-silapentane-5-sulfonic acid) (Wishart et al. 1995b). PANAV (Wang et al. 2010), CheckShift (Ginzinger et al. 2009) and SHIFTX were also used to check the quality of the protein chemical shift assignments and to identify certain types of gross assignment errors (i.e. “flipped” assignments from folded spectra). Within the accepted set of structures and assignments we further excluded certain chemical shifts from the dataset that seemed to be extreme outliers (beyond four standard deviations) based on the expected shifts of their atom type, residue type or observed secondary structure. These outliers were identified by CheckShift (Ginzinger et al. 2009) and PANAV (Wang et al. 2010). Finally all of the X-ray structures were “protonated” (i.e. H atoms added) using the program called REDUCE (Word et al. 1999). Consequently, the final training dataset consisted of 197 high resolution and high-quality protein structures (with computationally added hydrogen atoms) which had a total of 140,518 re-referenced backbone chemical shifts and 66,385 re-referenced side chain chemical shifts. A list of the training set’s protein names along with their BMRB accession numbers and PDB identifiers is provided in Table S1. The complete training data set (coordinates and assignments) is downloadable from the SHIFTX2 website.
In addition to this large training set, a separate “testing” dataset was assembled to assess the performance of both SHIFTX2 and other chemical shift prediction programs. This test set was constructed using the same criteria described above, but with the requirement that the proteins could not already be in the training or testing sets used by other programs (SHIFTX2, SHIFTX, SPARTA, CamShift). This was done to reduce any potential performance bias towards a single prediction program. The final testing dataset consisted of 61 high resolution protein structures corresponding to 47,514 re-referenced backbone chemical shifts and 24,933 re-referenced side chain chemical shifts. A list of the test set’s protein names along with their BMRB accession numbers and PDB identifiers is provided in Table S2. The complete testing data set (coordinates and assignments) is also downloadable from the SHIFTX2 website.
To develop our structure-based shift prediction algorithm (SHIFTX+) each protein structure in the training data set was further processed by VADAR (Willard et al. 2003), SHIFTX (Neal et al. 2003), PROSESS (Berjanskii et al. 2010) and other in-house programs. These programs calculate dozens of structural features from protein coordinate data, including backbone torsion angles, side chain torsion angles, hydrogen bond energies, hydrogen bond angles, hydrogen bond lengths, solvent exposure, secondary structure, etc. In addition to these structural features, other features pertaining to the pH, temperature and solvents were extracted from each protein’s BioMagResBank file. Likewise, experimentally derived random coil chemical shifts (Wishart et al. 1995a) and nearest-neighbour sequence information were also used as input features. In total, 97 atom-specific, residue-specific and protein-specific data features were compiled.
From this initial set of features, we applied machine-learning methods to develop a multiple-regression model that predicts protein chemical shifts from protein coordinate data. A variety of popular machine learning methods were tested including Support Vector Regression (SVR), Support Vector Machine (SVM), M5P/M5Rules, Artificial Neural Networks (ANNs), Bagging and others. These were evaluated using the WEKA suite of machine learning tools (Frank 2004; Hall et al. 2009). We found that while several machine learning algorithms gave reasonable prediction accuracy, ensemble methods usually achieved the best prediction performance. In particular, we found that when we combined several machine learning methods together, somewhat higher prediction accuracy could be achieved. These ensemble methods exploit the advantages of different machine learning algorithms (“base learners”) by combining them to build a more accurate model. There are two popular ensemble methods: Bagging (Breiman 1996) and Boosting (Schapire 1990) or its variants (e.g. AdaBoost) (Freund and Schapire 1997; Kotsiantis 2007; Opitz and Maclin 1999). Bagging trains “base learners” from a random sample (with replacement) of the original training dataset and then averages the predictions from all the base learners to make a final prediction. In contrast, Boosting trains subsequent base learners on the mistakes of the previous base learner. Boosting will only use the next base learner if the previous base learners are uncertain about their predictions. In SHIFTX+, we combined Boosting with Bagging. In particular, SHIFTX+ uses an additive regression (a Boosting method) model which employs a series of Bagging learners as its base learner; each Bagging learner uses a series of regression trees (i.e. the REPTree method) as its base learners (see Fig. 1). Therefore SHIFTX+’s architecture consists of six models for predicting backbone shifts and 34 models for predicting side chain shifts. The total number of side chain models used by SHIFTX+ was dictated by the number of shifts available for training (a minimum of 100). Too few shifts were available for 9 side chain 15N atoms, eight 1H atoms (HD21, HD22, HE21, HE22, HH11, HH12, HH21 and HH22) and four carbon atoms (CE3, CH2, CZ2, CZ3).

A flow chart explaining the design of the SHIFTX2 program
Once the optimal machine learning method was identified, SHIFTX+ was then further refined through a process known as feature selection. In machine learning, a high quality feature set is particularly important for improving the accuracy of any given predictor. Generally speaking, optimal accuracy may only be obtained by retaining the most important features. To select the best input features, we initially used as many features as possible to train our predictor. We then progressively examined each feature and retained it only if the exclusion of such a feature decreased the prediction accuracy of the model. This feature selection process was repeated several times using different orderings of input features. From this initial set of 97 features, our feature selection process reduced this list to a final set of 63 “useful” features. These features are listed in Table S3 (which is also available on the SHIFTX2 website). The performance of the final version of SHIFTX+ was assessed against both its training set (via tenfold cross-validation) and the testing dataset. This was done to determine the robustness of the predictor and to check if any over-training had occurred.
As noted earlier, SHIFTX2 is composed of two components, a structure-based component (SHIFTX+) and a sequence-based component (SHIFTY+). SHIFTY+ is essentially an enhanced version of SHIFTY (Wishart et al. 1997). Both SHIFTY and SHIFTY+ predict 1H, 13C and 15N chemical shifts based on sequence matching and alignment of a query protein against a database of previously assigned proteins (RefDB or BMRB). Sufficiently high scoring matches (>40% sequence identity) are aligned together and the chemical shifts of the database protein(s) are transferred to the chemical shifts of the query protein using appropriate residue-specific corrections. In developing SHIFTY+, a number of improvements were made including the use of BLAST (Altschul et al. 1990) to identify sequence matches instead of the slower Needleman-Wunsch algorithm, the expansion of the chemical shift database by a factor 27.5% to include 1,903 assigned proteins, the correction of numerous chemical shift referencing errors in the database (via CheckShift and SHIFTCOR), and the elimination of erroneous or questionable assignments among the reference database’s collection of shifts (via PANAV and CheckShift).
Sequence-based methods tend to outperform structure-based methods, especially when a good homologue is found (Wishart and Nip 1998; Wishart et al. 1997). However, if no suitable homologue exists sequence-based methods will obviously do much worse than structure-based methods. Even when homologues are found, sequence-based methods make a potentially dangerous assumption that the structure of the matching homologue is always similar to the query protein. This is not always true. In NMR it is certainly possible to have identical sequences but completely different chemical shifts (i.e. folded and unfolded versions of the same protein). In these (rare) situations sequence-based methods cannot distinguish whether the folded or unfolded form is correct. Likewise, sequence-based methods are not sensitive to subtle conformational changes arising from mutations, deletions, structure refinement or the existence of “excited” states that are conformationally different from the database’s homologues. On the other hand, structure-based methods are not limited by these kinds of constraints. Therefore, by intelligently combining structure-based methods (SHIFTX+) with sequence based methods (SHIFTY+) we should be able to exploit the high prediction accuracy of sequence-based methods with the broad prediction coverage of structure-based methods.
To properly combine output from SHIFTX+ and SHIFTY+, we compared their relative performance using various sequence identity cut-offs. It was determined that using a 40% (or above) sequence identity cut-off for SHIFTY+ consistently generated more accurate predictions than SHIFTX+. Therefore in the combined SHIFTX2 program, any SHIFTY+ prediction derived from a homologue having >40% sequence identity is combined with any shift predictions from SHIFTX+. Below this sequence cutoff, no SHIFTY+ data is used in making a chemical shift prediction. SHIFTX2 combines the predictions of SHIFTX+ and SHIFTY+ according to the magnitude of the atom-by-atom difference between their predictions. When the difference is sufficiently small, SHIFTY+ overrules SHIFTX+; otherwise the predictions are combined in a simple linear fashion with increasing weight for SHIFTX+ predictions as the difference grows. This combination rule is given by the following equations:
where \( \sigma_{\Updelta \delta } \) is the standard deviation (calculated using the SHIFTX+ training dataset) of the observed secondary chemical shift for a given atom type; d represents the difference between SHIFTX+ and SHIFTY+ predictions versus the standard deviation; and SDmin and SDmax are two parameters controlling the weight w we assign to the SHIFTX+ predictions. We experimented with various values of SDmin and SDmax ranging from 0.5 to 5 in increments of 0.5. From these tests we found that the best prediction results were achieved with SDmin = 0.5 and SDmax = 1.5. The resulting blended program (SHIFTX2) is able to function much like a structure-based chemical shift predictor. Hence when a protein structure is completely unfolded, SHIFTX2 biases itself towards SHIFTX+ predictions (large differences between SHIFTX+ and SHIFTY+ predictions); whereas when the protein is near its native structure, SHIFTX2 biases itself towards using SHIFTY+ predictions (small differences between SHIFTX+ and SHIFTY+ predictions).
SHIFTX2 was written in C, Java and Python is available as a standalone program, as an online web server and as a VMWare version. All of these versions are available at http://www.shiftx2.ca. SHIFTX2 has been compiled and tested on Ubuntu Linux 10.04LTS; however, if properly configured, the SHIFTX2 program should run under most UNIX-like environments including Debian/GNU and Mandriva Linux, openSUSE, OpenSolaris, OpenBSD and Mac OS X. Despite having many more computationally intensive components than the original SHIFTY or SHIFTX programs, a number of code optimizations were also implemented to make SHIFTX2 sufficiently fast so that it could be used in chemical shift refinement or incorporated into chemical-shift-based structure generation programs such as CS23D (Wishart et al. 2008), CSRosetta (Shen et al. 2008) or GeNMR (Berjanskii et al. 2009) without any loss in speed.
Results and discussion
Assessment criteria
To fully assess SHIFTX2, we initially studied the performance of each of its component programs (SHIFTX+ and SHIFTY+). First, we evaluated SHIFTX+ on its training (197 proteins) dataset using tenfold cross validation. This was done to test the general robustness of the predictor. Second, we evaluated SHIFTX+ on a separate testing (61 proteins) dataset. This was done to obtain an independent measure of SHIFTX+’s performance. Third we evaluated SHIFTY+ on the combined training/testing dataset (235 unique proteins) by excluding any exact database matches from the SHIFTY+ predictions. The exclusion of exact database matches was done to avoid predicting chemical shifts for proteins that had already been assigned and to simulate more realistic prediction scenarios. These results were used to assess SHIFTY+’s performance relative to SHIFTX+ and to get an estimate of its coverage (i.e. rate of prediction). Fourth, we assessed SHIFTY+ on the full set of proteins in RefDB (1,903 proteins) to obtain a more precise estimate of SHIFTY+’s expected coverage or probability of prediction for any new query protein.
After obtaining estimates of the performance and coverage of the component programs we then evaluated the performance of the combined program—SHIFTX2. This assessment involved comparing the performance of SHIFTX2 to its component parts (SHIFTX+ and SHIFTY+) and to other state-of-the-art protein chemical shift predictors (SHIFTX, CamShift, SPARTA, PROSHIFT, SHIFTS, SPARTA +) using our independent test set of 61 proteins. All seven programs were evaluated on the basis of: (1) their correlation coefficients (between observed and predicted shifts); (2) their root mean square deviation (RMSD) or RMS error; (3) their coverage (proportion of proteins or residues in the test set that were predicted); (4) their comprehensiveness (number of atoms or atom types predicted); and (5) their speed (CPU seconds or processing time to return an answer).
Component testing
Table S4 (also available on the SHIFTX2 website) shows the correlation coefficients and RMSDs of the backbone chemical shifts achieved for SHIFTX+ both for the training dataset and the testing dataset. As noted earlier, the training dataset performance was assessed using cross-validation. Cross-validation is a standard method in machine learning for evaluating almost any prediction model. In tenfold cross validation, 10% of data is randomly extracted to test a model from the training set, the algorithm is trained on the remaining 90% data and then evaluated on the test set. This process is repeated ten times and the results are averaged. If the algorithm has not been over-trained, the performance for the tenfold cross validation should match closely with performance on the independent test set. As seen in Table S4, this is indeed the case. This result certainly gives us a high level of confidence that the SHIFTX+ algorithm is robust and that the regression model has not been over-trained. Overall, SHIFTX+ is able to attain correlation coefficients (R) of 0.9149, 0.9842, 0.9970, 0.8939, 0.8103, and 0.9226 for 15N, 13Cα, 13Cβ, 13C′, 1HN, 1Hα shifts with corresponding RMS errors of 2.2878, 0.8743, 1.0099, 0.9945, 0.4356, and 0.2152 ppm, respectively. Table S5 (also available on the SHIFTX2 website) shows SHIFTX+’s prediction accuracy for side chain atoms. The correlation coefficients between SHIFTX+’s predicted and observed side chain chemical shifts are 0.9769 (13C) and 0.9321 (1H) with RMS errors of 0.9903 and 0.2238 ppm, respectively.
Because SHIFTY+ is not based on machine learning techniques but on sequence alignment, its performance can be assessed much more simply. Table S6 (see the SHIFTX2 website) provides the prediction accuracy data for SHIFTY+ for the 235 non-redundant proteins in the training and testing datasets. As noted before, exact matches of the database proteins to the query protein were excluded from the performance calculations to simulate more realistic prediction scenarios. This “forced” SHIFTY+ to predict shifts using only homologous proteins or protein fragments. Using a sequence identity cutoff of 40%, we found that up to 74.5% (175/235) of the proteins could have at least one class of chemical shifts predicted by SHIFTY+. Because there is considerable variability in the type and number of protein assignments deposited in chemical shifts databases (some report on 1H shifts, others report only 13C shifts and still others report all shifts), there will naturally be some variability in the chemical shift coverage that SHIFTY+ can achieve. In particular, SHIFTY+’s coverage ranged from a low of 38% (for HE3) to a high of 74% (for 1HN), with an average of 57% over all atom types. This means that SHIFTY+ was able to generate nearly complete assignments for about 57% of the query proteins or, alternately, that SHIFTY+ predicted shifts for 57% of the residues it processed. For those chemical shifts that SHIFTY+ did predict in the 235 protein testing/training set, it achieved correlation coefficients between predicted and observed backbone chemical shifts of 0.9800, 0.9925, 0.9991, 0.9638, 0.9610, and 0.9677 for 15N, 13Cα, 13Cβ, 13C′, 1HN, 1Hα atoms with corresponding RMS errors of 1.1352, 0.6127, 0.5562, 0.5784, 0.2097, and 0.1411 ppm, respectively. The correlation coefficient between SHIFTY+’s predicted and observed side chain proton chemical shifts was 0.9628 (1H) with and RMS error of 0.1393 ppm. The performance for SHIFTY+ was slightly better for the 61 protein testing dataset (for which it predicted shifts for 46 proteins). In particular, SHIFTY+ achieved correlation coefficients between predicted and observed backbone chemical shifts of 0.9974, 0.9991, 0.9999, 0.9961, 0.9964, and 0.9882 for 15N, 13Cα, 13Cβ, 13C′, 1HN, 1Hα atoms with corresponding RMS errors of 0.4115, 0.2087, 0.2136, 0.1847, 0.0630, and 0.0845 ppm, respectively. While the coverage of SHIFTY+ is certainly not as comprehensive as SHIFTX+, it is clear that for the ~57% of residues it could predict, SHIFTY+ is somewhat more accurate.
Expanding the SHIFTY+ testing dataset to include all 1903 proteins in the RefDB/BMRB database revealed that very similar levels of coverage and accuracy could be obtained. In particular a total of 1,270 out of 1,903 proteins (66.7%) could have at least one class of backbone and/or side chain chemical shifts predicted by SHIFTY+. Averaged over all atom types, SHIFTY+ achieved a residue coverage of 55%. In terms of protein coverage (76% vs. 67%) or residue coverage (57% vs. 55%) these numbers are almost identical to those found with the smaller (235 protein) testing/training set. Likewise, as seen in Table S7, the correlation coefficients and RMS errors for the backbone and side-chain shifts are essentially identical to those seen in Table S6. These data suggest that sequence-based methods should routinely work about 70% of the time for any new query protein. Assessing SHIFTY+’s performance with different sizes of the RefDB showed a clear correlation between the size of the reference database and the level of coverage as well as the quality of the predictions (see Table S8 and the SHIFTX2 website for more details). Based on the size and current growth rate of the BMRB and RefDB (about 300 proteins/year) we expect that the proportion of proteins predictable by SHIFTY+ should climb at a rate of about 3–5% per year. This coverage projection was calculated by fitting the data in Table S8 to the following equation: Coverage = 0.84 − 390/NRefDB + 45,000/(N 2RefDB ) where NRefDB is the number of proteins in RefDB.
Comparative performance of SHIFTX2
To evaluate the performance of SHIFTX2 relative to its two component programs (SHIFTX+ and SHIFTY+) we used all three programs to calculate correlation coefficients and RMS errors for both the backbone and side chain chemical shifts on the full testing dataset of 61 proteins. The results are summarized in Fig. 2 and Table S9 (see the SHIFTX2 website). From these data it is clear that SHIFTX2 achieves higher correlation coefficients and lower RMS errors than SHIFTX+. In fact, for the complete set of 235 proteins, SHIFTX2 achieves correlation coefficients of 0.9800, 0.9959, 0.9992, 0.9676, 0.9714, and 0.9744 for 15N, 13Cα, 13Cβ, 13C′, 1HN, and 1Hα shifts with RMS errors of 1.1169, 0.4412, 0.5163, 0.5330, 0.1711, and 0.1231 ppm, respectively.

The backbone chemical shift prediction performance of SHIFTX+, SHIFTY+, and SHIFTX2 as evaluated on a test of 61 protein structures using correlation coefficients (a) and RMS error (b). The statistics for SHIFTY+ were calculated using 46, 46, 44, 39, 46, and 39 homologous proteins for 15N, 13Cα, 13Cβ, 13C′, 1HN, and 1Hα shifts, respectively
Relative to SHIFTX+, SHIFTX2 routinely performs about 6% better (as measured by correlation coefficients), with the highest performance gain being seen for amide 1HN shifts (17.8%). For those proteins (~46) where SHIFTY+ is able to make predictions, the performance of SHIFTY+ and SHIFTX2 is identical. However, when the performance of SHIFTX2 for the complete set of 61 proteins is compared to the performance of SHIFTY+ for its partial set of 46 proteins, SHIFTX2 performs only slightly worse (~1.3% as measured by average correlation coefficient). On the other hand, SHIFTX2’s coverage (percentage of proteins or residues predicted) is more than 24% greater than SHIFTY+’s coverage. These data clearly show that SHIFTX2 is superior to both SHIFTX+ and SHIFTY+.
To compare the performance of SHIFTX2 with other state-of-the-art shift predictors, we ran our test dataset of 61 proteins on six publicly available chemical shift prediction programs or web servers, including SHIFTS, SHIFTX, PROSHIFT, CamShift, SPARTA and SPARTA+. All seven programs were evaluated on the basis of: (1) their correlation coefficients (between observed and predicted shifts); (2) their root mean square deviation (RMSD); (3) their coverage (proportion of proteins or residues in the test set that were predicted); (4) their comprehensiveness (number of atoms or atom types predicted); and (5) their speed (CPU seconds or processing time to return an answer).
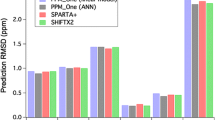
The performance (correlation coefficient and RMSD) of all seven chemical shift predictors for backbone chemical shifts is shown in Fig. 3 and Table 1. To simplify the comparisons between programs, the 1Hα shifts of glycine were averaged (both predicted and observed) and incorporated into the 1Hα evaluation. Based on these performance assessments, the programs appear to fall into three categories. SHIFTS and PROSHIFT form one group, SPARTA, SPARTA+, CAMSHIFT and SHIFTX form another group and SHIFTX2 seems to stand on its own. Overall, SHIFTX2 is substantially more accurate (up to 26% better by correlation coefficient with an RMS error that is up to 3.3× smaller) than the next best performing program. SHIFTX2 appears to be particularly good at predicting proton and nitrogen chemical shifts. This may be due to its use of sequence-based prediction methods and its integration of more detailed descriptors or features associated with hydrogen bond geometry. For those proteins in the test set (~75%) that had sequence homologues in RefDB, SHIFTX2 did somewhat better (~5% in terms of correlation coefficient) than for those that didn’t have homologues. Given this performance differential, SHIFTX2 “flags” those proteins for which it has identified a sequence homologue so that users can easily differentiate chemical shift predictions that will be slightly better than average. Nevertheless, as we showed earlier, this overall performance appears to accurately reflect the “average” result for a SHIFTX2 prediction as the average query will have ~70% probability of being homologous to at least one protein in the RefDB database. Indeed it might be argued that these numbers underestimate the true performance of SHIFTX2 because a significant number of proteins that are being solved today are identical or highly homologous to previously solved proteins.

Bar graphs showing the correlation coefficients (a) and RMSD (b) between the observed and predicted backbone chemical shifts as measured for seven different chemical shift prediction programs using a standard test set of 61 proteins
While most state-of-the-art protein chemical shift predictors predict backbone chemical shifts, only four attempt to predict a subset of side chain shifts (SHIFTX, SHIFTX2, SHIFTS and PROSHIFT) and only two attempt to predict all possible side chain shifts (SHIFTX2 and PROSHIFT). Given the enormous amount of structural information contained in side chain chemical shifts (especially with respect to the influence of ring current effects and other long-range effects) it is surprising that more effort is not directed towards studying this class of chemical shifts. Indeed, ignoring side chain chemical shifts for proteins is a bit like ignoring side chain NOEs. Certainly most protein structures could not be solved or at least solved accurately without the inclusion of side chain NOEs. Similarly any effort directed at refining or solving protein structures using only backbone chemical shifts would no doubt lead to somewhat middling or ambiguous results.
Table 2 presents the correlation coefficients and RMS errors for the complete set of 27 measurable (1H and 13C) side chain chemical shifts as well as the two 1Hα shifts for glycine calculated via PROSHIFT, SHIFTS, SHIFTX and SHIFTX2. The average correlation coefficient for 13C side chain chemical shifts calculated via SHIFTX2 is 0.9783, versus 0.9560 via PROSHIFT. More importantly, the average RMS error for 13C side chain chemical shifts calculated via SHIFTX2 is just 0.9614 ppm, versus 2.6308 ppm via PROSHIFT. Likewise the average correlation coefficient for 1H side chain chemical shifts calculated via SHIFTX2 is 0.9504, versus 0.8785 (PROSHIFT), 0.8786 (SHIFTX), or 0.8602 (SHIFTS—excluding the 18% of shifts that SHIFTS could not predict). Based on these numbers it is clear that SHIFTX2 is between 2 and 9% better (by correlation coefficient) and between 1.6× and 2.7× better (by RMS error) than SHIFTX, SHIFTS or PROSHIFT.
In addition to comparing or assessing the accuracy (via correlation and RMSD) of these different chemical shift predictors, it is also important to assess their coverage (proportion of proteins or residues that could be predicted), their comprehensiveness (number of atoms or atom types predicted) and their speed (CPU seconds or processing time to return an answer). Somewhat surprisingly we found that a number of popular programs were unable to make predictions for a significant number of residues or protein structures (Table 3). For example, SHIFTS typically makes no predictions for about 10% of backbone 1H atoms and 18% of side chain 1H atoms. CamShift makes no predictions for about 5% of backbone atoms while SPARTA and SPARTA+ make no predictions for about 2 and 0.03% of backbone atoms, respectively. Given the variability in PDB file structures and the difficulty in writing robust PDB file parsers, a small percentage of file reading errors is not entirely unexpected. In other cases, it appears that the programs were specifically designed to ignore certain residues or atom types. Table 3 describes the chemical shift coverage, both in terms of the number of shifts and the number of proteins that could be analyzed by each of the seven programs used in this study. As seen in this table, only SHIFTX and SHIFTX2 achieve near 100% coverage. Note that for the performance comparisons given in Tables 1 and 2, we used only the atoms, residues and/or proteins in the 61-protein test set where all seven programs were able to calculate a chemical shift. Certainly if the unpredicted (i.e. null) shifts were included in the calculations shown in Tables 1 and 2 then the relative performance of SHIFTX2 against most other programs would be somewhat better than reported.
Table 4 summarizes the comprehensiveness (number of atom types predicted) and the computational speed (limited to backbone shifts) of each of the seven different chemical shift predictors. In terms of comprehensiveness, only SHIFTX2 and PROSHIFT provide complete coverage (all 40 atom types). SHIFTS and SHIFTX provide coverage for 78 and 68% (respectively) of all atom types while SPARTA/SPARTA+ and CamShift provide coverage for only 15% of all atom types.
In terms of computational speed, there is obviously considerable variability among the seven programs. SPARTA appears to be the slowest program, with an average speed of 17.92 s per 100 residues. PROSHIFT is the next slowest (12.87 s per 100 residues) while SHIFTS is approximately four times faster with an average speed of 3.66 s per 100 residues. The fastest program is SHIFTX, which averages 0.59 s per 100 residues. Of the seven programs, SHIFTX2 appears to be the third fastest program with an average speed of 2.10 s per 100 residues. All of the computational speed tests for SPARTA, SPARTA+, SHIFTS, CamShift, SHIFTX and SHIFTX2 were performed on the same computer (an Intel Core™2 Duo CPU 1.83 GHz processor with 1.6 GB RAM) using the same set of proteins. The calculation speed reported for PROSHIFT is based on the response rate of its web server. Without knowing the architecture of the PROSHIFT server it is difficult to know whether PROSHIFT numbers are comparable to the values generated on our test CPU processor.
Influence of different parameters on different chemical shifts
One of the goals of this study was to identify which protein structural parameters appear to most significantly influence the chemical shifts seen for specific nuclei. Our earlier work in 2003 identified a number of structural factors for 1H, 13C and 15N backbone shifts and ranked them in a qualitative fashion (Wishart et al. 1997). This early study highlighted the importance of ring currents in determining 1H shifts and the influence of nearest-neighbour interactions in determining 15N shifts. Since this study was first published a number of other structural parameters pertaining to torsion angles and hydrogen bond geometry (kappa and theta angles) have been determined to influence chemical shifts. To quantify the impact of these and other factors in our structure-based model of chemical shifts we conducted a simple leave-one-out feature analysis. Specifically, by removing a single feature at a time from the SHIFTX+ model and quantifying the increase that this missing feature brings to the predictor’s RMS error it is possible to estimate the importance of this feature to the predictor. To get a more robust, quantitative assessment of the impact of each feature, we then averaged the RMS change using tenfold cross validation. Table 5 lists the top 20 most influential features for each backbone nucleus. Table S10 (see the SHIFTX2 website) provides the weighting for all features for each backbone nucleus. As might be expected, the most influential features for proton chemical shifts were found to be backbone torsion angles, ring currents, electric field effects and hydrogen bonding effects while for carbon and nitrogen shifts, the most influential features are the backbone and side chain dihedral angles.
Assessing SHIFTX2 for chemical shift refinement
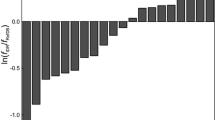
One of the main drivers for developing better protein chemical shift predictors has been the hope that they could be routinely used in protein structure determination and protein structure refinement. Obviously, the faster and more accurate a chemical shift predictor can be, the better it would be at refining or defining protein structures. To evaluate SHIFTX2 in the context of protein structure refinement, we simulated the structure refinement process by generating 15 randomly perturbed structures for ubiquitin (PDB entry 1UBQ) by progressively altering the backbone ϕ/ψ angles of the native protein. This led to the creation of 16 different ubiquitin-like structures ranging from a completely unfolded structure (structure #1, with an RMSD between the native structure of 68.9 Angstroms) to the properly folded native structure (structure #16, with an RMSD of 0 Angstroms). We then predicted the backbone chemical shifts using SHIFTX+, SHIFTY+ and SHIFTX2 for each of the structures and calculated their average correlation coefficient with the observed chemical shifts (BMRB 5387). The result is illustrated in Fig. 4, with the protein structures progressively arrayed from the most dissimilar (most unfolded) on the left to the most similar (or native-like) on the right. This figure illustrates three important points. First, it can be seen that the sequence-based predictor (SHIFTY+) is not sensitive to conformational changes while the structure-based predictor (SHIFTX+) clearly is. Second, by combining the two predictors it is possible to get a single predictor that is both sensitive to conformational changes and actually more accurate than any of the constituent predictors. Third, it is also evident that as the RMSD between each of the models and the actual structure gets smaller, the correlation coefficient progressively (and smoothly) climbs higher. This “smoothness” certainly suggests that chemical shifts have the appropriate characteristics (i.e. the sensitivity to both gross and subtle conformational changes) to be used in refining and even determining protein structures. As expected when the protein structure changes from the completely unfolded state to the properly folded state, SHIFTX2 achieves increasingly higher correlation coefficient (and low RMSD) with the observed chemical shifts. This is because SHIFTX2 reflects characteristics of the structure (via SHIFTX+) and characteristics of the sequence (via SHIFTY+).

A plot illustrating the change in the combined (backbone + sidechain) 1H chemical shift correlation coefficient (predicted vs. observed) relative to the similarity (measured by RMSD) of ubiquitin to its native state. This graph illustrates the correlation coefficients calculated via SHIFTX + , SHIFTY+ and SHIFTX2 using 16 different 1UBQ structures (15 randomized and 1 native structure). Sample structures are shown below the RMSD axis to illustrate how the RMSD values relate to observable structural changes. Note that SHIFTY+ is not sensitive to the structure changes and so it is not useful (on its own) for chemical shift refinement
Caveats and limitations
While we have presented a substantial body of data showing that SHIFTX2 has achieved a significant improvement in protein chemical shift prediction accuracy, it is important to be aware of its limitations. In particular, it is essential to remember that the high correlation coefficients and low RMS errors reported here will typically be better (1–2%) than what one will get using an “average” protein. This is because the test set of 61 proteins used to assess SHIFTX2’s (and all of the other predictors’) performance was specially selected for their exceptionally high resolution and high quality. If one were to choose lower quality structures (low resolution X-ray or NMR) then the agreement between observed and predicted shifts would obviously be lower—regardless of which program is chosen. Chemical shifts are exquisitely sensitive to small coordinate errors or small coordinate displacements (Iwadate et al. 1999; Kohlhoff et al. 2009; Meiler 2003; Moon and Case 2007; Neal et al. 2003; Shen and Bax 2007, 2010; Vila et al. 2009; Xu and Case 2001). Therefore any errors or lack of precision in coordinate data will always be reflected in any set of predicted chemical shifts. In other words, “garbage in = garbage out”. For instance, if one were to use a low resolution or a poor quality structure to attempt to predict chemical shifts for assignment purposes, then it is likely that a number of assignment errors will ensue. On the other hand, if one finds that the calculated shifts for a given structure disagree with the observed shifts by more than what is quoted in Tables 1 or 2, then this is likely a good indication that the structure is in need of further refinement. As shown in Fig. 4, and as advocated in many other recent publications (Kohlhoff et al. 2009; Meiler 2003; Neal et al. 2003), using chemical shifts to assist with the structure refinement process would certainly help improve the quality of many NMR-generated structures.
It is also important to remember that most protein chemical shift predictors are designed to predict chemical shifts of diamagnetic proteins in aqueous conditions at moderate temperatures and at moderate pH values. Therefore, attempting to use SHIFTX2 (or most other programs) on paramagnetic proteins or on proteins dissolved in non-aqueous buffers or at extreme temperatures or at extremes of pH will likely lead to poor results. While SHIFTX2 can be used to calculate chemical shifts of protein–protein complexes, it is not capable of accurately predicting shifts of amino acid residues in close proximity to DNA, RNA or certain small molecule co-factors (heme rings, NAD, FAD, etc.). This is because the characteristic ring current and charge models for these non-proteinaceous molecules are not included in the current SHIFTX2 model.
Conclusion
In this report we have described SHIFTX2, a novel, hybrid chemical shift predictor that is capable of rapidly and accurately calculating diamagnetic 1H, 13C and 15N chemical shifts from protein coordinate data. Comparison’s of SHIFTX2 against many state-of-the art predictors clearly show that the program is substantially more accurate (up to 26% better by correlation coefficient with an RMS error that is up to 3.3× smaller) than the next best performing program. It also provides significantly more coverage (up to 10% more), is significantly faster (up to 8.5×) and capable of calculating a wider variety of backbone and side chain chemical shifts (up to 6×) than many other shift predictors. We were able to achieve this high level of performance by carefully training and testing each of SHIFTX2’s component programs (SHIFTY+ and SHIFTX+) on a set of large and very accurate databases. By utilizing advanced machine learning techniques and by incorporating many more features in our machine learning model we were able to substantially improve SHIFTX2’s structure-based predictor (SHIFTX+). By carefully preparing a large reference sequence/shift database (RefDB) and enhancing the sequence alignment algorithm we were also able to substantially improve SHIFTX2’s sequence-based predictor (SHIFTY+). By combining the results of these two programs using an automated differential weighting scheme we were able to get the benefits of both shift prediction techniques.
While the results we have obtained with SHIFTX2 are impressive and the improvements over existing methods are significant, it is likely that the predictive performance of protein chemical shift predictors is now nearing its limit. No doubt as databases continue to expand and as more methods are intelligently combined, it may be possible to improve shift prediction accuracy by another 1 or 2%. However, once this level is reached, the inherent imprecision of atomic coordinates and the inherent conformational differences between proteins in the solid state (crystals) versus those in solution will probably become the largest contributors to any observed chemical shift discrepancies. In other words, it will be impossible to get perfect chemical shift predictions. Perhaps the only way to get around this “atomic precision” barrier may be to start including conformational ensembles determined from molecular dynamic simulations or generated via chemical shift refinement (Lehtivarjo et al. 2009; Markwick et al. 2010). Certainly a number of recent studies have suggested that chemical shifts calculated over carefully weighted ensembles of protein structures appear to give better agreement to observed shifts than those generated from just a single protein structure.
Despite these caveats, we believe that SHIFTX2, with is high level of accuracy and broad chemical shift coverage, should open the door to many long-anticipated applications of chemical shift prediction. Indeed SHIFTX2 should be particularly useful in refining and assessing protein structures, validating and adjusting chemical shift assignments, and ultimately, for generating protein structures using only chemical shift data alone.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Andrec M, Snyder DA, Zhou Z, Young J, Montelione GT, Levy RM (2007) A large data set comparison of protein structures determined by crystallography and NMR: statistical test for structural differences and the effect of crystal packing. Proteins 69:449–465
Berjanskii MV, Wishart DS (2005) A simple method to predict protein flexibility using secondary chemical shifts. J Am Chem Soc 127:14970–14971
Berjanskii M, Tang P, Liang J, Cruz JA, Zhou J, Zhou Y, Bassett E, MacDonell C, Lu P, Lin G, Wishart DS (2009) GeNMR: a web server for rapid NMR-bsed protein structure determination. Nucleic Acids Res 37:W670–W677
Berjanskii M, Liang Y, Zhou J, Tang P, Stothard P, Zhou Y, Cruz J, MacDonell C, Lin G, Lu P, Wishart DS (2010) PROSESS: a protein structure evaluation suite and server. Nucleic Acids Res 38:W633–W640
Breiman L (1996) Bagging predictors. Mach Learn 24:123–140
Frank E (2004) Data mining in bioinformatics using Weka. Bioinformatics 20:2479–2481
Freund Y, Schapire RE (1997) A decision-theoretic generalization of on-line learning and an application to boosting. J Comput Syst Sci 55:119–139
Ginzinger SW, Skočibušić M, Heun V (2009) CheckShift improved: fast chemical shift reference correction with high accuracy. J Biomol NMR 44:207–211
Ginzinger SW, Weichenberger CX, Sippl MJ (2010) Detection of unrealistic molecular environments in protein structures based on expected electron densities. J Biomol NMR 47:33–40
Gronwald W, Willard L, Jellard T, Boyko RF, Rajarathnam K, Wishart DS, Sönnichsen FD, Sykes BD (1998) CAMRA: chemical shift based computer aided protein NMR assignments. J Biomol NMR 12:395–405
Hall M, Frank E, Holmes G, Pfahringer B, Reutemann P, Witten IH (2009) The WEKA data mining software: an update. SIGKDD Explor 11:10–18
Hooft RWW, Vriend G, Sander C, Abola EE (1996) Errors in protein structures. Nature 381:272
Iwadate M, Asakura T, Williamson MP (1999) Cα and Cβ carbon-13 chemical shifts in proteins from an empirical database. J Biomol NMR 13:199–211
Kohlhoff KJ, Robustelli P, Cavalli A, Salvatella X, Vendruscolo M (2009) Fast and accurate predictions of protein NMR chemical shifts from interatomic distances. J Am Chem Soc 131:13894–13895
Kotsiantis SB (2007) Combining bagging and additive regression. Int J Comput Math Sci 1:61–67
Laskowski RA, Rullmann JAC, MacArthur MW, Kaptein R, Thornton JM (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR 8:477–486
Lehtivarjo J, Hassinen T, Korhonen SP, Peräkylä M, Laatikainen R (2009) 4D prediction of protein 1H chemical shifts. J Biomol NMR 45:413–426
Markwick PR, Cervantes CF, Abel BL, Komives EA, Blackledge M, McCammon JA (2010) Enhanced conformational space sampling improves the prediction of chemical shifts in proteins. J Am Chem Soc 132:1220–1221
Meiler J (2003) PROSHIFT: protein chemical shift prediction using artificial neural networks. J Biomol NMR 26:25–37
Moon S, Case DA (2007) A new model for chemical shifts of amide hydrogens in proteins. J Biomol NMR 38:139–150
Neal S, Nip AM, Zhang H, Wishart DS (2003) Rapid and accurate calculation of protein 1H, 13C and 15 N chemical shifts. J Biomol NMR 26:215–240
Opitz D, Maclin R (1999) Popular ensemble methods: an empirical study. J Artif Intell Res 11:169–198
Osapay K, Case DA (1991) A new analysis of proton chemical shifts in proteins. J Am Chem Soc 113:9436–9444
Pastore A, Saudek V (1990) The relationship between chemical shift and secondary structure in proteins. J Magn Reson 90:165–176
Perkins SJ, Dwek RA (1980) Comparisons of ring-current shifts calculated from the crystal structure of egg white lysozyme of hen with the proton nuclear magnetic resonance spectrum of lysozyme in solution. Biochemistry 19:245–258
Potts BCM, Chazin WJ (1998) Chemical shift homology in proteins. J Biomol NMR 11:45–57
Schapire RE (1990) The strength of weak learnability. Mach Learn 5:197–227
Seavey BR, Farr EA, Westler WM, Markley JL (1991) A relational database for sequence-specific protein NMR data. J Biomol NMR 1:217–236
Sharma D, Rajarathnam K (2000) C-13 NMR chemical shifts can predict disulfide bond formation. J Biomol NMR 18:165–171
Shen Y, Bax A (2007) Protein backbone chemical shifts predicted from searching a database for torsion angle and sequence homology. J Biomol NMR 38:289–302
Shen Y, Bax A (2010) SPARTA+: a modest improvement in empirical NMR chemical shift prediction by means of an artificial neural network. J Biomol NMR 48:13–22
Shen Y, Lange O, Delaglio F, Rossi P, Aramini JM, Liu G, Eletsky A, Wu Y, Singarapu KK, Lemak A, Ignatchenko A, Arrowsmith CH, Szyperski T, Montelione GT, Baker D, Bax A (2008) Consistent blind protein structure generation from NMR chemical shift data. Proc Natl Acad Sci U S A 105:4685–4690
Spera S, Bax A (1991) Empirical correlation between protein backbone conformation and C-alpha and C-beta 13C nuclear magnetic resonance chemical shifts. J Am Chem Soc 113:5490–5492
Sternlicht H, Wilson D (1967) Magnetic resonance studies of macromolecules. I. Aromatic-methyl interactions and helical structure effects in lysozyme. Biochemistry 6:2881–2892
Vila JA, Arnautova YA, Martin OA, Scheraga HA (2009) Quantum-mechanics-derived 13C chemical shift server (CheShift) for protein structure validation. Proc Natl Acad Sci USA 106:16972–16977
Vranken WF, Rieping W (2009) Relationship between chemical shift value and accessible surface area for all amino acid atoms. BMC Struct Biol 9:20
Wang B, Wang Y, Wishart DS (2010) A probabilistic approach for validating protein NMR chemical shift assignments. J Biomol NMR 47:85–99
Wiederstein M, Sippl MJ (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 35:W407–W410
Willard L, Ranjan A, Zhang H, Monzai H, Boyko RF, Sykes BD, Wishart DS (2003) VADAR: a web server for quantitative evaluation of protein structure quality. Nucleic Acids Res 31:3316–3319
Williamson MP (1990) Secondary-structure dependent chemical shifts in proteins. Biopolymers 29:1428–1431
Wishart DS, Nip AM (1998) Protein chemical shift analysis: a practical guide. Biochem Cell Biol 76:153–163
Wishart DS, Sykes BD, Richards FM (1991) Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J Mol Biol 222:311–333
Wishart DS, Bigam CG, Holm A, Hodges RS, Sykes BD (1995a) H-1, C-13 and N-15 random coil NMR chemical shifts of the common amino acids. 1. Investigations of nearest-neighbor effects. J Biomol NMR 5:67–81
Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD (1995b) 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J Biomol NMR 6:135–140
Wishart DS, Watson MS, Boyko RF, Sykes BD (1997) Automated 1H and 13C chemical shift prediction using the BioMagResBank. J Biomol NMR 10:329–336
Wishart DS, Arndt D, Berjanskii M, Tang P, Zhou J, Lin G (2008) CS23D: web server for rapid protein structure generation using NMR chemical shifts and sequence data. Nucleic Acids Res 36:W496–W502
Word JM, Lovell SC, Richardson JS, Richardson DC (1999) Asparagine and glutamine: using hydrogen atom contacts in the choice of sidechain amide orientation. J Mol Biol 285:1735–1747
Xu XP, Case DA (2001) Automated prediction of 15 N, 13Cα, 13Cβ and 13C′ chemical shifts in proteins using a density functional database. J Biomol NMR 21:321–333
Zhang H, Neal S, Wishart DS (2003) RefDB: a database of uniformly referenced protein chemical shifts. J Biomol NMR 25:173–195
Acknowledgments
The authors would like to thank the Alberta Prion Research Institute (APRI), PrioNet, the Natural Sciences and Engineering Research Council (NSERC), Genome Canada and the Austrian Science Fund (FWF), grant P21294-B12 for financial support.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Han, B., Liu, Y., Ginzinger, S.W. et al. SHIFTX2: significantly improved protein chemical shift prediction. J Biomol NMR 50, 43–57 (2011). https://doi.org/10.1007/s10858-011-9478-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-011-9478-4